
Abstract
Patient diagnosis and care would be significantly improved by understanding the mechanisms underlying platinum and taxane resistance in ovarian cancer. Here, we aim to establish a gene signature that can identify molecular pathways/transcription factors involved in ovarian cancer progression, poor clinical outcome, and chemotherapy resistance. To validate the robustness of the gene signature, a meta-analysis approach was applied to 1,020 patients from 7 datasets. A 97-gene signature was identified as an independent predictor of patient survival in association with other clinicopathological factors in univariate [hazard ratio (HR): 3.0, 95% Confidence Interval (CI) 1.66–5.44, p = 2.7E-4] and multivariate [HR: 2.88, 95% CI 1.57–5.2, p = 0.001] analyses. Subset analyses demonstrated that the signature could predict patients who would attain complete or partial remission or no-response to first-line chemotherapy. Pathway analyses revealed that the signature was regulated by HIF1α and TP53 and included nine HIF1α-regulated genes, which were highly expressed in non-responders and partial remission patients than in complete remission patients. We present the 97-gene signature as an accurate prognostic predictor of overall survival and chemoresponse. Our signature also provides information on potential candidate target genes for future treatment efforts in ovarian cancer.
Similar content being viewed by others


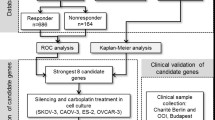
Introduction
Ovarian cancer has the highest mortality rate of all gynecological cancers and the fifth highest mortality rate of all cancers in the world1. Because the early stage of the disease is asymptomatic, ovarian cancer is rarely diagnosed until it reaches a late stage2. Primary debulking surgery followed by first-line chemotherapy (platinum-taxane) is the standard treatment in advanced ovarian cancer3, 4. Approximately 70% of patients achieve complete remission (CR) after the current standard treatment; however, 30% to 40% of these patients relapse within 12 months5, 6. The overall 5-year survival still remains at 30%7, 8, necessitating the need for better prognostic prediction of chemoresponse in ovarian cancer.
Clinicopathological characteristics such as histological grade and the International Federation of Gynecology and Obstetrics (FIGO) staging system are considered as the most important prognostic indicators in ovarian cancer. However, they are insufficient for predicting survival time and chemoresponse9, 10. Currently, there is no fully validated and clinically applicable test to guide treatment decisions in ovarian cancer.
Several gene signatures associated with overall survival and response to chemotherapy have been established in ovarian cancer11,12,13,14,15, but none are in clinical use yet due to discrepancies in technology and disease heterogeneity. Therefore, identification of a prognostic gene signature that captures the key pathways/regulators leading to platinum and taxane resistance in ovarian cancer is critical for prediction of patient outcome and overall survival (OS).
In this study, we established a novel prognostic gene signature to distinguish low- and high-risk patients, using a gene expression profiling technique, by analyzing seven microarray datasets and one RNA-seq dataset. Furthermore, we assessed the association of the identified prognostic gene signature with clinicopathological factors and investigated whether it could predict chemoresponse. Our findings suggest an optimal treatment based on the molecular characteristics of ovarian cancer patients.
Results
Seven microarray datasets (GSE49997, GSE14764, GSE26712, GSE63885, GSE19829, GSE30161, The Cancer Genome Atlas – TCGA) and one TCGA RNA-seq dataset were used in this study. Data selection was based on availability of clinicopathological information such as OS time and event, grade, stage, chemoresponse, and related chip types. Detailed information for these datasets is described in the Materials and Methods (Supplementary Table S1).
The end point of this study was the identification of a gene signature prognostic of overall survival after first-line chemotherapy in both low- and high-risk groups. GSE49997 was used as the training dataset. A supervised method of risk prediction, with a stringent threshold cut off (p < 0.01 and 2-fold difference) was applied to the training dataset. After filtering for probe set intensity, 5,218 genes were analyzed using univariate Cox proportional hazard regression (p < 0.01) and 137 genes associated with OS were identified. However, only 97 genes were measured in the training dataset and all validation datasets (Fig. 1a). The 97-gene signature defined a threshold that split the training dataset into low- and high-risk groups with 91 and 93 patients, respectively. To evaluate the prognosis, Kaplan-Meier survival curves were plotted and the log-rank test showed a significant difference in OS (p = 1.2E-4; Fig. 1b) and progression-free survival (PFS) (p = 0.026; Fig. 1c). The expression pattern of the 97-gene signature divided low- and high-risk patients into two clusters (Fig. 1d).

Training set survival analysis (GSE49997). (a) Schematic overview of the procedure used in the construction of the 97 gene signature based on gene expression data. (b) Kaplan-Meier plots depicting overall survival (OS) into low and high risk groups in the training data set predicted by compound covariate predictor (CCP). (c) Kaplan-Meier plots depicting progression- free survival (PFS) of patients into low and high risk groups in the training data set. The p values were computed by the log-rank test. (d) The heat map of the median centered 97 genes expression profiles (red, relative high expression; blue, relative low expression) between low and high risk groups.
Prognostic significance of the 97-gene signature and clinical relevance in the training dataset
To investigate the prognostic significance of the 97-gene signature in association with other covariates in ovarian cancer, univariate and multivariate Cox regression analyses were performed on variables including age, stage, and grade in the training dataset. Both in univariate and multivariate analyses, the 97-gene signature showed the strongest prognostic ability in association with OS (HR 3.008, 95% Cl 1.6–5.4, p = 2.7E-4 and HR 2.885, 95% CI 1.57–5.27, p = 0.001, respectively; Table 1).
Kaplan-Meier plots were made to show which patients were low- or high-risk, based on grade and FIGO staging system. Grades 1 & 2 were combined as low grade (well differentiated, p = 0.0191; Supplementary Fig. S1a) and grade 3 was considered high grade (poorly differentiated, p = 8.1E-5; Supplementary Fig. S1b). Because ovarian cancer is usually discovered at a late stage, most of the patients in the training dataset were in stage III (p = 0.00185; Supplementary Fig. S1c) and stage IV (p = 0.0191; Supplementary Fig. S1d).
Validation of the 97-gene signature in independent datasets
To further assess outcome prediction, we combined all of the validation datasets (total of 836 patients) to evaluate the robustness of the 97-gene signature. We further validated the prognostic significance of the gene signature in an external TCGA RNA-seq dataset. A flow chart of the procedure used to validate external datasets is depicted in Fig. 2a. Leave-one-out cross-validation (LOOCV) specificity and sensitivity used was 0.972 and 0.932 in Compound Covariate Prediction (CCP), respectively. The gene signature significantly stratified patients in all combined validations into low- and high-risk groups (p = 1.18E-08; Fig. 2b). Most of the independent datasets had a small number of patients. Therefore, to assess the generalizability of our gene signature, validation datasets were merged according to chip type as follows: GSE14764 and GSE26712 as GPL96; GSE63885, GSE19829 and GSE30161 as GPL570; and TCGA GPL96. The merged validation datasets’ Kaplan- Meier plots predicted significant differences in prognosis: GPL96 (p = 8.1E-5; Fig. 2c), GPL570 (p = 0.00478; Fig. 2d), TCGA GPL96 (p = 0.0319; Fig. 2e) and TCGA RNA-seq as an independent validation (p = 0.044; Fig. 2f). Kaplan-Meier graphs for each independent validation dataset demonstrated a decrease in the overall survival of the high-risk group versus the low-risk group, but some datasets were statistically insignificant because of the small number of patients (Supplementary Fig. S3).

The prognostic significance of the 97 gene signature in all combined validation data sets. (a) A schematic overview of the strategy used in the construction of the prediction model and evaluation of predicted outcomes in the merged validation data sets. (b) Overall survival Kaplan-Meier plots stratified patients into low and high risk groups in all combined validation data set. (c) Overall survival Kaplan-Meier curves dichotomized patients into low and high risk groups in GPL96 data set (GSE14764, GSE26712). (d) Kaplan-Meier curves stratified patients in GPL 570 data set into low and high risk groups (GSE 63885, GSE19829, and GSE30161). (e) Overall survival Kaplan-Meier curves of patients in TCGA GPL96 data set classified into low and high risk groups. (f) Kaplan-Meier for overall survival of patients classified into low and high risk groups in TCGA RNA seq data set, predicted by compound covariate predictor (CPP). The p values were computed by log-rank test.
Prognostic subclassification of patients in relation to current clinical covariates
To investigate the prognostic significance of the 97-gene signature in association with stage and grade in all validation datasets, univariate and multivariate Cox regression analyses were performed. In both univariate and multivariate analyses, the 97-gene signature proved to be a stronger predictor of OS than grade and stage (HR 1.778, 95% CI 1.4–2.2, p = 1.2 E-6; and HR 1.749, 95% Cl 1.385–2.209, p = 2.6E-6 respectively; Table 2).
To assess whether the 97-gene signature could stratify patients in the low grade into low- and high-risk groups, patients in grades 1 and 2 were merged because only 5patients were in grade 1. The 97-gene signature significantly stratified patients into low- and high-risk groups (p = 0.00171; Fig. 3a). The gene signature also separated patients in grade 3 into low- and high-risk groups (p = 1.11e-06; Fig. 3b).

Prognostic significance of the 97 gene signature in relation to grade and FIGO stage in the combined validation data set. Patients from all combined validations merged according to grade and stage. Kaplan-Meier plots were predicted by compound covariate predictor (CPP). The p values were computed by the log-rank test. (a) Overall survival Kaplan-Meier plots of patients in low and high risk groups in grade 1 & 2. (b) Kaplan-Meier plots for overall survival of patients in low and high risk groups in grade 3. (c) Overall survival Kaplan-Meier plots showing patients in low and high risk groups in FIGO stage III. (d) Kaplan-Meier plots depicting overall survival of patients into low and high risk groups in FIGO stage IV. FIGO; International Federation of gynecology and obstetrics.
Further, to evaluate whether the 97-gene signature could classify patients in each stage into low- and high-risk groups, all patients from the validation datasets were merged according to stage: I (n = 20), II (n = 27), III (n = 548) and IV (n = 222). The signature clearly stratified patients in stage III (p = 0.000151; Fig. 3c) and stage IV (p = 2.02E-07; Fig. 3d).
Prognostic value of the 97-gene signature in association with chemoresponse
All patients in our study received first-line platinum and taxane combined chemotherapy. Patients were considered to be in complete remission (CR) if all detectable tumors disappeared after first-line chemotherapy, partial remission (PR) if the tumor significantly reduced in volume, and non-responder (NR) or stable disease (SD) if the tumor continued to grow or new tumors appeared. To evaluate whether our gene signature could accurately predict response to first-line chemotherapy, subset analyses were performed on TCGA (n = 326) and GSE30161 (n = 58) datasets, for which chemoresponse information was available. The signature clearly classified low-risk patients into CR and PR groups (p = 9.1E-18, Fig. 4a) and high-risk patients into CR and PR groups (p = 2.04E-11; Fig. 4b). The signature also dichotomized low- and high-risk patients into responders and non-responders (p = 1.12E-05 and p = 0.0309, respectively; Fig. 4c,d). To consolidate these results regarding chemosensitivity, we sorted both CR and PR case groups into low- and high-risk. Our gene signature showed that the low-risk group had better prognosis both in PR (Fig. 4e) and CR (Fig. 4f).

Significant association of the 97 gene signature with combined platinum and taxane based chemotherapy. Patients from the combined validation dataset with available first line chemotherapy information were included for the analysis predicted by compound covariate predictor (CPP). The p values were computed by the log rank test. (a) Overall survival Kaplan-Meier plots for patients in high risk group classified into complete and partial remission groups. (b) Kaplan-Meier overall survival analysis stratified patients in low risk group into complete or partial remission. (c) Overall survival Kaplan-Meier plots classified patients in high risk group into responders and non- responders. (d) Kaplan-Meir survival analyses stratified patients in low risk group into responders and non-responders to first line chemotherapy. (e,f) Kaplan-Meier plots depicting low and high risk in partial and complete remission subgroups, with patients in low risk group showing better prognosis. The p values were computed by log-rank test.
Gene Ontology term enrichment analysis and visualization of the 97-gene signature
We performed Gene Ontology enrichment analysis in DAVID to identify biological functions of the genes in the 97-gene signature and identified 20 significant terms (biological processes), including metastasis, migration, growth factor binding, angiogenesis, regulation of apoptosis, and regulation of epithelial cell proliferation. False discovery rates were estimated using the procedure of Benjamin (p < 0.05, Supplementary Table S2).
Transcription factors and pathways associated with the 97-gene signature
We investigated transcription factors and pathways associated with our gene signature, using Ingenuity Pathway Analysis software. TP53 and HIF1α were found to be the most activated transcription factors. TP53 regulated genes involved in cell proliferation, angiogenesis, and metastasis (Fig. 5), and HIF1α regulated important genes involved in tumor cell growth, angiogenesis, and blood vessel morphology (Fig. 4). Furthermore, we showed that genes from the 97-gene signature that were involved in angiogenesis and metastasis were the most upregulated (Supplementary Fig. S3). We also demonstrated interactions using the STRING database to elaborate physical and functional association amongst the genes in the signature (Supplementary Fig. S4).

Significant association of the 97 gene signature with transcription factors TP53 and HIF1α. IPA analysis of gene networks from the 97 gene signature significantly associated with TP53 and HIF1α pathways. Identified gene networks were ranked to score (Z-score = 02). TP53, p = 0.000267 and HIF1α p = 0.00103. IPA; Ingenuity Pathway Analysis. TP53; Tumor protein p53. HIF1α; Hypoxia-inducible factor 1-alpha.
Significant association of the HIF1α-regulated genes with platinum and taxane
To understand the molecular mechanisms behind the three chemoresponse groups CR, PR, and NR, individual box plots for each of the nine genes were conducted (Fig. 6). HIF1α-regulated genes were found to be highly expressed in NR and PR but less expressed in CR cases.

HIF1α nine genes box plots in CR, PR and NR. Individual box plots depicting expression levels for HIF1α regulated genes in complete remission, partial remission and non-responders groups. The left vertical represents the expression of each gene in SPSS and the p values are indicated on the graphs. CR; Complete Remission. PR; Partial Remission. NR; No-responder. SPSS; Statistical Package for the Social Sciences.
Discussion
Several reports have attempted to show that gene signatures are capable of predicting individual clinical outcomes in ovarian cancer16,17,18,19,20,21,22,23. Prognostic gene signatures based on chemoresistance, molecular subtyping, and debulking tumors in ovarian cancer have been reported by analysis of homogenous datasets24,25,26,27. However, none of these are in clinical use yet because of difficulties caused by ovarian cancer heterogeneity. Hence, precise prognostic prediction for chemoresponse and mortality after first-line chemotherapy remains a critical issue in ovarian cancer treatment. Here, we developed a novel risk classification system based on a 97-gene signature from ovarian cancer patients27. Patients from various study groups were merged in order to get insight into distinctive endpoints of ovarian cancers and to overcome challenges reported by previous studies, such as homogeneity or small patient size. A supervised method was used to construct the gene signature that was then refined by LOOCV. Furthermore, a meta-analysis approach based on six Affymetrix microarray datasets (n = 836) and one RNA-seq dataset (n = 419) was applied to validate the prognostic significance of the gene signature in association with overall survival. The reproducibility of the gene signature was improved using only Affymetrix validation datasets. Univariate and multivariate analyses with adjustment for clinical parameters showed a significant association of our prognostic gene signature with survival rate. The 97-gene signature was also able to predict chemoresponse in both low- and high-risk patients. Moreover, our signature revealed nine HIF1α-regulated genes to be predictive of response to first-line platinum and taxane chemotherapy in ovarian cancer.
In clinical oncology, grade and stage are the current diagnostic criteria. However, the heterogeneity observed in therapeutic response within the same grade or stage indicates that histological classification is not an adequate predictor of survival9. In our study, univariate and multivariate Cox proportional hazards regression analyses of clinical variables based on OS in both the training and validation datasets also indicted that the 97-gene signature was a more reliable prognostic indicator than current clinical variables. Taken together, we think that the integration of the 97-gene signature with grade and stage could be a better approach for determining patient prognosis in clinical practice.
The precise prediction of chemoresponse would greatly improve ovarian cancer patient prognosis by guiding chemotherapy choices. However, it is difficult to determine the response to first-line chemotherapy because of the complexity of ovarian cancer genetic heterogeneity20. Several investigators previously reported gene signatures associated with chemoresponse in ovarian cancer28, 29. However, most of these studies were performed on small numbers of patients and were only prognostic, not predictive, of first-line chemotherapy response. The 97-gene signature could predict patients who would attain complete or partial remission after first-line chemotherapy in both low- and high-risk groups. In addition, the gene signature was also predictive of non-responders (NR) to first-line chemotherapy in both low- and high-risk groups. This predictability of our gene signature could benefit PR and NR patients by directing the choice of effective alternate and/or additional therapeutic options early in the disease.
Some of the genes in the 97-gene signature have been reported to play critical roles in biological processes, including angiogenesis, metastasis, and proliferation in ovarian cancer. These genes include: CLIP, COL3A1, COL10A1, LAMB1, INHBA, NBL, F3, PDGFRA, PDGFRB, PLAU, ROR2, SCG2, and ZFHX430; COL11A1, COL5A2, THBS2, TIMP3 and SINAI231; EGR232, 33 and FOS34. Moreover, we investigated transcription factors included in the 97-gene signature. Consistent with previous studies, p5335, 36 and HIF1α22 were the upregulated transcription factors. However, a previous study reported that a large phase III trial of adenoviral wild-type p53 delivery combined with taxol and carboplatinum did not show any positive results in ovarian cancer37. To date, there is no accepted explanation of why this trial failed38. It is well known that the p53 signaling pathway has many links to ovarian cancer and is triggered by various stress signals39.
We investigated our novel nine HIF1α-regulated genes (ACTA2, EPOR, CTGF, FOS, SCG2, IGF1, CYR61, NT5E and NR4A). Interestingly, these genes had higher expression in NR and PR but lower expression in CR. Several studies reported ACTA2 upregulation in high-grade ovarian cancer40, 41. Western blot analysis by McBroom JW demonstrated increased EPOR expression in multiple ovarian cancer cell lines42. CTGF was reported to be a target therapeutic gene in high-grade ovarian cancer43. FOS has also been reported as a molecular predictor of progression and survival in ovarian cancer44. SCG2 was mentioned in a systematic review of genes reported to play a role in chemotherapy resistance in ovarian cancer30. IGF1 is well known to be involved in the development and progression of several types of tumors, including ovarian cancer45. Overexpression of CYR61 has been shown to be associated with development and poor prognosis of ovarian carcinomas46. NT5E-expressing cancers were reported to be resistant to platinum-based therapy47, while NR42A has been reported to be highly expressed in ovarian cancer tumors with poor prognosis48. We found that these HIF1α-regulated genes play significant roles in ovarian cancer first-line chemotherapy resistance, leading to disease progression and poor clinical outcome. These results suggest the possibility of developing and testing anti-HIF1α target based therapies for non-responders and partial remission patients.
In conclusion, we propose that this 97- prognostic gene signature could predict patient outcome in ovarian cancer. This gene signature can stratify subgroups of ovarian cancers into low- and high-risk groups in a reliable and reproducible manner across combined and independent patient cohorts. Our data suggest that this classifier may have considerable clinical relevance, especially in predicting first-line chemoresponse. This study will enable additional efforts to move ovarian cancer signatures closer to clinical relevance, and not only offer insight into mechanisms of chemoresistance but also provide information on potential target genes for future therapies.
Materials and Methods
Our evaluation was performed in four major steps: selection of both eligible genomic datasets and eligible prognostic models, transparent reimplementation of risk prediction models identified by literature review, selection of the gene signature from the training dataset, and multi-study validation of the selected gene signature using meta-analytic methods.
Eligibility Criteria and Implementation of the Prognostic Model
All datasets that had prognostic value in epithelial ovarian cancer were considered. Dataset GSE49997 from Austria was selected as the training dataset. Although it had 204 patients, only 184 patients were used. Twenty patients were excluded because they lacked OS information (Supplementary Table S1). Probe set identifiers were translated into gene symbols27.
Eligibility criteria for datasets and samples
Data in the public domain that provided expression information about primary patient tumors, OS time and event, and related chip types were assessed. The analysis was repeated to exclude samples that did not fit the above mentioned criteria.
Information sources
Prognostic models were identified through PubMed searches. The following terms were used to search PubMed: (“ovarian cancer” OR “Ovary tumor” OR “ovary carcinoma” OR “ovarian tumor” OR “ovarian carcinoma” OR “microarray” OR “expression array” OR gene expression AND ovarian cancer gene signature).
All datasets used were downloaded from National Center for Biotechnology Information Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo) and Cancer Genome Atlas database (http://cancergenome.nih.gov/). The robust multi-array averaging (RMA) method was used to average raw files. Seven different datasets were used with a total of 1,020 patients (Supplementary Table S1).
Datasets GSE14764 (n = 80; GPL96, Charite University Hospital, Berlin)49, GSE26712 (n = 185; GPL96, National Cancer Institute, USA)24, 50, GSE63885 (n = 75; GPL570, Memorial Cancer Center and Institute of Oncology, Poland)26, GSE19829 (n = 28; GPL570, Harvard Medical School, USA)25, GSE30161 (n = 58; GPL570, University of Virginia)51, and TCGA (n = 410; GPL96) were merged (n = 836) for a combined validation (Supplementary Table S1). In addition, RNA-seq data from TCGA (n = 419; Illumina) was used as an external validation dataset. (https://genome-cancer.ucsc.edu).
Development of the prognostic gene expression signature
To develop a prognostic gene expression signature, dataset GSE49997 was used as the training dataset. Overall survival and gene expression data were combined to build a gene expression profiling based survival classifier52. The 5,218 genes measured were filtered by at least 2 absolute values of log2 scale, which represented the same gene expression level. Univariate cox proportional hazard regression (P < 0.01) was then used to identify an OS associated gene expression signature from the discovery data set. For prognosis prediction, genes from the survival signature were applied in the survival risk prediction analysis. The prognostic index was computed by the formula Σ i w i x i − 0.467581 where w i and x i are the weight and logged gene expression for the i-th gene. A sample was predicted as high (low) risk if their prognostic index was larger than (smaller than or equal to) the median prognostic index of −0.038061. Initially, 137 genes associated with survival were discovered in the training dataset, however, only 97 genes were measured by both U133 plus 2.0 and U133A, and so these genes were used in all validations as the 97 gene signature (Supplementary Table S3).
Validation of the prognostic signature
Six independent datasets were used to validate the gene signature. Gene expression from different datasets was adjusted individually by subtracting the median expression value from all samples before merging. CCP was used as the class prediction algorithm to further refine this model and sub-stratify the predicted outcomes. Robustness was estimated by the misclassification rate that was determined in the training dataset during LOOCV21.
Kaplan-Meier survival analyses were performed after classification of patients into two risk groups, and Chi-square (χ2) and log-rank tests were used to evaluate the survival risk between the two predicted subgroups of patients. Univariate and multivariate Cox proportional hazard regression analyses were performed to evaluate independent prognostic factors associated with survival. The gene signature, age, tumor grade, and FIGO stage were employed as covariates.
Pathway analysis
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) bioinformatics tool was used for Gene Ontology (GO) biological process enrichment analysis, P values of less than 0.05 was used to define significant pathways. Ingenuity Pathways Analysis (IPA) was also used for transcription factor and gene network analyses (http://www.ingenuity.com)53. Identified gene networks were ranked to score (Z-score = 02).
STRING analysis
To predict protein-protein interactions, Search Tool for Retrieval of Interacting Genes/Proteins (STRING) database V10.0 was used (http://www.string-db.org/). Proteins were linked based on the following six criteria: neighborhood, gene fusion, co-occurrence, co- expression, experimental evidence, and existing databases54.
Statistical methods for microarray data
Microarray data and heatmaps were analyzed using BRB-Array Tools Version 3.0 (http://linus.nci.nih.gov/BRB-ArrayTools.html)55. All other statistical analyses were accomplished in the R language environment (www.r-project.org) and Statistical Package for Social Sciences (SPSS) software (version 20, SPSS Inc., Chicago, IL, USA). In all statistical analyses, P values of less than 0.05 was considered significant.
References
Jemal, A. et al. Cancer statistics, 2007. CA: A Cancer Journal for Clinicians 57, 43–66 (2007).
Cannistra, S. A. Cancer of the ovary. New England Journal of Medicine 351, 2519–2529, doi:10.1056/NEJMra041842 (2004).
McGuire, W. P. et al. Cyclophosphamide and cisplatin versus paclitaxel and cisplatin: a phase III randomized trial in patients with suboptimal stage III/IV ovarian cancer (from the Gynecologic Oncology Group). Seminars in Oncology 23, 40–47 (1996).
McGuire, W. P. et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. New England Journal of Medicine 334, 1–6, doi:10.1056/NEJM199601043340101 (1996).
Levanon, K., Crum, C. & Drapkin, R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. Journal of Clinical Oncology 26, 5284–5293, doi:10.1200/JCO.2008.18.1107 (2008).
Landrum, L. M. et al. Prognostic factors for stage III epithelial ovarian cancer treated with intraperitoneal chemotherapy: a Gynecologic Oncology Group study. Gynecologic Oncology 130, 12–18, doi:10.1016/j.ygyno.2013.04.001 (2013).
du Bois, A. et al. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer 115, 1234–1244, doi:10.1002/cncr.24149 (2009).
Ozols, R. F. et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 21, 3194–3200, doi:10.1200/JCO.2003.02.153 (2003).
Bristow, R. E., Tomacruz, R. S., Armstrong, D. K., Trimble, E. L. & Montz, F. J. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. Journal of Clinical Oncology 20, 1248–1259, doi:10.1200/JCO.2002.20.5.1248 (2002).
Dressman, H. K. et al. An integrated genomic-based approach to individualized treatment of patients with advanced-stage ovarian cancer. Journal of Clinical Oncology 25, 517–525, doi:10.1200/JCO.2006.06.3743 (2007).
Kupryjanczyk, J. et al. TP53 status determines clinical significance of ERBB2 expression in ovarian cancer. British Journal of Cancer 91, 1916–1923, doi:10.1038/sj.bjc.6602238 (2004).
Grendys, E. C. Jr. et al. Overview of a chemoresponse assay in ovarian cancer. Clin Transl Oncol 16, 761–769, doi:10.1007/s12094-014-1192-8 (2014).
Krivak, T. C. et al. A chemoresponse assay for prediction of platinum resistance in primary ovarian cancer. American Journal of Obstetrics and Gynecology 211(68), e61–68, doi:10.1016/j.ajog.2014.02.009 (2014).
Muraji, M. et al. Histopathology predicts clinical outcome in advanced epithelial ovarian cancer patients treated with neoadjuvant chemotherapy and debulking surgery. Gynecologic Oncology 131, 531–534, doi:10.1016/j.ygyno.2013.09.030 (2013).
da Costa Miranda, V. et al. Neoadjuvant chemotherapy with six cycles of carboplatin and paclitaxel in advanced ovarian cancer patients unsuitable for primary surgery: Safety and effectiveness. Gynecologic Oncology 132, 287–291, doi:10.1016/j.ygyno.2013.12.002 (2014).
Berchuck, A. et al. Prediction of optimal versus suboptimal cytoreduction of advanced-stage serous ovarian cancer with the use of microarrays. American Journal of Obstetrics and Gynecology 190, 910–925, doi:10.1016/j.ajog.2004.02.005 (2004).
Jochumsen, K. M. et al. Gene expression profiles as prognostic markers in women with ovarian cancer. Int J Gynecol Cancer 19, 1205–1213, doi:10.1111/IGC.0b013e3181a3cf55 (2009).
Spentzos, D. et al. Unique gene expression profile based on pathologic response in epithelial ovarian cancer. Journal of Clinical Oncology 23, 7911–7918, doi:10.1200/JCO.2005.02.9363 (2005).
Spentzos, D. et al. Gene expression signature with independent prognostic significance in epithelial ovarian cancer. Journal of Clinical Oncology 22, 4700–4710, doi:10.1200/JCO.2004.04.070 (2004).
Tan, T. Z. et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol Med 5, 1051–1066, doi:10.1002/emmm.201201823 (2013).
Nguyen, M. N. et al. CRC-113 gene expression signature for predicting prognosis in patients with colorectal cancer. Oncotarget 6, 31674–31692, doi:10.18632/oncotarget.5183 (2015).
Yin, X. et al. A VEGF-dependent gene signature enriched in mesenchymal ovarian cancer predicts patient prognosis. Sci Rep 6, 31079, doi:10.1038/srep31079 (2016).
Waldron, L. et al. Comparative meta-analysis of prognostic gene signatures for late-stage ovarian cancer. Journal of the National Cancer Institute 106, doi:10.1093/jnci/dju049 (2014).
Bonome, T. et al. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Research 68, 5478–5486, doi:10.1158/0008-5472.CAN-07-6595 (2008).
Konstantinopoulos, P. A. et al. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. Journal of Clinical Oncology 28, 3555–3561, doi:10.1200/JCO.2009.27.5719 (2010).
Lisowska, K. M. et al. Gene expression analysis in ovarian cancer - faults and hints from DNA microarray study. Front Oncol 4, 6, doi:10.3389/fonc.2014.00006 (2014).
Pils, D. et al. Validating the impact of a molecular subtype in ovarian cancer on outcomes: a study of the OVCAD Consortium. Cancer Sci 103, 1334–1341, doi:10.1111/j.1349-7006.2012.02306.x (2012).
Kinose, Y., Sawada, K., Nakamura, K. & Kimura, T. The role of microRNAs in ovarian cancer. Biomed Res Int 2014, 249393, doi:10.1155/2014/249393 (2014).
Langhe, R. et al. A novel serum microRNA panel to discriminate benign from malignant ovarian disease. Cancer Letters 356, 628–636, doi:10.1016/j.canlet.2014.10.010 (2015).
Lloyd, K. L., Cree, I. A. & Savage, R. S. Prediction of resistance to chemotherapy in ovarian cancer: a systematic review. BMC Cancer 15, 117, doi:10.1186/s12885-015-1101-8 (2015).
Cheon, D. J. et al. A collagen-remodeling gene signature regulated by TGF-beta signaling is associated with metastasis and poor survival in serous ovarian cancer. Clinical Cancer Research 20, 711–723, doi:10.1158/1078-0432.CCR-13-1256 (2014).
Nielsen, J. S., Jakobsen, E., Holund, B., Bertelsen, K. & Jakobsen, A. Prognostic significance of p53, Her-2, and EGFR overexpression in borderline and epithelial ovarian cancer. Int J Gynecol Cancer 14, 1086–1096, doi:10.1111/j.1048-891X.2004.14606.x (2004).
Skirnisdottir, I., Seidal, T. & Sorbe, B. A new prognostic model comprising p53, EGFR, and tumor grade in early stage epithelial ovarian carcinoma and avoiding the problem of inaccurate surgical staging. Int J Gynecol Cancer 14, 259–270, doi:10.1111/j.1048-891X.2004.014209.x (2004).
Chen, F. & Liu, N. A 10-gene expression signature of Notch pathway predicts recurrence in ovarian carcinoma. Oncol Lett 10, 1704–1708, doi:10.3892/ol.2015.3382 (2015).
Mullany, L. K. et al. Specific TP53 Mutants Overrepresented in Ovarian Cancer Impact CNV, TP53 Activity, Responses to Nutlin-3a, and Cell Survival. Neoplasia 17, 789–803, doi:10.1016/j.neo.2015.10.003 (2015).
Mir, R. et al. Mdm2 antagonists induce apoptosis and synergize with cisplatin overcoming chemoresistance in TP53 wild-type ovarian cancer cells. International Journal of Cancer 132, 1525–1536, doi:10.1002/ijc.27832 (2013).
Buller, R. E. et al. A phase I/II trial of rAd/p53 (SCH 58500) gene replacement in recurrent ovarian cancer. Cancer Gene Therapy 9, 553–566, doi:10.1038/sj.cgt.7700472 (2002).
Zeimet, A. G. & Marth, C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol 4, 415–422 (2003).
Bernardini, M. Q. et al. Expression signatures of TP53 mutations in serous ovarian cancers. BMC Cancer 10, 237, doi:10.1186/1471-2407-10-237 (2010).
Anastassiou, D. et al. Human cancer cells express Slug-based epithelial-mesenchymal transition gene expression signature obtained in vivo. BMC Cancer 11, 529, doi:10.1186/1471-2407-11-529 (2011).
Martins, F. C. et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biol 15, 526, doi:10.1186/s13059-014-0526-8 (2014).
McBroom, J. W. et al. Erythropoietin receptor function and expression in epithelial ovarian carcinoma. Gynecologic Oncology 99, 571–577, doi:10.1016/j.ygyno.2005.06.038 (2005).
Moran-Jones, K. et al. Connective tissue growth factor as a novel therapeutic target in high grade serous ovarian cancer. Oncotarget 6, 44551–44562, doi:10.18632/oncotarget.6082 (2015).
Mahner, S. et al. C-Fos expression is a molecular predictor of progression and survival in epithelial ovarian carcinoma. British Journal of Cancer 99, 1269–1275, doi:10.1038/sj.bjc.6604650 (2008).
Brokaw, J. et al. IGF-I in epithelial ovarian cancer and its role in disease progression. Growth Factors 25, 346–354, doi:10.1080/08977190701838402 (2007).
Shen, H. et al. CYR61 overexpression associated with the development and poor prognosis of ovarian carcinoma. Med Oncol 31, 117, doi:10.1007/s12032-014-0117-2 (2014).
Nevedomskaya, E. et al. A Systems Oncology Approach Identifies NT5E as a Key Metabolic Regulator in Tumor Cells and Modulator of Platinum Sensitivity. J Proteome Res 15, 280–290, doi:10.1021/acs.jproteome.5b00793 (2016).
Delgado, E. et al. High expression of orphan nuclear receptor NR4A1 in a subset of ovarian tumors with worse outcome. Gynecologic Oncology 141, 348–356, doi:10.1016/j.ygyno.2016.02.030 (2016).
Denkert, C. et al. A prognostic gene expression index in ovarian cancer - validation across different independent data sets. Journal of Pathology 218, 273–280, doi:10.1002/path.2547 (2009).
Vathipadiekal, V. et al. Creation of a Human Secretome: A Novel Composite Library of Human Secreted Proteins: Validation Using Ovarian Cancer Gene Expression Data and a Virtual Secretome Array. Clinical Cancer Research 21, 4960–4969, doi:10.1158/1078-0432.CCR-14-3173 (2015).
Ferriss, J. S. et al. Multi-gene expression predictors of single drug responses to adjuvant chemotherapy in ovarian carcinoma: predicting platinum resistance. PLoS One 7, e30550, doi:10.1371/journal.pone.0030550 (2012).
Shahid, M. et al. An 8-gene signature for prediction of prognosis and chemoresponse in non-small cell lung cancer. Oncotarget 7, 86561–86572, doi:10.18632/oncotarget.13357 (2016).
Shahid, M. et al. Prognostic value and their clinical implication of 89-gene signature in glioma. Oncotarget. doi:10.18632/oncotarget.9983 (2016).
Akter, S. et al. Prognostic value of a 92-probe signature in breast cancer. Oncotarget 6, 15662–15680, doi:10.18632/oncotarget.3525 (2015).
Simon, R. et al. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform 3, 11–17 (2007).
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 2011-0030072).
Author information
Authors and Affiliations
Contributions
A.M. wrote the manuscript and processed the data. J.Y. collected and directed validation of the data. M.S., T.C., M.N., N.N., and S.A. contributed to validation of the data. I.K., J.H., J.L., and C.M. directed the writing of the manuscript. M.S., S.K. and J.K. provided current clinical gynecological expertise. S.K. directed the research project and writing of the manuscript. All authors read and approved this manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matondo, A., Jo, Y.H., Shahid, M. et al. The Prognostic 97 Chemoresponse Gene Signature in Ovarian Cancer. Sci Rep 7, 9689 (2017). https://doi.org/10.1038/s41598-017-08766-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08766-5
- Springer Nature Limited
This article is cited by
-
Residual ANTXR1+ myofibroblasts after chemotherapy inhibit anti-tumor immunity via YAP1 signaling pathway
Nature Communications (2024)
-
A prognostic model for ovarian neoplasms established by an integrated analysis of 1580 transcriptomic profiles
Scientific Reports (2023)
-
The R-enantiomer of ketorolac reduces ovarian cancer tumor burden in vivo
BMC Cancer (2021)
-
Stiffness increases with myofibroblast content and collagen density in mesenchymal high grade serous ovarian cancer
Scientific Reports (2021)
-
Gene expression profile association with poor prognosis in epithelial ovarian cancer patients
Scientific Reports (2021)