
Abstract
Multiple Sclerosis (MS) is an immune-mediated demyelinating disease of the human central nervous system (CNS). Memory impairments and hippocampal demyelination are common features in MS patients. Our previous data have shown that demyelination alters neuronal gene expression in the hippocampus. DNA methylation is a common epigenetic modifier of gene expression. In this study, we investigated whether DNA methylation is altered in MS hippocampus following demyelination. Our results show that mRNA levels of DNA methyltransferase were increased in demyelinated MS hippocampus, while de-methylation enzymes were decreased. Comparative methylation profiling identify hypo-methylation within upstream sequences of 6 genes and hyper-methylation of 10 genes in demyelinated MS hippocampus. Genes identified in the current study were also validated in an independent microarray dataset generated from MS hippocampus. Independent validation using RT-PCR revealed that DNA methylation inversely correlated with mRNA levels of the candidate genes. Queries across cell-specific databases revealed that a majority of the candidate genes are expressed by astrocytes and neurons in mouse and human CNS. Taken together, our results expands the list of genes previously identified in MS hippocampus and establish DNA methylation as a mechanism of altered gene expression in MS hippocampus.
Similar content being viewed by others

Introduction
Multiple sclerosis (MS) is an inflammatory, demyelinating, and neurodegenerative disease of the central nervous system (CNS) that affects more than two million people worldwide1, 2. Among the spectrum of cognitive impairments, memory dysfunction is most common among MS patients3, 4. Hippocampal demyelination is extensive in individuals with MS and modulates expression of neuronal genes involved in synaptic plasticity and memory function5, 6.
Large-scale genome-wide association studies (GWAS) have identified MS susceptibility loci, including human leukocyte antigen loci and other immune-function related genes7,8,9,10; however, their functional significance related to MS pathogenesis is still unknown. The relatively low concordance rate of single-nucleotide polymorphisms in monozygotic twins11, the presence of a strong gender bias, and the influence of migration on disease onset collectively suggest that the pathogenesis of MS likely results from a combination of both genetic and epigenetic factors12, 13. Epigenetic modifications, including DNA methylation, histone modification, chromatin remodeling, and noncoding RNA regulation have been reported to regulate gene expression and to participate in the etiology of MS14, 15.
DNA methylation occurs in special genomic regions called CpG islands, which contain greater than 50% cytosine and guanine nucleotides. It plays a major role in aberrant expression of genes that are important in several neurological diseases16, 17 as well as in memory formation and maintenance18, 19. We previously compared and identified several genes and microRNAs in MS hippocampus that correlate with synaptic changes, memory dysfunction, and hippocampal demyelination5, 20.
In this study, we investigated additional epigenetic mechanisms that alter gene expression in MS hippocampus. We found significant increases in mRNA levels of key DNA methyltransferase enzymes (DNMTs). Interestingly, the mRNA levels of the three DNA de-methylation enzymes (ten-eleven translocation methylcytosine dioxygenase 1–3; TET1-TET3), which catalyze hydroxy-methylation as well as the total level of hydroxy-methylated residues, were significantly decreased in MS demyelinated hippocampus. Several differentially methylated positions (DMPs) were also identified by comparing MS myelinated to demyelinated hippocampus. The methylation status of DMPs inversely correlated with mRNA levels of target genes that have been associated with neuronal survival and memory function. As methylation patterns in different cell types may contribute to overall methylation patterns21, human and mouse cell-specific databases indicated that a majority of the target genes were localized to neurons and astrocytes. Collectively, our results provide evidence that DNA methylation could play a major role in controlling gene expression in MS hippocampus.
Results
Demyelination correlates with changes in DNA methylation and de-methylation enzymes in MS hippocampus
In order to study DNA methylation, we measured levels of DNMT enzymes responsible for inserting and maintaining DNA methylation (DNMT1, DNMT3A, and DNMT3B) in myelinated and demyelinated MS hippocampi. Demyelination led to significant increases in mRNA levels of all three DNMTs in MS hippocampus (Fig. 1A). Immunohistochemical analysis showed that DNMT1 (Fig. 1B,C), DNMT3A (Fig. 1D,E) and DNMT3B (Fig. 1F,G) were primarily associated with hippocampal neurons in MS myelinated (Fig. 1B,D,F) and MS demyelinated (Fig. 1C,E,G) hippocampus.

DNA methyltransferase (DNMT) expression in hippocampi from multiple sclerosis (MS) brains. RT-PCR analysis shows significant increases in mRNA levels of DNMT 1, DNMT3A, and DNMT3B in demyelinated hippocampus (n = 4) compared to myelinated hippocampus (n = 4) (A). Immunohistochemistry showing cellular expression of DNMT1, DNMT3A, and DNMT3B in myelinated (B,D,F) and demyelinated hippocampus (C,E,G), with predominant expression in hippocampal neurons. Scale Bars: B–G: 30 μm; Error bars indicate + S.E.M.; *p < 0.05.
In concert with DNMT enzymes inserting and maintaining methylation patterns, the TET family of de-methylation enzymes oxidizes methylcytosine (5 mC) to hydroxymethylcytosine (5 hmC) to remove methylation22. Three TET proteins (TET 1–3) have been identified in humans23, which all convert 5 mC to 5 hmC. Given the relationship between methylation and TET enzymes, we also quantified mRNA levels of TET1–3 in MS hippocampus (Fig. 2A). The results showed a significant decrease in mRNA levels of all three TET genes in demyelinated hippocampi from individuals with MS. In order to test whether the lower mRNA levels of TET enzymes also correlated with levels of 5hmC residues, we measured the global levels of 5hmC within DNA isolated from MS myelinated and demyelinated hippocampi. The results showed that there was a significant decrease in the overall level of 5hmC within demyelinated hippocampus that correlated with the decreased mRNA levels of TET1–3 (Fig. 2B). We then conducted histochemical analysis to determine the cellular expression of 5hmC and we determined that neurons were the major cell type containing 5hmC in MS myelinated (Fig. 2C) and demyelinated hippocampus (Fig. 2D).

Demyelination in MS brains leads to lower expression of TET enzyme mRNA and hydroxymethyl residues. RT-PCR analysis shows significant decreases in mRNA levels of TET 1, TET 2, and TET 3 in demyelinated hippocampus (n = 4) compared to myelinated hippocampus (n = 4) (A). Compared to myelinated hippocampus (n = 8), demyelination (n = 7) led to a significant decrease in levels of hydroxymethyl content (hmC) (B). Immunohistochemistry analysis using a 5hmC antibody shows neurons as the major cell type with 5 hmC expression in myelinated (C) and demyelinated hippocampus (D). Scale Bars: C–D: 30 μm; Error bars indicate + S.E.M.; *p < 0.05.
Loss of myelin correlates with DNA methylation changes within transcription start sites
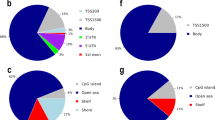
Imprinted gene expression is regulated by epigenetic mechanisms, particularly DNA methylation at DMPs. We did not detect any significant changes in overall 5mC levels in DNA from myelinated or demyelinated MS hippocampus (data not shown). We therefore used a more sensitive global array-based approach to identify DMPs by comparing myelinated (n = 8) and demyelinated (n = 7) MS hippocampus (Fig. 3A) using an Infinium HumanMethylation 450 K array (Illumina Inc, USA). Patient details are in Table 1. The 450 k array contains 485,512 probes covering 99% of RefSeq genes. The probes interrogate 19,755 unique CpG islands with additional coverage in shore regions as well as 3091 probes at non-CpG sites24. Resultant methylation assays and global clustering showed that the methylation pattern could reliably differentiate between myelinated and demyelinated MS hippocampus (Fig. 3A). Demyelination resulted in identification of 144 DMPs (62 hypermethylated, 82 hypomethylated) within the assessed CpG sites (adjusted p < 0.05, delta β 20%; Fig. 3B). Localization of these DMPs in the context of genomic location was classified into different categories (Fig. 3C): CpG islands, flanking CpG Islands (CGIs) (shores and shelves; 2–4 kb from CGIs), and open sea (not related to CpGs). We found 29 out of 62 (46.7%) DMPs detected within the CpG islands had increased methylation, whereas 26.8% (22 out of 82) of the DMPs within the CpG islands showed decreased methylation. We next asked whether these DMPs fall within the genomic location of well-characterized genes (UCSD genome browser hg119). The DMPs were mapped to either the ‘1st exon’, 3′ and 5′ UTRs, or the ‘body’ (non-exonic) of the gene, or within 1500 bp of the transcription start site (TSS). The results showed that 92 out of 144 altered DMPs (65%) were mapped to 75 annotated genes (UCSD genome browser hg119). Several of the identified genes had multiple DMPs associated with their sequences (Table 2). To further validate our findings, we compared these identified genes with results obtained from an independent gene microarray database using MS hippocampus5. The results showed that a significant number of genes (43 out of 75) identified in the current study were also altered at the mRNA expression level in the previous study (gene symbols in bold letters, Tables 2 and 3).

Global methylation profiles are different between myelinated and demyelinated MS hippocampus. Global DNA methylation was assessed using the Illumina 450 K array. Resultant clustering analysis shows that the MS demyelinated samples (shown in red) are separated from MS myelinated samples (A). Compared to myelinated MS hippocampus, 144 DMPs (62 hyper-methylated, 82 hypo-methylated) were identified in MS demyelinated hippocampus (B). Localization of these DMPs based on CpG islands, flanking CpG Islands (CGIs) (shores and shelves; 2–4 kb from CGIs), and open sea (non-related to CpGs) are shown (C). DMPs mapped onto genomic features (‘1st exon’, 3′ and 5′ UTRs, ‘body’ of the gene, within 1500 bp of the transcription start site (TSS) of well characterized genes (UCSD genome browser hg19) show differences between hypo- and hyper-methylated targets.
Identification of cell types expressing transcripts containing DMPs in mouse CNS
After we identified altered DMPs and their target genes in MS hippocampus, we compared the cellular expression of these target genes in mouse CNS using published mouse and human RNA-seq databases25, 26. This comparison revealed that DMPs are associated with genes expressed by the four major cell types (astrocytes, neurons, oligodendrocytes, and microglia) in mouse and human CNS (Table 2). Among identified hypomethylated targets (within UTR, gene body, and exon), 41% were specific to astrocytes, whereas 44% (13 out of 29) of the target genes were expressed by neurons in mouse CNS. The results also showed that 34% of the target genes were expressed by microglia/macrophage lineage cells in mouse (Table 2). Interestingly, hypomethylation identified 1 gene in mouse oligodendrocytes (F Box and WD repeat domain 8) and 4 in human oligodendrocytes (Small Nuclear Ribonucleoprotein 40 kDA; Bestrophin 3; Meis Homeobox 1; and Transmembrane Protein 165). None of the genes were expressed by both mouse and human oligodendrocytes. Among the hypermethylated transcripts, (within UTR, gene body, and exon), 26% of the genes were expressed by astrocytes and neurons (8 out of 30), while 20% were also expressed by microglia/macrophage cells in mouse brain (Table 2). Four hypermethylated genes were oligodendrocyte-specific in mouse brain (McKusick-Kaufmna Syndrome; Inscuteable Homolog (Drosophila); Neurofascin; and Topoisomerase (DNA) I, Mitochondrial) and three (Chromosome 1 open reading frame 106; Neurofascin; and Shisa Family Member 2) were localized to human oligodendrocytes. Cellular analysis revealed that the hypermethylated, oligodendrocyte-specific Neurofascin 155, which is involved in maintenance of axoglial junctions27, is expressed by both mouse and human oligodendrocytes.
As the presence of DMPs within the promoter and TSS has the greatest possibility of affecting mRNA expression14, we identified the genes where a DMP was identified within 1500 bp of the TSS (Table 2). Our results showed that there were 14 DMPs (targeting 6 genes) that showed decreased methylation, while 10 DMPs (identified with 10 genes) showed increased methylation. Due to the presence of DMPs within 1500 bp of the TSS, we determined the cellular identification of the targets in both mouse and human RNA databases25, 26. The comparative results identified several genes that are differentially expressed by mouse and human CNS cells (Table 3). As brain tissue is a heterogeneous mixture of several cell types, these results not only provide clues as to the cellular specificity of each of the target genes, but they also provide options for further genetic manipulations in mouse models.
DNA methylation changes within transcription start sites of genes inversely correlate with mRNA levels of target genes
DNA methylation at the proximity of the TSS is generally considered to be a potent epigenetic modification that prohibits transcription factor (TF) recruitment, resulting in transcription suppression28, 29. We therefore validated whether the presence of DMPs within 1500bp of the TSS affects mRNA levels in 4 hyper-methylated and 4 hypo-methylated target genes. The results showed (Fig. 4) that hypomethylation within the TSS1500 of AT-hook transcription factor (AKNA), Emopamil Binding Protein Like (EBPL), HECT domain, RCC1-like domain containing protein 6 (HERC6), and Secreted frizzled related protein 1 (SFRP1) led to significant increases in mRNA levels of these genes following demyelination. In contrast, hyper-methylation identified within the TSS1500 of Nescient helix-loop-helix 2 (NHLH2), Phospholipase C eta 1 (PLCH1), Transmembrane protein 132B (TMEM132B), and WD repeat domain 81 (WDR81) correlated with the decrease in mRNA levels of these genes following demyelination in MS hippocampus. Immunohistochemical analysis to confirm cellular localization, showed that SFRP1 (Fig. 4B,C), and PLCH1 (Fig. 4D,E) were primarily associated with hippocampal neurons in MS myelinated (Fig. 4B,D) and MS demyelinated (Fig. 4C,E) hippocampus.

Inverse correlation between DMP and mRNA levels of target genes. RT-PCR analysis shows significant increases in mRNA levels of AT-hook transcription factor (AKNA), Emopamil Binding Protein Like (EBPL), HECT domain and RCC1-like domain containing protein 6 (HERC6), and Secreted frizzled related protein 1 (SFRP1) in demyelinated MS hippocampus (n = 4) compared to myelinated MS hippocampus (n = 4). Hyper-methylation within Nescient helix-loop-helix 2 (NHLH2), Phospholipase C eta 1 (PLCH1), Transmembrane protein 132B (TMEM132B), and WD repeat domain 81 (WDR81) following demyelination led to significant decreases in mRNA levels. Immunohistochemistry showing cellular expression of SFRP1 and PLCH1 in myelinated (B,D) and demyelinated hippocampus (C,E), with predominant expression in hippocampal neurons. Scale Bars: B–E: 30 μm; Error bars indicate + S.E.M.; * p < 0.05, **p < 0.005, ***p < 0.0005, *****p < 0.000005.
Discussion
Epigenetic targets of gene regulation provide interesting targets for therapeutics because they are modifiable. The present study identifies DNA methylation as a correlate of demyelination and gene expression in MS hippocampus. Demyelination coincides with increases in mRNA levels of DNA methylation enzymes, with concomitant decreases in levels of DNA demethylation enzymes. Global methylation analysis identified 144 DMPs in MS hippocampus targeting several genes expressed by the major cell types in the human brain. As human tissue is not amenable to experimental manipulations, we also performed a comparative database search to determine the cellular specificity of the candidate genes in mouse brain. These data were further screened to identify the 25 DMPs localized within 1500 bp of TSS of 16 genes. Independent validation revealed that mRNA levels of the identified genes inversely correlated with DNA methylation status. Previous studies have shown that hippocampal demyelination is common in MS patients, which leads to loss of synaptic density and can influence expression of synaptic and neuronal genes as well as regulate the expression of neuronal miRNAs5, 20. In this study, we identified candidate genes that are altered by methylation changes following demyelination in MS hippocampus and that may play a role in altering synaptic plasticity, memory performance, and neuronal survival in MS brains.
The presence/loss of methylation within the TSS alters mRNA levels of the target genes. We identified 6 genes where demyelination led to decreased methylation. Among these, mRNA levels of AKNA were significantly increased following demyelination. AKNA is a major regulator of CD40 and CD40 ligand30 and is reported in RNA seq databases to be expressed by microglia/macrophage cells (Table 3) in both mouse and human. Increases in microglial CD40 expression and interactions with its ligand CD40L have been shown to induce/stimulate the expression of tumor necrosis factor-alpha (TNF-α) and to initiate neuronal death31. Following demyelination, we detected a significant increase in methylation within the TSS of WDR81, a gene involved in neuronal survival32. Increased levels of AKNA in conjunction with decreased WDR81 following demyelination could therefore lead to increased levels of TNF-α and neuronal injury. In addition, we also identified hypomethylation and a significant increase in mRNA levels of SFRP1 following demyelination. SFRP1 is an inhibitor of the WNT signaling proteins, which regulate learning and memory as well as synaptic plasticity at central synapses33, 34. Our comparative methylation analysis also identified hypermethylation (near the TSS) and decreased mRNA levels of NHLH2 and PLCH1 (Fig. 4). NHLH2 is a positive regulator of melanocortin receptors and modulates memory and learning35, 36, while PLCH1 loss is generally associated with impaired working memory37. Differential methylation and concomitant increased levels of SFRP1 as well as decreased NHLH2 and PLCH1 could therefore lead to the decreased synaptic density and memory performance that have been previously reported following demyelination5, 20.
The ongoing discovery of epigenetic factors underlying pathogenesis in MS is substantial14. These factors are relatively easily modifiable, in many cases utilizing readily available pharmacological agents targeting epigenetic modifiers. The study of DNA methylation in MS pathogenesis has been largely limited to comparisons of peripheral blood cells14, 15, 38. Using the same platform used in this study, 74 methylation sites (29 associated with either the MHC or the human leukocyte antigen (HLA-DRB1 region) were identified in a comparison between CD4+ T cells from patients with MS and healthy individuals39. In addition, significant differences in the methylation patterns associated with CD4 and CD8 T cells were detected in MS patients compared to healthy controls40. A genome-wide study of methylation in normal appearing white matter from MS patients identified 319 significantly hypermethylated and 220 significantly hypomethylated regions, which mapped to genes involved in oligodendrocyte survival and immune responses41. Our study is the first to provide insight into the landscape of methylation changes following demyelination in MS hippocampus. In addition, none of the genes identified in the current study was common to the candidates described in our previous collaborative study using MS normal appearing white matter41. This supports the need to develop datasets of gene changes from different gray matter regions in MS brains. Of great interest is the fact that the identified genes are expressed by different cell types in human and mouse brain. While similar DNA methylation studies in mouse are not currently possible due to the lack of a global mouse methylation profile chip, our species-specific cell identity provides the option to query, validate, and manipulate candidate genes for their possible role in memory function using animal models. Future tissue-, region-, and cell-specific analyses of epigenetic changes in MS brains need to be conducted in order to gain further insight into the pathogenesis of MS.
Materials and Methods
Human subjects and regulatory compliance
All brains were collected as part of the tissue procurement program approved by the Cleveland Clinic Institutional Review Board. Patient anonymity was strictly maintained and all tissue samples were handled in a coded fashion. All donors or their surrogates gave informed consent for their brains to be used for research studies. All experiments were carried out in accordance with the relevant Cleveland Clinic Institutional regulations and guidelines.
Tissue collection and characterization
Brains were removed according to a rapid autopsy protocol, sliced (1 cm thick), and then either fixed in 4% paraformaldehyde for morphological studies or rapidly frozen for biochemical analysis. Patient demographics are listed in Table 1. All hippocampi were characterized for demyelination by immunostaining using proteolipid protein (PLP) as described previously5, 20. Briefly, frozen 30 µm sections were cut for characterization by immunohistochemistry and for assessment of demyelination. This was followed by collection of 3–4 subsequent sections for DNA isolation. No significant differences in disease duration (27.2 yrs vs 23.2 yrs; p = 0.60) or postmortem interval (7.1 hr vs 7.5 hr; p = 0.87) were detected between the myelinated and demyelinated MS patients.
Methylation profiling
Genomic DNA was isolated from frozen tissue sections corresponding to regions of myelinated (n = 8) or demyelinated MS hippocampus (n = 7). Genomic DNA was isolated using a QIAamp DNA mini kit (Qiagen Inc, USA) following the manufacturer’s instructions. Purified genomic DNA was processed for bisulfide conversion and subsequent methylation assays. DNA samples were delivered to the Case Western Reserve University Genomics Core Facility, where 1.5 µg of DNA was bisulphite-treated (Zymo EZ DNA methylation gold) per manufacturer’s instructions. Genome-wide methylation profiles were generated using Illumina 450 K methylation arrays. Isolated DNA (100 ng) was used to measure DNA hydroxymethylation with MethylFlash™ Global DNA Hydroxymethylation (5hmC) ELISAs (Epigentek Inc, USA) using a 5hmC mAb-based detection complex.
Data analysis and cell type identification
Raw data (idat format) were generated through Illumina’s Genome Studio software. Raw data were preprocessed, normalized, and analyses was carried out in the R environment using the ChAMP package, which integrates currently available 450 k analysis methods and also offers its own novel functionality42. After running basic quality control metrics, we performed a beta mixture quantile normalization method to adjust for bias introduced by the Infinium type 2 probe design42. All DNA methylation data files will be deposited at the GEO (https://www.ncbi.nlm.nih.gov/GEO) and can be accessed through the accession number GSE101658. As part of routine analysis of DNA methylation datasets in our study, the probes on the X chromosome were normalized for males and females separately and independently of autosomal probes because X chromosome inactivation causes significant gender differences in methylation patterns. In addition, to account for age-related DNA methylation changes, we employed the methods described by Horvath et al.43. Briefly, this multi-tissue predictor of age allows for the estimation of the DNA methylation age of most tissues and cell types. The predictor, which is freely available, was developed using 8,000 samples from 82 Illumina DNA methylation array datasets, encompassing 51 healthy tissues and cell types. Using these methods, we did not detect any differences in DNA methylation for predicted and actual age between MS samples. To determine the cellular identity of the methylation target, identified genes were queried against mouse and human cell-specific RNA sequencing databases25, 26. To further validate the results and cellular specificity, we only selected genes that were above the 50th percentile of expression level across all cell types.
Immunohistochemistry
Sections (30 μm thick) from corresponding (to frozen sections) fixed blocks of the hippocampus were cut on a sliding microtome, microwaved in 10 mM citric acid buffer (pH 6.0) for 5 minutes, incubated in 3% hydrogen peroxide and 1% Triton X-100 in phosphate-buffered saline for 30 minutes, and immunostained by the avidin-biotin complex procedure with diaminobenzidine (DAB) for myelin PLP, MHC Class II, HuR, or other antibodies as described previously5, 20. The extent of demyelination and neuronal status were determined from PLP and HuR staining. Using the same protocol, sections were stained using antibodies specific to DNMT1 (1:250; HPA002694; Sigma Aldrich, St. Louis, MO), DNMT3A (1:250; HPA02588; Sigma Aldrich, St. Louis, MO), DNMT3B (1:250; HPA001595; Sigma Aldrich, St. Louis, MO), and α-5-hydroxymethylcytosine (1:500; ab106918; Abcam Inc. Cambridge MA), PLCH1 (1:250; GTX108612; GeneTex Inc. Irvine CA), SFRP1 (1:250; ab94942; Abcam Inc. Cambridge MA).
Real-time PCR
RT-PCR was performed using a standard TaqMan PCR kit protocol on an Applied Biosystems 7500HT Sequence Detection System using 0.2 μM TaqMan probes. Probes specific to DNMT1 (Hs00154749_m1), DNMT3A Hs01027166_m1) DNMT3B (Hs00171876_m1), TET1 (Hs00286756_m1), TET2 (Hs00325999_m1), TET3 (Hs00379125_m1), PLP1(Hs00166914_m1), AKNA (Hs00363936_m1), WDR81 (Hs00912091_m1), TMEM132B (Hs00287113_m1), EBPL (Hs00831100_s1), HERC6 (Hs00215555_m1), SFRP1 (Hs00610060_m1), NHLH2 (Hs00271585_s1), and PLCH1 (Hs00324566_m1) were used in triplicate reactions. Total RNA was reverse transcribed using a high capacity cDNA reverse transcription kit (Applied Biosystems, Carlsbad, CA). Reverse transcription reactions contained hippocampal RNA, 1X RT random primers (Applied Biosystems, Carlsbad, CA), 0.25 mM dNTP mix, 50 U MultiScripe reverse transcriptase (Applied Biosystems, Carlsbad, CA), and 1 U/μL RNase inhibitor (Applied Biosystems, Carlsbad, CA). TaqMan gene expression assays for multiplex reactions were performed following the manufacturer’s protocol. Each RT-PCR reaction contained 100ng of cDNA product, 1X TaqMan gene expression mix, 100nM GAPDH primers (Applied Biosystems, ID# 4310884E), and 100 nM FAM-labeled target probes for methylated target genes (Applied Biosystems, Carlsbad, CA). Resultant Ct values were normalized and quantitative data are expressed as mean ± S.E.M. The statistical significance of differences between groups in RT-PCR were determined using previously published methods20.
References
Hauser, S. L. & Oksenberg, J. R. The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron 52, 61–76 (2006).
Trapp, B. D. & Nave, K. A. Multiple sclerosis: an immune or neurodegenerative disorder? Annu. Rev. Neurosci. 31, 247–269 (2008).
Benedict, R. H. & Zivadinov, R. Risk factors for and management of cognitive dysfunction in multiple sclerosis. Nat. Rev. Neurol. 7, 332–342 (2011).
Chiaravalloti, N. D. & DeLuca, J. Cognitive impairment in multiple sclerosis. Lancet Neurol. 7, 1139–1151 (2008).
Dutta, R. et al. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann. Neurol. 69, 445–454 (2011).
Geurts, J. J. et al. Extensive hippocampal demyelination in multiple sclerosis. J. Neuropathol. Exp. Neurol. 66, 819–827 (2007).
Oksenberg, J. R. & Hauser, S. L. Decoding multiple sclerosis. Ann. Neurol. 70, A5–A7 (2011).
Ramagopalan, S. V., Dobson, R., Meier, U. C. & Giovannoni, G. Multiple sclerosis: risk factors, prodromes, and potential causal pathways. Lancet Neurol. 9, 727–739 (2010).
Sawcer, S. et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011).
Sawcer, S., Franklin, R. J. & Ban, M. Multiple sclerosis genetics. Lancet Neurol. 13, 700–709 (2014).
Baranzini, S. E. et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 464, 1351–1356 (2010).
Ascherio, A. Environmental factors in multiple sclerosis. Expert. Rev. Neurother. 13, 3–9 (2013).
Handel, A. E., Giovannoni, G., Ebers, G. C. & Ramagopalan, S. V. Environmental factors and their timing in adult-onset multiple sclerosis. Nat. Rev. Neurol. 6, 156–166 (2010).
Huynh, J. L. & Casaccia, P. Epigenetic mechanisms in multiple sclerosis: implications for pathogenesis and treatment. Lancet Neurol. 12, 195–206 (2013).
Koch, M. W., Metz, L. M. & Kovalchuk, O. Epigenetic changes in patients with multiple sclerosis. Nat. Rev. Neurol. 9, 35–43 (2013).
Jakovcevski, M. & Akbarian, S. Epigenetic mechanisms in neurological disease. Nat. Med. 18, 1194–1204 (2012).
Zoghbi, H. Y. & Beaudet, A. L. Epigenetics and Human Disease. Cold Spring Harb. Perspect. Biol. 8, a019497 (2016).
Jarome, T. J. & Lubin, F. D. Epigenetic mechanisms of memory formation and reconsolidation. Neurobiol. Learn. Mem. 115, 116–127 (2014).
Zovkic, I. B., Guzman-Karlsson, M. C. & Sweatt, J. D. Epigenetic regulation of memory formation and maintenance. Learn. Mem. 20, 61–74 (2013).
Dutta, R. et al. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann. Neurol. 73, 637–645 (2013).
Iwamoto, K. et al. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 21, 688–696 (2011).
Tahiliani, M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009).
Wu, H. & Zhang, Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 25, 2436–2452 (2011).
Bibikova, M. et al. High density DNA methylation array with single CpG site resolution. Genomics 98, 288–295 (2011).
Zhang, Y. et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53 (2016).
Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 (2014).
Pillai, A. M. et al. Spatiotemporal ablation of myelinating glia-specific neurofascin (Nfasc NF155) in mice reveals gradual loss of paranodal axoglial junctions and concomitant disorganization of axonal domains. J. Neurosci. Res. 87, 1773–1793 (2009).
Jin, J. et al. The effects of cytosine methylation on general transcription factors. Sci. Rep. 6, 29119 (2016).
Hu, S. et al. DNA methylation presents distinct binding sites for human transcription factors. Elife. 2, e00726 (2013).
Siddiqa, A. et al. Regulation of CD40 and CD40 ligand by the AT-hook transcription factor AKNA. Nature 410, 383–387 (2001).
Tan, J. et al. Activation of microglial cells by the CD40 pathway: relevance to multiple sclerosis. J. Neuroimmunol. 97, 77–85 (1999).
Traka, M. et al. WDR81 is necessary for purkinje and photoreceptor cell survival. J. Neurosci. 33, 6834–6844 (2013).
Tabatadze, N., McGonigal, R., Neve, R. L. & Routtenberg, A. Activity-dependent Wnt 7 dendritic targeting in hippocampal neurons: plasticity- and tagging-related retrograde signaling mechanism? Hippocampus 24, 455–465 (2014).
Inestrosa, N. C. & Arenas, E. Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 11, 77–86 (2010).
Cui, H. et al. Melanocortin 4 receptor signaling in dopamine 1 receptor neurons is required for procedural memory learning. Physiol Behav. 106, 201–210 (2012).
Wankhade, U. D. & Good, D. J. Melanocortin 4 receptor is a transcriptional target of nescient helix-loop-helix-2. Mol. Cell Endocrinol. 341, 39–47 (2011).
Kim, S. W. et al. Knockdown of phospholipase C-beta1 in the medial prefrontal cortex of male mice impairs working memory among multiple schizophrenia endophenotypes. J. Psychiatry Neurosci. 40, 78–88 (2015).
Aslani, S. et al. Epigenetic Modifications and Therapy in Multiple Sclerosis. Neuromolecular. Med (2016).
Graves, M. C. et al. Methylation differences at the HLA-DRB1 locus in CD4+ T-Cells are associated with multiple sclerosis. Mult. Scler. 20, 1033–1041 (2014).
Bos, S. D. et al. Genome-wide DNA methylation profiles indicate CD8+ T cell hypermethylation in multiple sclerosis. PLoS. One. 10, e0117403 (2015).
Huynh, J. L. et al. Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat. Neurosci. 17, 121–130 (2014).
Morris, T. J. et al. ChAMP: 450 k Chip Analysis Methylation Pipeline. Bioinformatics. 30, 428–430 (2014).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Acknowledgements
We thank Dr. Christopher Nelson for editorial assistance. We also thank the MS tissue collection program and the rapid autopsy team at the Cleveland Clinic. The work is supported by grants from the NIH, NINDS (NS096148) and the National Multiple Sclerosis Society, USA (RG 5298) to RD. The MS tissue collection is supported by NIH, NINDS (NS091683) grant to BDT.
Author information
Authors and Affiliations
Contributions
A.C. conducted the DNA methylation studies and data analysis. C.V., S.A.D. helped with the tissue characterization and immunohistochemistry experiments. A.T. compared the hippocampus and white matter DNA methylation datasets. R.J.F. and B.D.T. helped in the procurement of tissues and review of MS diagnosis. R.D. designed the study, performed data interpretation, and drafted the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chomyk, A.M., Volsko, C., Tripathi, A. et al. DNA methylation in demyelinated multiple sclerosis hippocampus. Sci Rep 7, 8696 (2017). https://doi.org/10.1038/s41598-017-08623-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-08623-5
- Springer Nature Limited
This article is cited by
-
The neuropathobiology of multiple sclerosis
Nature Reviews Neuroscience (2024)
-
Epigenetic Orchestration of Neurodegenerative Disorders: A Possible Target for Curcumin as a Therapeutic
Neurochemical Research (2024)
-
Myelin in Alzheimer’s disease: culprit or bystander?
Acta Neuropathologica Communications (2023)
-
From methylation to myelination: epigenomic and transcriptomic profiling of chronic inactive demyelinated multiple sclerosis lesions
Acta Neuropathologica (2023)
-
N6-Methyladenosine RNA modification in cerebrospinal fluid as a novel potential diagnostic biomarker for progressive multiple sclerosis
Journal of Translational Medicine (2021)