
Abstract
Purinergic P2Y 2 receptors, G-protein coupled receptors that primarily couple with Gαq/11-proteins, are activated equipotently by adenosine-5′-triphosphate (ATP) and uridine-5′-triphosphate. Evidence suggests that P2Y 2 agonists make potential drug candidates for the treatment of cardiovascular diseases. However, selective non-nucleotide, small-molecule P2Y 2 agonists have yet to be developed. In this report, we discuss Compound 89, a novel non-nucleotide allosteric P2Y 2 agonist that was active in signal transduction and gene induction, and in our in vitro cardiac hypertrophy model. Compound 89 exhibited selective P2Y 2 agonistic activity and potentiated responses to the endogenous agonist ATP, while exhibiting no agonistic activities for four other Gαq/11-coupled human P2Y (hP2Y) receptors and one representative Gαi/o-coupled hP2Y12 receptor. Its P2Y 2 agonistic effect on mouse P2Y 2 receptors suggested non-species-specific activity. Compound 89 acted as a pure positive allosteric modulator in a Ca2+ mobilization assay of neonatal rat cardiomyocytes; it potentiated ATP-induced expression of genes in the nuclear receptor 4A family (negative regulators of hypertrophic stimuli in cardiomyocytes). Additionally, Compound 89 attenuated isoproterenol-induced cardiac hypertrophy, presumably through dose-dependent interaction with pericellular ATP. These results indicate that Compound 89 is potentially efficacious against cardiomyocytes and therefore a good proof-of-concept tool for elucidating the therapeutic potential of P2Y2 activation in various cardiovascular diseases.
Similar content being viewed by others

Introduction
P2Y receptors, G-protein coupled receptors (GPCRs) that are activated by extracellular adenine/uridine nucleotides and nucleotide sugars, mediate various physiological functions in virtually all tissues and cells1. Studies have shown that among them, P2Y2 receptors are broadly and highly expressed in the eyes, lungs, intestine, immune system, and skeletal muscles, as well as tissues associated with cardiovascular homeostasis (i.e. blood vessels, heart, kidneys, and brain)2,3,4,5. Increasing evidence indicates that P2Y 2 receptors could function as cell-surface ‘mechano-sensors’ for the autocrine or paracrine release of adenosine-5′-triphosphate (ATP) and/or uridine-5′-triphosphate (UTP) from different cellular sources6,7,8,9 in order to primarily mediate Gαq/11 signalling, resulting in downstream transcriptional regulation via nuclear inositol 1,4,5-trisphosphate receptor-induced local Ca2+ signalling in muscle fibres and cardiac myocytes10,11,12. Such regulation of transcriptional activity through local Ca2+ signal transduction is known as excitation-transcription coupling, as opposed to the excitation-contraction coupling that links global increases in intracellular Ca2+ more closely with muscle contractions.
A growing number of studies have suggested that P2Y 2 receptors potentially improve cardiovascular dysfunction in various ways, such as through endogenous UTP-mediated cardioprotection during ischemia13, 14, ATP-mediated antihypertrophic effects in the heart2, 15, control of shear stress-induced blood pressure and vessel arteriogenesis16, 17, and maintenance of normal renal functions18. Thus, the pharmacological activation of P2Y 2 receptors can provide symptomatic relief of cardiovascular dysfunction.
Several nucleotide-based P2Y 2 agonists and an allosteric partial agonist have been identified and synthesized through extensive library screening in conjunction with molecular modelling19,20,21. One of the most advanced compounds is diquafosol, an analogue of UTP for the treatment of dry eye22. However, that compound is not suitable for oral administration because of high polarity and low intestinal absorption. Non-nucleotide P2Y 2 antagonists such as Reactive Blue 223, suramin24, AR-C11892525, 26 (a thiouracil derivative), anilinoanthraquinone derivatives27, and some flavonoids28 have been reported, but novel non-nucleotide P2Y2 selective agonists—compounds that could help elucidate the effects of P2Y2 activation in peripheral tissues—had yet to be discovered until now. Indeed, while the recent rapid advances in GPCR and ion channel crystallography have enhanced the practical understanding of purinergic receptors such as A2AAR, P2X4, P2Y1, and P2Y12 29,30,31,32,33,34, a structure-based approach to ligand design that is capable of predicting ligand selectivity and functionality remains under development35.
In this study, we investigated a novel highly selective non-nucleotide P2Y2 allosteric agonist: Compound 89, a 4(1H)-quinolinone derivative with the chemical name 2-((ethyl(4-fluorobenzyl)amino)methyl)-7,8-dimethylquinolin-4(1H)-one. Compound 89 was selected from our compound library based on its effect on Ca2+ mobilization in the 1321N1 human astrocytoma cell line. The 1321N1cells are useful for identifying P2Y isoform-specific agents, because they lack P2 receptors and are unresponsive to nucleotides36, 37. We also studied the effects of Compound 89 on Ca2+ signalling, nuclear receptor 4A (NR4A) gene induction, and isoproterenol-induced cardiac hypertrophy in neonatal rat cardiomyocytes (NRCMs).
Results
Compound 89 is a highly selective agonist for P2Y2 receptors relative to other P2Y receptors in P2Y-overexpressing 1321N1 cells
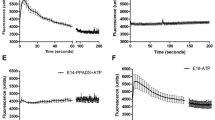
By using the 1321N1 human astrocytoma cell line, we were able to identify Compound 89 (Fig. 1a) from our compound library as a potential P2Y2 selective agonist. In our study, a FLIPR Ca2+ mobilization assay of hP2Y2–1321N1 cells revealed that Compound 89 had agonistic activity against human P2Y2, although it was less potent than ATP (Fig. 1b). Compound 89 also exhibited agonistic activity against mouse P2Y2 in mP2Y2-1321N1 cells (Fig. 1b), suggesting that agonistic activity is conserved across species. The calculated EC50 values (potency) for Compound 89 were 10.5 μM and 2.7 μM for human and mouse P2Y2 receptors, respectively; and the extrapolated maximum efficacies for Compound 89 with respect to 10 μM ATP were 65.8% and 50.9% for human and mouse P2Y2 receptors, respectively (Fig. 1b).

Compound 89 displays selective agonist activity for P2Y2 in P2Y-overexpressing 1321N1 cells. (a) The chemical structure of the 4(1H)-quinolinone derivative Compound 89: 2-((ethyl(4-fluorobenzyl)amino)methyl)-7,8-dimethylquinolin-4(1H)-one. (b) Agonistic activity against human and mouse P2Y2 receptors in a FLIPR Ca2+ mobilization assay using 1321N1 cells that were transiently-expressing hP2Y2 receptors (hP2Y2-1321N1) and stably-expressing mP2Y2 receptors (mP2Y2-1321N1). The activity of 10 μM ATP was normalized to 100%. (c) Agonistic activity against human P2Y1, P2Y4, P2Y6, and P2Y11 receptors in FLIPR Ca2+ mobilization assays utilising their respective transiently-expressing 1321N1 cells. The agonistic activity of 10 μM ADP for hP2Y1, UTP for hP2Y4, UDP for hP2Y6, and ATP for hP2Y11 was normalized to 100%. (d) Agonistic activity against human P2Y12 receptor in a cAMP accumulation assay using stably-expressing hP2Y12-1321N1 cells. The activity of 1 μM ADP was normalized to 100%. All data points represent the mean in duplicate or quadruplicate and comparable results were obtained from another independent experiment.
The selectivity of Compound 89 for P2Y2 was analysed in hP2Ys-1321N1 cells. The P2Y receptors were grouped into two subgroups: five Gαq/11-coupled receptors (P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11) and three Gαi/o-coupled receptors (P2Y12, P2Y13, and P2Y14)1, 11. While endogenous nucleotide ligands showed clear dose-response patterns, Compound 89 (at up to 30 µM) lacked agonistic activity against the remaining four Gαq/11-coupled hP2Y receptors in the FLIPR Ca2+ mobilization assay (P2Y1, P2Y4, P2Y6 and P2Y11), and one representative Gαi/o-coupled hP2Y12 receptor in the cAMP accumulation assay (Fig. 1c and d). Thus, Compound 89 appeared to selectively activate hP2Y2 receptors. For comparison, transfection experiments confirmed that 10 μM concentrations of endogenous P2Y ligands (ATP, UTP, adenosine-5′-diphosphate [ADP], and uridine-5′-diphosphate [UDP]) lacked activity in empty vector-transfected parental 1321N1 cells (data not shown), which is consistent with the previous reports36, 37.
Compound 89 exhibits allosteric modulation of ATP-induced Ca2+ mobilization in P2Y2-overexpressing 1321N1 cells and neonatal rat cardiomyocytes
To explore the interaction between Compound 89 and endogenous ATP during P2Y2 activation, we investigated how Compound 89 affects the agonistic activity of ATP and vice versa. In a FLIPR Ca2+ mobilization assay utilizing hP2Y2-1321N1 cells, the concentration-response curves of three physiological agonists (ATP, UTP, and diadenosine tetraphosphate [Ap4A]) and the synthesized agonist 2-thio-UTP (a P2Y2 preferential agonist)38, 39 were evaluated (Fig. 2a, left). Addition of Compound 89 caused an upward and leftward shift in the ATP concentration-response curve, implying positive allosteric modulation of P2Y2 by ATP (an orthosteric agonist) and Compound 89 (Fig. 2b, left). The EC50 values for ATP decreased from 15.1 nM to 2.4 nM in the presence of 3 nM to 30 µM Compound 89, respectively. In comparison, addition of increasing concentrations of ATP caused a progressive upward and leftward shift followed by an upper plateau in the Compound 89 concentration-response curve. The EC50 value for Compound 89 decreased from 53.9 μM in the absence of ATP, to 2.5 μM in the presence of 10 nM ATP (Fig. 2b, right). Similar findings were observed with the endogenous agonists UTP (an orthosteric agonist) and Ap4A (Supplementary Fig. S1). These results demonstrate that in hP2Y2-expressing human astrocytoma 1321N1 cells, Compound 89 behaves as a probe-independent, highly selective P2Y2 positive allosteric modulator with intrinsic agonistic activity.

Compound 89 exhibits allosteric modulatory effects on ATP-induced Ca2+ mobilization in P2Y2-overexpressing 1321N1 cells and in cardiomyocytes. (a) Concentration-response curves of physiological agonists in a FLIPR Ca2+ mobilization assay using hP2Y2 stably-expressing 1321N1 cells and NRCMs. (b) Allosteric modulatory effect of Compound 89 on the ATP concentration-response curve (left), and allosteric modulatory effect of ATP on the Compound 89 concentration-response curve (right) in hP2Y2 stably-expressing 1321N1 cells. (c) Allosteric modulatory effect of Compound 89 on the ATP concentration-response curve (left), and the allosteric modulatory effect of ATP on the Compound 89 concentration-response curve (right) in NRCMs. The activity of 10 μM ATP was normalized to 100%. All data points represent the mean of experiments performed in duplicate or triplicate, and comparable results were obtained by another independent experiment.
We next examined the potency of Compound 89 under physiological conditions using NRCMs. In a FLIPR Ca2+ mobilization assay, ATP and 2-thio-UTP exhibited comparable concentration-response curves and maximum efficacies (Fig. 2a, right), implying that NRCMs can be used to estimate Gαq/11-mediated Ca2+ signalling downstream from P2Y2. Then, we evaluated how the agonistic activity of ATP was affected by the presence of Compound 89 in NRCMs. Compound 89 caused a left- and upward shift in the ATP concentration-response curve, indicating that the compound behaves as a positive allosteric modulator of P2Y2 even in NRCMs (Fig. 2c, left). In addition, the EC50 values for ATP decreased from 2.6 μM in the absence of Compound 89, to 0.53 μM in the presence of 10 μM Compound 89; and the maximum efficacy in the presence of 10 μM Compound 89 reached 130.7%. Likewise, a progressive upward and leftward shift in the Compound 89 concentration-response curve was obtained by the addition of increasing concentrations of ATP (Fig. 2c, right). The EC50 values for Compound 89 decreased from 1.3 μM in the presence of 1 μM ATP, to 0.11 μM in the presence of 30 μM ATP; and the maximum efficacy in the presence of 30 μM ATP reached 130.5%. Of note, Compound 89 displayed no intrinsic agonistic activity in NRCMs, which was not the case in hP2Y2-1321N1 cells (Fig. 2b and c, right), suggesting that the compound acts as a pure P2Y2 positive allosteric modulator in NRCMs.
Compound 89 allosterically enhances ATP-induced gene expression in cardiomyocytes
We sought to determine how P2Y2 activation in cardiomyocytes regulates the expression of the NR4A subfamily of orphan nuclear receptors, consisting of NR4A1 (Nur77), NR4A2 (Nurr1), and NR4A3 (Nor-1). These receptors, previously reported as negative-feedback regulators for hypertrophic stimuli in NRCMs40, are rapidly induced by a broad spectrum of stimuli41. We stimulated the receptors with endogenous agonist ATP alone and in combination with Compound 89 over 90 min and examined the time-course changes in the mRNA expression of Nr4a1, Nr4a2, Nr4a3 and another immediate early gene (IEG), c-fos. Stimulation with ATP at 10 μM increased the expression of all Nr4a genes steadily after 30 min, while c-fos expression was transiently induced with a single peak evident at 30 min (Fig. 3, upper). Co-administration of the compound at 10 and 30 μM enhanced ATP-induced gene expression changes in a concentration-dependent manner (Fig. 3, middle and lower). Interestingly, Compound 89 itself did not induce any changes in gene expression. These results suggest that Compound 89 serves as a pure positive allosteric regulator of P2Y2 that potentiates gene expression changes induced by ATP, which is consistent with results from our signalling assay (Fig. 2c).

Compound 89 allosterically potentiates ATP-induced changes in IEG gene expression in cardiomyocytes. Gene expression changes of IEGs, c-fos and the NR4A subfamily, in a time-dependent manner. Inductions by ATP (0, 1 or 10 μM) in the presence of Compound 89 (0, 10 or 30 μM) were plotted. Expression levels are normalized to Rplp0 and shown as means ± SEM (n = 3).
Compound 89 reverses cardiac hypertrophy induced by β-adrenergic agonists in cardiac myocytes
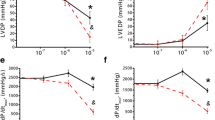
Earlier studies have demonstrated that exogenous ATP stimulation and NR4A1 induction have anti-hypertrophic effects on cardiac myocytes15, 40. Because Compound 89 can potentiate the expression of genes in the NR4A family, we speculated that it could exert an anti-hypertrophic effect in cardiac hypertrophy. To test this hypothesis, we evaluated the effects of Compound 89 on isoproterenol-induced cardiac hypertrophy in NRCMs. Before starting, we confirmed that Compound 89 does not affect β-adrenergic receptor-mediated cAMP signalling in parental 1321N1 cells, which have been reported to express endogenous β-adrenergic receptors42, 43 (Supplementary Fig. S2). Isoproterenol-induced cardiac hypertrophy was attenuated by Compound 89 in a concentration-dependent manner (Fig. 4a and b). In comparison, treatment with ATP produced no significant effects (P = 0.568, Fig. 4c). To interpret these results, we also analysed the changes in expression of Nr4a1 mRNA at 2 h, as well as the resulting hypertrophic scores at 48 h after the start of stimulation. As shown in Fig. 4d, the β-adrenergic agonist isoproterenol significantly induced Nr4a1 at 2 h (P = 5 × 10−4), in line with a previous report40. Induction of Nr4a1 was greater with Compound 89 than with isoproterenol alone, while ATP did not affect induction (Fig. 4d). These changes in expression corresponded well with alterations in cardiac hypertrophy. Complete attenuation of hypertrophy was observed after 48 h of treatment with Compound 89 (Fig. 4d), suggesting that early induction of NR4As determines the antihypertrophic phenotype for isoproterenol-induced cardiac hypertrophy.

Compound 89 exerts anti-hypertrophic effect in cardiomyocytes. (a) Fluorescent microscopic images of filamentous actin in NRCMs labelled with phalloidin-FITC, and imaged with 10 × 0.45 NA objective on a 96-well plate. (b,c) Quantification of the images and effects of (b) Compound 89 (1 to 30 μM) and (c) ATP (10 and 100 μM). Hypertrophic scores were calculated as the product of FITC-fluorescence intensity and cell area per cell. Data are shown as means ± SD (n = 3). # P < 0.025 versus 10 μM isoproterenol (ISO) alone, one-tailed Williams’ test. **P < 0.01 versus vehicle, Student’s t-test. (d) Changes in expression of Nr4a1 mRNA at 2 h, and hypertrophic scores at 48 h after P2Y2 activation by 100 μM ATP and/or 30 μM Compound 89 in the presence of 10 μM isoproterenol. Data are shown as mean ± SD (n = 4). **P < 0.01 versus vehicle, Aspin-Welch test (Nr4a1 mRNA) or Student’s t-test (hypertrophic scores). $$ P < 0.01 versus 10 μM isoproterenol (ISO) alone, Dunnett’s test. n.s., not significant.
Discussion
In this study, we demonstrated that Compound 89 is an allosteric agonist for P2Y2 that potentiates ATP-induced downstream responses, including Ca2+ influx and gene induction. Three key features distinguish Compound 89 from other compounds: structure, specificity, and allosteric agonistic properties. First, Compound 89 has a 4(1H)-quinolinone-based non-nucleotide structure (Fig. 1a). Given the difficulties of producing nucleotide analogues in medicinal chemistry (i.e. difficulties with high yield, reproducible synthesis, purification, etc.)44, the development of non-nucleotide P2Ys ligands is worth pursuing. Second, Compound 89 was capable of distinguishing between human P2Y2 and P2Y4 (Fig. 1b and c), the two closest phylogenetic subtypes1. In addition, its agonistic activity against mouse P2Y2 receptors (Fig. 1b) implied that Compound 89’s binding capacity is not species-specific, a speculation supported by the high (90%) similarity in amino acid sequence among human, mouse, and rat P2Y2 receptors. Finally, although Compound 89 displayed partial agonistic activity as well as positive allosteric modulation of ATP responses in P2Y2-overexpressing 1321N1 cells, it served as a purely positive allosteric modulator in NRCMs (Fig. 2b and c). Considering that partial agonistic activity is dependent on receptor expression levels and coupling efficacy45,46,47, the difference may be due to the variation in P2Y2 expression levels between hP2Y2-1321N1 cells and NRCMs.
A number of allosteric modulators have been discovered for class A GPCRs, such as adenosine receptors, free fatty acid receptors, and muscarinic receptors, some of which are supported by the X-ray co-crystallography48,49,50. In addition, a series of nucleotide-derivative P2Y2 allosteric agonists have previously been reported21; but as a non-nucleotide allosteric modulator of P2Y2, Compound 89 is in a potentially novel class. In a study by Van Poecke et al.21, a series of synthesized phosphonate analogues at their highest concentrations induced ~10% of the maximum response of UTP in hP2Y4 stably expressing 1321N1 cells, suggesting weak agonistic activity against human P2Y4 receptors. In our present study, Compound 89 showed agonistic activity against P2Y2, but not against P2Y1, P2Y4, P2Y6, and P2Y11 at up to 30 μM (Fig. 1b and c). Although subtle differences between assay systems exist, our results indicate that Compound 89 may be a truly selective P2Y2 allosteric agonist.
To clarify the role of ATP/P2Y2-mediated signalling in NRCMs, we investigated the regulation of putative target genes downstream of P2Y2. The NR4A subfamily, consisting of NR4A1 (Nur77), NR4A2 (Nurr1), and NR4A3 (Nor-1), is a group of transcriptional regulators involved in the control of tissue metabolism and energy balance. The functional significance of the NR4A family in the heart has not been elucidated well in comparison to other tissues such as the liver, skeletal muscles, and adipose tissues. However, the NR4A family has been implicated in cardiac remodelling that occurs in response to β-adrenergic stimulation51. In addition, NR4A1 has recently been reported to function as a negative-feedback regulator for hypertrophic stimuli in cardiomyocytes40. Although NR4As are rapidly and transiently induced by diverse stimuli such as receptor agonists, activators for intracellular mediators, mechanical and chemical factors, and exercise41, P2Y2-mediated upregulation of NR4As has not been fully described. Given the multiplicity of NR4A stimulators and their high sensitivity, it is surprising that Compound 89 alone did not induce expression of NR4As, but only enhanced ATP-induced NR4A responses, thus reflecting Compound 89’s pharmacological property as a pure positive allosteric modulator of P2Y2 in NRCMs (Figs 2c and 3).
Compound 89 inhibited isoproterenol-induced hypertrophy in NRCMs in a concentration-dependent manner (Fig. 4a and b). Mechanistically, Zheng, et al.15 previously reported that ATP signalling, presumably at least partly mediated via P2Y2, could induce transcriptional activation that is somewhat distinct from that induced by other Gαq/11-mediating neurohumoral hypertrophic factors such as norepinephrine and phenylephrine. Such a difference would result in a unique anti-hypertrophic effect, although further mechanistic analysis would be required. In the present study, Compound 89’s anti-hypertrophic activity was not affected by the presence of ATP (Fig. 4d). One possible explanation is that ATP released by isoproterenol-induced contractions of NRCMs reached a plateau in the pericellular area, thus abrogating the effect of exogenous ATP stimulation. This hypothesis is supported by the fact that intracellular ATP is released into pericellular space in response to various mechanical and biochemical stimuli6. Aside from skeletal and cardiac muscles themselves, multiple potential sources exist for interstitial ATP including the surrounding nerve endings, blood cells, and endothelium, via vesicular and conductive mechanisms of exocytosis, apoptosis, Cl− channels and pannexin hemichannels52, 53.
To date, the autocrine release of ATP via pannexin 1 hemichannels from cardiac myocytes has been substantiated through the combined use of cultured cardiac myocytes and bioluminescent probes54, 55. Those reports have estimated extracellular ATP levels in the mid-nanomolar range, which is likely to fall short of EC50 values (low micromolar range; Fig. 2a). However, the actual ATP concentration very close to the cell membrane surface could be much higher, although almost impossible to measure accurately considering the nucleotide-degrading system based on ectonucleotidases such as CD39 and CD738. ATeams, a new class of genetically encoded fluorescent ATP indicators, might help to overcome these limitations56. At any rate, it is possible that greater induction of NR4As by a combination of β-adrenergic stimulation and P2Y2 allosteric potentiation resulted in the attenuation of cardiac hypertrophy shown in Fig. 4d.
In summary, our results demonstrate that Compound 89, a 4(1H)-quinolinone derivative, is a potentially novel non-nucleotide allosteric P2Y2 agonist that inhibits the development of β-adrenergic agonist–induced cardiac hypertrophy through activation of NR4As. Our data provided clues for the development of a new series of P2Y2 allosteric agonists, which will help elucidate the therapeutic potential of P2Y2 activation in various cardiovascular diseases.
Materials and Methods
Reagents
Compound 89, also known as 2-((ethyl(4-fluorobenzyl)amino)methyl)-7,8-dimethylquinolin-4(1H)-one, was purchased from ChemDiv (San Diego, USA). Adenosine-5′-diphosphate monosodium salt, ATP disodium salt, UDP monosodium salt, and isoproterenol hydrochloride were purchased from Wako Pure Chemicals (Osaka, Japan). Uridine-5′-triphosphate was purchased from Sigma-Aldrich Japan (Tokyo, Japan). 2-Thio-UTP was purchased from Tocris Bioscience (Bristol, UK). Diadenosine 5′,5′″-P1,P4- tetraphosphate was purchased from Jena Bioscience (Jena, Germany).
Animals
Newborn pups (2–4 days old) born from female Wistar rats (CLEA Japan) were used for primary culturing of cardiomyocytes. The animals were housed in an environment with a 12 h dark-light cycle (lights on at 7:00 a.m.), and fed food (CE-2, CLEA Japan) and tap water ad libitum. All experiments were performed in accordance with protocols reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Takeda Pharmaceutical Company, Ltd.
Cell preparation
The human astrocytoma 1321N1 cell lines, which stably express human P2Y2, P2Y12, and mouse P2Y2 receptors (i.e. hP2Y2-, hP2Y12-, and mP2Y2-1321N1 cells), were established at the Takeda Pharmaceutical Company (Fujisawa, Japan). The 1321N1 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated foetal bovine serum, 100 IU/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific Japan). The aminoglycoside G-418 (0.5 mg/mL; Thermo Fisher Scientific) was added to culture media only when selection was made. For transient expression studies, human P2Ys/pcDNA3.1(+) plasmids were transfected into 1321N1 cells using Lipofectamine 3000 (Thermo Fisher Scientific).
Neonatal rat cardiomyocytes were isolated from the hearts of Wistar rats (2–4 days old) through enzymatic dissociation. In brief, isolated hearts were minced and digested by stirring in 1% collagenase II (Worthington, NJ, USA) at 37 °C for 30 min, then cultured at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air for 90 min to allow fibroblasts to attach to the dish. Floating NRCMs were then collected and seeded in a collagen I-coated 96-well plate using DMEM/Nutrient Mixture F-12 supplemented with 10% foetal bovine serum, penicillin (100 IU/mL), and streptomycin (100 μg/mL) until use.
Ca2+ mobilization assay
To assess intracellular Ca2+ levels in P2Ys-expressing 1321N1 cell lines, cells were seeded in 96- or 384-well plates, and incubated at 37 °C for 1 h in HEPES-buffered Hank’s balanced salt solution (HBSS; pH = 7.4) containing 2.5 mM probenecid, 0.1% fatty acid-free bovine serum albumin, and Ca2+ indicator dyes: a wash-type acetoxymethyl ester of Fluo-4 (Fluo-4 AM; Dojindo, Japan) or a homogenous no-wash type dye (Fluorometric Imaging Plate Reader [FLIPR] calcium assay kit; Molecular Devices Japan). For NRCMs, isolated cells were seeded in collagen I-coated 96-well plates and a FLIPR calcium assay kit was used to reduce cell damage. Intracellular Ca2+ levels over time were measured before and after the addition of ligands by using a FLIPR. Fluorescence intensity was defined as the difference between the maximum and minimum values during the measurement period, normalized to the intensity produced by submaximal concentrations of endogenous nucleotide ligands for individual P2Y receptors as indicated in each figure. The EC50 and Emax values for each curve were determined through data analysis and curve-fitting using a 4-parameter logistic equation in Prism 5 software (GraphPad Software, CA, USA).
cAMP accumulation assay
To measure the intracellular cyclic adenosine monophosphate (cAMP) levels in hP2Y12-1321N1 cells, a cAMP cell-based Dynamic 2 (d2) assay kit based on homogeneous time resolved fluorescence (HTRF Technology; Cisbio Bioassays, France) was used according to the attached manual. Suspended cells were stimulated with 1 μM forskolin and ligands, followed by incubation for 30 min at room temperature. Then, cAMP-d2 and anti-cAMP-cryptate reagents in lysis buffer were added. The lysate was incubated for 60 min at room temperature and then analysed using an EnVision plate reader (Thermo Fisher Scientific).
mRNA expression analysis
Neonatal rat cardiomyocytes were seeded in a collagen I-coated 96-well plate, then cultured and maintained for a week before the ligand stimulation assay. The culture medium was replaced by HBSS buffer containing 20 mM HEPES and then stimulated with the indicated ligands for 15 to 90 min. After stimulation, cDNA samples were synthesized from lysates using TaqMan Gene Expression Cells-to-Ct kits (Thermo Fisher Scientific) according to the manufacturer’s instructions, followed by quantitative real-time polymerase chain reaction analysis using a Prism 7900HT sequence detector (Thermo Fisher Scientific). Thermal cycling parameters were 2 min at 50 °C and 10 min at 95 °C, followed by 40 cycles at 95 °C for 15 sec and 60 °C for 1 min. The mRNA levels were analysed with the comparative Ct method (2−ΔΔCt) using Rplp0 as the housekeeping gene. The TaqMan Gene Expression assays (Thermo Fisher Scientific) used herein were as follows: Rn01533237_m1 (Nr4a1), Rn00570936_m1 (Nr4a2), Rn01354012_m1 (Nr4a3), Rn02396759_m1 (c-fos), and Rn03302271_gH (Rplp0).
Cardiac hypertrophy measurements
Neonatal rat cardiomyocytes were seeded in a 96-well collagen I plate at a density of 5000 cells/well. They were incubated overnight and then transferred to a serum-free medium for one day. Next, the cells were treated with the indicated concentrations of compounds and 10 μM isoproterenol, and then incubated for 2 days. To quantify cardiomyocyte hypertrophy via filamentous actin staining, NRCMs were fixed with 4% paraformaldehyde (Wako) and permeabilized with 0.1% Triton X-100 (MP Biomedicals, France), followed by blocking with 1% bovine serum albumin (Wako) and staining with 2 μM phalloidin–fluorescein isothiocyanate (phalloidin-FITC; Sigma-Aldrich, Japan) and 10 μM Hoechst 33258 (Dojindo). Quantification of FITC-fluorescence intensity and cell number was performed using an IN Cell Analyzer 6000 (GE Healthcare Japan). The signals from obtained images were quantitated by means of a customized image analysis algorithm using the IN Cell Developer Toolbox (GE Healthcare Japan). Hypertrophic scores were calculated as the product of FITC-fluorescence intensity and cell area per cell as programmed by GE Healthcare technical support experts.
Data analysis and statistics
The dose-response relationships in Fig. 4b and c were tested using a Williams or Shirley-Williams test conducted at a one-tailed significance level of P < 0.025, based on results of the homogeneity of variance test (Bartlett’s test). The Aspin-Welch test or Student’s t-test between two groups, and Dunnett’s multiple-comparison test were performed at a significance level of P < 0.05 to analyse for statistical significance in Fig. 4d. All statistical analyses were performed using Statistical Analysis System version 9.3 (SAS Institute, NC, USA).
Data availability
All datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Abbracchio, M. P. et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 58, 281–341, doi:10.1124/pr.58.3.3 (2006).
Erlinge, D. & Burnstock, G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. 4, 1–20, doi:10.1007/s11302-007-9078-7 (2008).
Regard, J. B., Sato, I. T. & Coughlin, S. R. Anatomical profiling of G protein-coupled receptor expression. Cell. 135, 561–571, doi:10.1016/j.cell.2008.08.040 (2008).
Idzko, M., Ferrari, D. & Eltzschig, H. K. Nucleotide signalling during inflammation. Nature. 509, 310–317, doi:10.1038/nature13085 (2014).
Moore, D. J. et al. Expression pattern of human P2Y receptor subtypes: a quantitative reverse transcription-polymerase chain reaction study. Biochim Biophys Acta. 1521, 107–119 (2001).
Corriden, R. & Insel, P. A. Basal release of ATP: an autocrine-paracrine mechanism for cell regulation. Sci Signal. 3, re1, doi:10.1126/scisignal.3104re1 (2010).
Erlinge, D. P2Y receptors in health and disease. Adv Pharmacol. 61, 417–439, doi:10.1016/B978-0-12-385526-8.00013-8 (2011).
Falzoni, S., Donvito, G. & Di Virgilio, F. Detecting adenosine triphosphate in the pericellular space. Interface Focus. 3, 20120101, doi:10.1098/rsfs.2012.0101 (2013).
Davies, P. F. Flow-mediated endothelial mechanotransduction. Physiol Rev. 75, 519–560 (1995).
Wu, X. et al. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 116, 675–682, doi:10.1172/JCI27374 (2006).
Erb, L. & Weisman, G. A. Coupling of P2Y receptors to G proteins and other signaling pathways. Wiley Interdiscip Rev Membr Transp Signal. 1, 789–803, doi:10.1002/wmts.62 (2012).
Cardenas, C. et al. Nuclear inositol 1,4,5-trisphosphate receptors regulate local Ca2+ transients and modulate cAMP response element binding protein phosphorylation. J Cell Sci. 118, 3131–3140, doi:10.1242/jcs.02446 (2005).
Djerada, Z., Feliu, C., Richard, V. & Millart, H. Current knowledge on the role of P2Y receptors in cardioprotection against ischemia-reperfusion. Pharmacol Res., doi:10.1016/j.phrs.2016.08.009 (2016).
Cohen, R. et al. UTP reduces infarct size and improves mice heart function after myocardial infarct via P2Y2 receptor. Biochem Pharmacol. 82, 1126–1133, doi:10.1016/j.bcp.2011.07.094 (2011).
Zheng, J. S. et al. Extracellular ATP inhibits adrenergic agonist-induced hypertrophy of neonatal cardiac myocytes. Circ Res. 78, 525–535 (1996).
Wang, S. et al. P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. J Clin Invest. 125, 3077–3086, doi:10.1172/JCI81067 (2015).
McEnaney, R. M., Shukla, A., Madigan, M. C., Sachdev, U. & Tzeng, E. P2Y2 nucleotide receptor mediates arteriogenesis in a murine model of hind limb ischemia. J Vasc Surg. 63, 216–225, doi:10.1016/j.jvs.2014.06.112 (2016).
Potthoff, S. A. et al. P2Y2 receptor deficiency aggravates chronic kidney disease progression. Front Physiol. 4, 234, doi:10.3389/fphys.2013.00234 (2013).
Jacobson, K. A. Structure-based approaches to ligands for G-protein-coupled adenosine and P2Y receptors, from small molecules to nanoconjugates. J Med Chem. 56, 3749–3767, doi:10.1021/jm400422s (2013).
Hillmann, P. et al. Key determinants of nucleotide-activated G protein-coupled P2Y2 receptor function revealed by chemical and pharmacological experiments, mutagenesis and homology modeling. J Med Chem. 52, 2762–2775, doi:10.1021/jm801442p (2009).
Van Poecke, S. et al. Synthesis and P2Y2 receptor agonist activities of uridine 5′-phosphonate analogues. Bioorg Med Chem. 20, 2304–2315, doi:10.1016/j.bmc.2012.02.012 (2012).
Lau, O. C., Samarawickrama, C. & Skalicky, S. E. P2Y2 receptor agonists for the treatment of dry eye disease: a review. Clin Ophthalmol. 8, 327–334, doi:10.2147/OPTH.S39699 (2014).
Brown, J. & Brown, C. A. Evaluation of reactive blue 2 derivatives as selective antagonists for P2Y receptors. Vascul Pharmacol. 39, 309–315 (2002).
Charlton, S. J. et al. PPADS and suramin as antagonists at cloned P2Y- and P2U-purinoceptors. Br J Pharmacol. 118, 704–710 (1996).
Rafehi, M., Burbiel, J. C., Attah, I. Y., Abdelrahman, A. & Muller, C. E. Synthesis, characterization, and in vitro evaluation of the selective P2Y2 receptor antagonist AR-C118925. Purinergic Signal. 13, 89–103, doi:10.1007/s11302-016-9542-3 (2017).
Meghani, P. The design of P2Y2 antagonists for the treatment of inflammatory diseases. Abstract of papers, 224th ACS National Meeting, Boston, MA. (2002).
Weyler, S. et al. Combinatorial synthesis of anilinoanthraquinone derivatives and evaluation as non-nucleotide-derived P2Y2 receptor antagonists. Bioorg Med Chem Lett. 18, 223–227, doi:10.1016/j.bmcl.2007.10.082 (2008).
Kaulich, M. Flavonoids - novel lead compounds for the development of P2Y2 receptor antagonists. Drug Devel Res. 59, 72–81 (2003).
Jaakola, V. P. et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 322, 1211–1217, doi:10.1126/science.1164772 (2008).
Xu, F. et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 332, 322–327, doi:10.1126/science.1202793 (2011).
Zhang, K. et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 509, 115–118, doi:10.1038/nature13083 (2014).
Zhang, J. et al. Agonist-bound structure of the human P2Y12 receptor. Nature. 509, 119–122, doi:10.1038/nature13288 (2014).
Zhang, D. et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature. 520, 317–321, doi:10.1038/nature14287 (2015).
Kawate, T., Michel, J. C., Birdsong, W. T. & Gouaux, E. Crystal structure of the ATP-gated P2X4 ion channel in the closed state. Nature. 460, 592–598, doi:10.1038/nature08198 (2009).
Jacobson, K. A. et al. John Daly Lecture: Structure-guided Drug Design for Adenosine and P2Y Receptors. Comput Struct Biotechnol J. 13, 286–298, doi:10.1016/j.csbj.2014.10.004 (2015).
Lazarowski, E. R., Watt, W. C., Stutts, M. J., Boucher, R. C. & Harden, T. K. Pharmacological selectivity of the cloned human P2U-purinoceptor: potent activation by diadenosine tetraphosphate. Br J Pharmacol. 116, 1619–1627 (1995).
Filtz, T. M., Li, Q., Boyer, J. L., Nicholas, R. A. & Harden, T. K. Expression of a cloned P2Y purinergic receptor that couples to phospholipase C. Mol Pharmacol. 46, 8–14 (1994).
Ko, H. et al. Synthesis and potency of novel uracil nucleotides and derivatives as P2Y2 and P2Y6 receptor agonists. Bioorg Med Chem. 16, 6319–6332, doi:10.1016/j.bmc.2008.05.013 (2008).
El-Tayeb, A., Qi, A. & Muller, C. E. Synthesis and structure-activity relationships of uracil nucleotide derivatives and analogues as agonists at human P2Y2, P2Y4, and P2Y6 receptors. J Med Chem. 49, 7076–7087, doi:10.1021/jm060848j (2006).
Yan, G. et al. Orphan Nuclear Receptor Nur77 Inhibits Cardiac Hypertrophic Response to Beta-Adrenergic Stimulation. Mol Cell Biol. 35, 3312–3323, doi:10.1128/MCB.00229-15 (2015).
Pearen, M. A. & Muscat, G. E. Minireview: Nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol. 24, 1891–1903, doi:10.1210/me.2010-0015 (2010).
Toews, M. L., Harden, T. K. & Perkins, J. P. High-affinity binding of agonists to beta-adrenergic receptors on intact cells. Proc Natl Acad Sci USA 80, 3553–3557 (1983).
Toll, L. et al. β2-adrenergic receptor agonists inhibit the proliferation of 1321N1 astrocytoma cells. J Pharmacol Exp Ther. 336, 524–532, doi:10.1124/jpet.110.173971 (2011).
Jacobson, K. A., Jayasekara, M. P. & Costanzi, S. Molecular Structure of P2Y Receptors: Mutagenesis, Modeling, and Chemical Probes. Wiley Interdiscip Rev Membr Transp Signal. 1, doi:10.1002/wmts.68 (2012).
Hoyer, D. & Boddeke, H. W. Partial agonists, full agonists, antagonists: dilemmas of definition. Trends Pharmacol Sci. 14, 270–275 (1993).
McDonald, J. et al. Partial agonist behaviour depends upon the level of nociceptin/orphanin FQ receptor expression: studies using the ecdysone-inducible mammalian expression system. Br J Pharmacol. 140, 61–70, doi:10.1038/sj.bjp.0705401 (2003).
Yabuki, C. et al. A novel antidiabetic drug, fasiglifam/TAK-875, acts as an ago-allosteric modulator of FFAR1. PLoS One. 8, e76280, doi:10.1371/journal.pone.0076280 (2013).
Kruse, A. C. et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 504, 101–106, doi:10.1038/nature12735 (2013).
Srivastava, A. et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature. 513, 124–127, doi:10.1038/nature13494 (2014).
Jacobson, K. A. New paradigms in GPCR drug discovery. Biochem Pharmacol. 98, 541–555, doi:10.1016/j.bcp.2015.08.085 (2015).
Myers, S. A., Eriksson, N., Burow, R., Wang, S. C. & Muscat, G. E. Beta-adrenergic signaling regulates NR4A nuclear receptor and metabolic gene expression in multiple tissues. Mol Cell Endocrinol. 309, 101–108, doi:10.1016/j.mce.2009.05.006 (2009).
Lazarowski, E. R. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 8, 359–373, doi:10.1007/s11302-012-9304-9 (2012).
D’Hondt, C. et al. Pannexin channels in ATP release and beyond: an unexpected rendezvous at the endoplasmic reticulum. Cell Signal. 23, 305–316, doi:10.1016/j.cellsig.2010.07.018 (2011).
Nishida, M. et al. P2Y6 receptor-Gα12/13 signalling in cardiomyocytes triggers pressure overload-induced cardiac fibrosis. EMBO J. 27, 3104–3115, doi:10.1038/emboj.2008.237 (2008).
Godecke, S., Stumpe, T., Schiller, H., Schnittler, H. J. & Schrader, J. Do rat cardiac myocytes release ATP on contraction? Am J Physiol Cell Physiol. 289, C609–616, doi:10.1152/ajpcell.00065.2005 (2005).
Imamura, H. et al. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci USA 106, 15651–15656, doi:10.1073/pnas.0904764106 (2009).
Acknowledgements
We thank Drs Minoru Maruyama, Yoshiyuki Tsujihata, Nobuyuki Matsunaga, and Masahiro Oka for valuable discussions and suggestions, and Dr. Tomoko Satomi, Hiroki Nagase, and Toshihiro Yamamoto for their technical assistance. We also thank Dr. Yukio Yamada for helpful discussions, suggestions, and support for this study, which was financially supported by the Takeda Pharmaceutical Company Limited.
Author information
Authors and Affiliations
Contributions
K.S. conceived the project, and designed, performed, and analysed the experiments. H.N. and T.O. performed and analysed the experiments. M.N. and S.I. conceived the project and designed the experiments. K.S. and S.I. wrote the manuscript. All authors reviewed and approved the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sakuma, K., Nakagawa, H., Oikawa, T. et al. Effects of 4(1H)-quinolinone derivative, a novel non-nucleotide allosteric purinergic P2Y 2 agonist, on cardiomyocytes in neonatal rats. Sci Rep 7, 6050 (2017). https://doi.org/10.1038/s41598-017-06481-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06481-9
- Springer Nature Limited
This article is cited by
-
Analysis of two novel 1–4 quinolinone structures with bromine and nitrobenzyl ligands
Journal of Molecular Modeling (2019)