
Abstract
Population structure was investigated in 990 Botswana individuals according to ethno-linguistics, Bantu and Khoisan, and geography (the nine administrative districts) using the Identifiler autosomal microsatellite markers. Genetic diversity and forensic parameters were calculated for the overall population, and according to ethno-linguistics and geography. The overall combined power of exclusion (CPE) was 0.9999965412 and the combined match probability 6,28 × 10−19. CPE was highest for the Khoisan Tuu ethnolinguistic group and the Northeast District at 0.9999582029 and 0.9999922652 respectively. CMP ranged from 6.28 × 10−19 (Khoisan Tuu) to 1,02 × 10−18 (Northwest district). Using pairwise genetic distances (FST), analysis of molecular variance (AMOVA), factorial correspondence analysis (FCA), and the unsupervised Bayesian clustering method found in STRUCTURE and TESS, ethno-linguistics were found to have a greater influence on population structure than geography. FCA showed clustering between Bantu and Khoisan, and within the Bantu. This Bantu sub-structuring was not seen with STRUCTURE and TESS, which detected clustering only between Bantu and Khoisan. The patterns of population structure revealed highlight the need for regional reference databases that include ethno-linguistic and geographic location information. These markers have important potential for bio-anthropological studies as well as for forensic applications.
Similar content being viewed by others


Introduction
Botswana is a landlocked country in Southern Africa. It has 25 languages belonging to the Niger-Congo and Khoisan African language phyla1. The Niger-Congo language phylum is widespread through-out sub-Saharan Africa; with languages belonging to its Bantoid branch collectively referred to as Bantu2. Botswana Bantu languages are members of the Central-K (Subiya and Mbukushu), Central-R (Yeyi), and Central-S (Kgalagadi, Tswana and Kalanga) Bantu sub-groups2. Khoisan languages have distinctive click consonants and are widely used by hunter-gatherer (San) and pastoralist (Khoi) populations of southern Africa3. The Khoe-Kwadi, Kx’a, and Tuu Khoisan languages in Botswana are members of the Southern African Khoisan (SAK) family4,5,6. In this study, the terms “Bantu” and “Khoisan” are applied as they relate to linguistics and ethnicity (ethno-linguistics).
Khoisan speakers were the first inhabitants of Botswana7. The Khoi pastoralists appear in the archaeological record of southern Africa around 2000–1,200 years ago having migrated from eastern Africa5, 8, 9. Bantu speakers originated around the Cameroon/Nigeria border region about 5000–3000 years ago, and only arrived in southern Africa not earlier than the Khoi pastoralists8. The Kgalagadi, Tswana and Kalanga of the Central-S Bantu ethno-linguistic sub-group entered present day Botswana between the 15th and the 17th century, the Kalanga from the north (present day Zimbabwe), and the Kgalagadi and Tswana from the south-east (present day South Africa)7. The Yeyi (Central-R Bantu ethno-linguistic sub-group) and Mbukushu (Central-K Bantu ethno-linguistic sub-group) moved towards the Okavango Delta (Northwest district) in the 18th century from southwest Zambia, the Caprivi Strip and the Kwando and Linyanti Rivers; while the Subiya (Central-R Bantu ethno-linguistic sub-group) fled into Botswana from the Caprivi Strip in 187510.
There is little information about the genetic and demographic processes that influenced the genetic landscape of Botswana population groups today. In southern Africa, the admixture dynamics between the Bantu farmers and foraging San and pastoralist Khoi played an important role in shaping the current patterns of genetic diversity11,12,13,14,15. As the Bantu farmers entered new areas and intermarried with autochthonous populations, the local languages, cultures and genetic composition were influenced or even replaced by the Bantu16,17,18. There is strong evidence of sex-biased gene flow from autochthonous populations to the Bantu19, 20, and admixture and assimilation partly involving now extinct autochthonous populations20. Besides anthropological evidence, linguistically, click consonants (distinctive to Khoisan languages) have been borrowed into southern African Bantu languages such as the Central-S Bantu Nguni (found in South Africa), and the Central-K Mbukushu and Central-R Yeyi from Botswana21, 22.
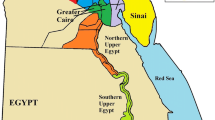
Today, Botswana has approximately two million inhabitants, of which over 90% are Bantu and only 3% are Khoisan and geographically, the country is divided into nine administrative districts: Central, Ghanzi, Kgalagadi, Kgatleng, Kweneng, Northeast, Northwest, Southeast, and Southern district (see Fig. 1). While the Bantu ethno-linguistic population groups are dispersed throughout the country, the Khoisan ethno-linguistic population groups are mainly concentrated in the sparsely populated central regions of the country near the Kalahari Desert with populations of less than 1 inhabitant per 10 km2 (see Supplementary Fig. S1). The majority of the population is concentrated in the south-east regions (Southern and Southeast districts) of the country with the Khoisan groups being concentrated in the Northwest and Ghanzi districts.

Botswana Administrative Districts and sampling locations made with R 3.2.479 (http://www.R-project.org) package ggplot2 (https://cran.r-project.org/web/packages/ggmap/citation.html)80 and package ggmap (https://cran.r-project.org/web/packages/ggplot2/citation.html)81.
The official country census makes no reference to ethnicity because at independence from the British in 1966 the government attempted to create an ethnic-blind state by placing all the Bantu ethno-linguistic groups found in Botswana under the umbrella of the Tswana23. Even though no identification of ethnicity is recorded, this information could potentially be inferred from individual genotypic data. Both historic and demographic factors had an influence on the current amount and distribution of genetic variation in Botswana (though the levels of their influence are uncertain)19, 20. Evidence of genetic structure between the Bantu and Khoisan has been detected using over 1, 000 nuclear microsatellites in 121 African populations24; 1 million autosomal markers in 103 Bantu and Khoisan southern African populations14; 900 complete mitochondrial DNA sequences in southern African populations25; however this has not been investigated with forensic markers.
Forensic markers have been poorly investigated in Botswana26, 27. Results from these few studies indicate high levels of polymorphism detected with the forensic kit AmpFLSTR Profiler Plus PCR Amplification Kit (Applied Biosystems)26 (commercial forensic kit preceding AmpFlSTR Identifiler PCR Amplification Kit) and AmpFLSTR Yfiler PCR Amplification Kit Y-STR markers27 indicating their potential for forensic and bio-anthropological studies. Analysis with Y-filer markers also revealed a lack of geographic, regional and ethnic variation in Botswana. The Botswana police has adopted the AmpFlSTR Identifiler PCR Amplification Kit (Identifiler) for forensic investigations. Identifiler is a commonly used commercial multiplex PCR kit designed to amplify the 13 core STR loci from the FBI Combined DNA Index System (CODIS), and two additional markers (D2S1338 and D19S433), plus a homologous region of the Amelogenin gene on the X and Y chromosomes. It has been used to generate relevant reference data on various worldwide and African populations28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43 as well as elucidating the population structure of samples from Rwanda28, Namibia34, Sudan38, and South Africa42, 43.
In this work, the widely utilized forensic kit Identifiler was used to investigate the population structure in Botswana, according to ethno-linguistics and geography. For this, we applied summary statistics, multivariate analysis, and model-based Bayesian clustering methods. These markers were further investigated for their ability to provide ancestry information of random individuals. These analyses of the patterns and distribution of genetic diversity are discussed in a forensic and bio-anthropological context.
Results and Discussion
Rare ‘variant’ alleles, ‘off-ladder’ alleles, and tri-allelic patterns
Rare ‘variant’ are alleles that differ from common variants which fall within virtual bins, while ‘off-ladder’ alleles are those that do not size the same as consensus alleles present in the allelic ladder and are not found in the bin set44. A total of 15 ‘rare variant’ alleles and 9 ‘off ladder’ alleles were found in this study (Supplementary Table S1). All rare variants have been previously reported (Supplementary Table S1).
Two types of tri-allelic patterns have been distinguished by Clayton et al.45: Type 1 with two alleles having different peak height intensity to the third allele, and Type 2 with peaks with even intensity. Tri-allelic patterns in this study were only detected at TPOX locus all Type 2 (Supplementary Figure S1). All these involved allele 10, and eight of the 10 samples were female. The extra allele 10 has been theorized as being a translocation of allele 10 onto the X-chromosome (X-linked) as a higher frequency of women have presented with tri-allelic patterns than men46, 47. The transmission of the tri-allelic genotype by mothers and fathers showed that fathers only transmitted the tri-allelic pattern to daughters, while the sons and daughters received the tri-allelic genotype equally from their mothers, evidence of X-chromosome inheritance46. Lane46 hypothesized that the translocation of the extra TPOX allele with the X-chromosome within African populations occurred prior to the Bantu expansions because these occurrences were found in South African, Namibian and Ghanaian populations. Ristow et al.43, suggested that the driving force of the high frequency of the tri-allelic genotype in South African populations is the cultural practice of polygamy, also practiced in Botswana.
Genetic diversity parameters
The genetic diversity parameters of the overall Botswana population, and according to ethno-linguistics and geography are shown in Supplementary Table S3 (A-P). Deviations from HWE were found at CSF1PO and D19S433 in the overall population and were due to heterozygote deficiency. The Central-R sub-group from the Bantu ethno-linguistic population groups displayed deviation from HWE at locus D13S317. The Khoisan ethno-linguistic sub-group Tuu showed deviation from HWE at the D19S433 and TPOX locus.
Deviations from HWE were observed for CSF1PO, D19S433, and FGA in the Ghanzi district, and for CSF1PO and D18S51 in the Kgalagadi and Southeast districts respectively. Locus D19S433 is the only locus in which deviations from HWE were found in both ethno-linguistic and geographic population groups. This observation can be linked to the high concentration of Khoisan in the Ghanzi district. Schlebusch et al.39 also detected departure from HWE for D19S433 in San populations of South Africa. The CSF1PO locus was also seen to deviate from HWE in populations from Uganda (Karamoja)35 and Angola37.
A review by Dakin and Avise48 revealed that silent alleles at frequencies normally reported in literature are unlikely to introduce serious biases in average exclusion probabilities. However, they can introduce errors that may lead to false exclusions of maternity or paternity in individual assessment. Amorim and Carneiro49 reported that the presence of silent alleles may lower paternity index (PI) ratios in trios. Therefore, the presence of silent alleles should be taken into account in all forensic analysis. The vWA locus was reported to show high frequencies of silent alleles in South African Bantu and Coloured populations50. Therefore, in absence of trio genotypes, we investigated the possibility of silent alleles applying the maximum likelihood method incorporated in ML-NullFreq51 (See Supplementary Table S4). No appreciable levels of silent alleles were detected at any locus except for the Khoisan Tuu, with a proportion of 0.17 at locus D19S433. Therefore, the presence of silent allele/s might explain the deviation from HWE and heterozygote deficiency in this ethno-linguistic sub-group.
Evaluation of population heterogeneity
Summary statistics, F ST and AMOVA, multivariate methods, and unsupervised model-based Bayesian clustering were used to evaluate population structure in Botswana. The comparative study based on F ST showed significant differences between the self-declared Bantu and Khoisan ethno-linguistic groups (n = 990; FST = 0.01213; P = 0.00000). Supplementary Table S5 shows F ST results comparing the language subgroups of the Bantu (Central-K, -R, -S n = 747) and Khoisan (Khoe-Kwadi, Kx’a, Tuu n = 223). There were significant differences between all the ethno-linguistic subgroups with the exception of Central-K and Central-R Bantus. The F ST analysis between the administrative districts revealed significant differences between the Ghanzi district and all other districts except the Kgalagadi and Kgatleng districts; between the Southern and Northwest district; and between the Northwest and the Kweneng and Central districts (Supplementary Table S6).
Non-hierarchical AMOVA testing for ethno-linguistic and geographical variation showed greater variation amongst self-declared ethno-linguistic groups than among the geographic groups at 3.30% and 1.54% respectively (Table 1 Test 1 and 3). Hierarchical AMOVA testing for ethno-linguistic variation between the self-declared Bantu and Khoisan ethno-linguistic population groups (n = 970) showed greater within population variation (95.63%) than among populations within groups (1.00%) (Table 1 Test 2). Hierarchical AMOVA testing for geographical variation between the northern and southern districts (n = 990) showed 0.12% variation amongst groups; and higher variation within populations at 98.39% than amongst populations within groups at 1.49% (Table 1 Test 4). Variation among populations within groups was higher when testing for geographic heterogeneity (1.49%) than for ethno-linguistic heterogeneity (1.00%).
The relationship between Bantu and Khoisan self-declared sub-language ethno-linguistic groups is further illustrated using factorial correspondence analysis (FCA) (Fig. 2a) Bantu Central-S is an outlier. The Bantu Central-K and-R cluster together separated from the Khoisan sub-groups. This is a result of their common origin (southwest Zambia) and subsequent gene flow as they settled in the same geographical region (Northwest district)10 The FCA comparing the nine administrative districts illustrated in Fig. 2b shows the Ghanzi district is an outlier. The Northwest, Ghanzi, and Kgalagadi districts are slightly separated from the cluster formed by the remaining six administrative districts. This result illustrates both the close relationship of the Bantu Central-K and -R as well as the districts with Khoisan can be seen as outliers.

(a) Factorial correspondence analysis (FCA) of the language subgroups of the Bantu (Central-K, -R and -S) and Khoisan (Tuu, Kx’a, and Khoe-Kwadi) speaking people of Botswana. (b) Factorial correspondence analysis (FCA) of the nine administrative districts of Botswana.
F ST , AMOVA, and FCA show evidence of the greater influence of ethno-linguistics on population structure than geography. The partition of genetic variation within Botswana is due to ethno-linguistics which consequently influences the geographic distribution of genetic variation due to the geographic pattern of genetic distribution as evidenced by F ST , AMOVA and FCA data.
The results of the model-based cluster analysis method in STRUCTURE are shown in Fig. 3, assuming K = 1–6 ancestral components. The ΔK method52 identified three populations as having the highest level of genetic structuring. Three genetic clusters were also identified by InP(D | K) as the most likely K using the Evanno method53. TESS results indicate that there are two district genetic clusters in the Botswana population as seen in the plateau seen in the deviance information criterion (DIC) plot from the no-admixture model implemented in TESS. The ethno-linguistic population groups Bantu and Khoisan are distinguishable at K = 2 up until K = 6. Clustering analysis indicates that the largest concentration of the Khoisan ethno-linguistic group is in the Ghanzi district (Fig. 3). This is in accordance with the geographical data of this study (see Supplementary Table S7). Figure 3 also show two distinct Khoisan clusters in the Northwest district that correspond to two sampling sites on the Okavango Delta (Gudigwa and Xaixai) that are dense in Khoe-Kwadi and Kx’a people respectively. Using TESS results, we were able to show a geographical representation of the admixture coefficients through spatial kriging (Fig. 4). The Bantu and Khoisan ethno-linguistic population groups form distinct clusters with regions of admixture.

STRUCTURE analysis of Botswana individuals with Identifiler assuming K = 2 to 6. Colours represent the inferred ancestry from K ancestral populations and vertical black lines indicate the nine administrative districts.

Geographical representation of the admixture coefficients through spatial kriging with low (cool colours) to high (hot colours) representing mean (TESS) admixture proportions. Individuals are classed from the non-admixture analysis in TESS with different colour and/or shapes representing different clusters. The green represents the Bantu and the pink represents the Khoisan. Map created using R 3.0.372 (http://www.R-project.org) package spatial 7.3.97173 and maps 2.3.97274.
STRUCTURE and TESS analysis did not detect substructure within the Bantu ethno-linguistic population groups. However, FCA indicates substructure within the Botswana Bantu ethno-linguistic groups. Even though central and southern African Bantu speaking groups have been found to be genetically similar, the exact patterns of dispersal are still under study. Furthermore, Tishkoff et al.24 also found that ethno-linguistics and geography explained a significant proportion of the genetic differentiation found in African populations, most of the genetic variation was explained by ethno-linguistics. This is also evident in Botswana, which is consistent with the recent migration of Bantu speakers. The high degree of admixture evidenced in Botswana from the interaction between the Bantu farmers, foraging San, and pastoralist Khoi played an important role in shaping the current patterns of genetic diversity in the country11,12,13. This indicates that these markers have important potential for bio-anthropological studies as well as for forensic applications.
Assignment and Ancestry
The efficiency of Identifiler to identify ethnic origin (assignment) of the Botswana individuals to the two major ethno-linguistic groups was evaluated using two strategies: log likelihoods using WHICHRUN and proportions of ancestral components from STRUCTURE (Table 2). The ranked log likelihood ratios obtained with each individual are shown in Fig. 5, suggesting an important process of admixture from the Khoisan into the Bantu ethno-linguistic population group. The lowest rate of correct assignment was obtained with STRUCTURE which detected a higher proportion of admixed individuals (597) than WHICHRUN (124). Both methods detected a higher proportion of admixed individuals among self-declared the Bantu (87.7% for STRUCTURE and 91.9% with WHICHRUN) than in the Khoisan (12.1% with STRUCTURE and 8.77% with WHICHRUN). The highest proportions of admixed individuals are from the Central (28.6% using STRUCTURE 29.03% with WHICHRUN) and Northwest (28.8% with STRUCTURE and 34.7% with WHICHRUN) districts.

Log likelihood ratios of assignment for all 990 Botswana individuals to Bantu and Khoisan ethno-linguistic groups. The cut off range for Bantu was ≥0.477 (log103) and for Khoisan it was <−0.477. The self-declared Bantu individuals are indicated in blue and the Khoisan in maroon. The samples that fall within the purple dashes represent admixed individuals.
The most informative locus for the inference of ancestry was locus CSF1PO (I n = 0.154), with D7S820 (I n = 0.036) being the least informative (Table 3). The mean I n across the markers (and standard deviation) I n was 0.078 (0.035). This is lower than the average I n found for the Sudanese populations at 0.167 (0.070)38. These low I n values indicate that individually these markers are not best suited for the inference of ancestry.
The possibility of assignment of individuals to population groups is beneficial in forensic investigations54,55,56,57,58,59. Autosomal STRs are the markers of choice due to their highly polymorphic nature and significant potential for distinguishing individual identity32. These markers have also been used infer the ancestry of profiles54, 60,61,62. The accuracy of assignment depends on marker efficiency and the number of markers. The Identifiler STRs and other forensic markers were not selected for the inference of ancestry but for their ability to distinguish individuals. Algee-Hewitt63 found that markers with high individual identifiability also possess high population identifiably and that CODIS loci contain higher ancestry information than randomly chosen STRs.
This study has shown that the Identifiler markers were able to distinguish between the Bantu and Khoisan ethno-linguistic population groups in Botswana. The log-likelihood method is more efficient and faster at assignment than STRUCTURE, resulting in reduced computational time. However, this should be taken carefully due to the important levels of admixture detected in Botswana as a high proportion of individuals who self-declared to belong to the Bantu or Khoisan ethno-linguistic groups were found to be admixed. These results showed the influence of gene flow in the distribution of genetic diversity in Botswana, with a very important incorporation of the Khoisan into the Bantu gene pool seen mostly in the Northwest District. This study has found that the Identifiler markers contain information on population structure supporting findings by Phillips et al.61, Babiker et al.38, and Pereira et al.62.
Forensic parameters
Forensic parameters for each locus were estimated overall Botswana populations, and according to ethno-linguistics, and geography (Supplementary Table S9). The overall combined power of exclusion (CPE) was 0,9999965412. CPE according to ethno-linguistics ranged from 0,9999582029 (Khoisan Tuu) to 0,999998666 (Khoisan Kx’a); and according to districts ranged from 0,9999922652 (Northeast District) to 0,9999992679 (Kweneng District). Also noticeable is the fact that the Kgalagadi district genotypes did not show any homozygotes and therefore had a CPE value of 1. Compared to other African populations, the CPE was higher for the overall Botswana population than two east African populations (Somalia36 and Sudan)38 and the South African40 populations. It was lower than the southern African Namibia (Ovambo)34 and Angola30, and the east African Uganda (Buganda41 and Karamoja)35 populations.
The probability of obtaining a random match between individuals, the combined match probability (CMP), for the overall Botswana population as a whole was 6,28 × 10−19. CMP ranged from 6.28 × 10−19 (Khoisan Tuu) to 5.91 × 10−14 (Bantu Central-K) when tested according to ethno-linguistics; and ranged from 1,02 × 10−18 (Northwest district) to 5,12 × 10−15 (Kweneng district) when calculated according to geography. The highest CPM was higher than that detected in the South African39, 40, Sudanese38, and Ugandan (Buganda41 and Karamoja)35 populations, but lower than that found for the Equatorial Guinea population29. Forensic summary statistics results are comparable to other African populations29,30,31, 33,34,35,36,37,38,39,40,41.
Conclusions
This study shows the possibility of investigating bio-anthropological processes, admixture, gene flow and major ethnic affiliation of individuals using Identifiler. These markers a suitable in the identification of individuals. This is a step towards closing the gap in understanding the amount and distribution of genetic diversity in Botswana and understand their contributing factors in order to provide recommendations for the application of these markers for the Botswana police. The population structure found in Botswana illustrates the need for regional reference databases that includes ethno-linguistic and geographic location instead of a single national reference database of voluntary donors. This is a much needed step towards creating a regional or national reference DNA database.
Methods
Population samples
A total of 990 unrelated voluntary donors (Supplementary Table S7) from the nine administrative districts of Botswana (Fig. 1) were sampled for the study. Of these samples 752 self-declared as Bantu and 238 Khoisan speakers (Supplementary Table S8). Written informed consent was obtained from all the voluntary donors who participated in the study. Blood samples were collected using Whatman FTA cards (Whatman, Maidstone, Kent, UK). DNA was extracted using the Chelex 100 extraction protocol (Bio-Rad) following the manufacturer’s instructions. Approval for this study was provided by the University of Botswana ethics committee and the Ministry of Health Research and Development Committee of Botswana and were carried out in accordance with approved guidelines.
Genotyping
The samples were amplified using the AmpFlSTR Identifiler (Applied Biosystems, Foster City, CA) kit, containing the loci D8S1179, D21S11, D7S820, CSF1P0, D3S1358, TH01, D13S317, D16S539, D2S1338, D19S433, vWA, TPOX, D18S51, D5S818, FGA, and the Amelogenin locus for sex typing following the user’s manual recommendations. Fragment sizes were detected using the Applied Biosystems ABI 3100 genetic analyser and sized with GeneScan500-LIZ internal size standard (Applied Biosystems, Foster City, CA) following manufacturer’s protocols. Allele calling was performed using GeneMapper ID v 1.1 software (Applied Biosystems, Foster City, CA).
Analysis of genotype data
Genetic Diversity parameters
Genetic diversity parameters such as allele frequency were estimated using Genepop vs 4.2.264. Departures from Hardy-Weinberg equilibrium (HWE), observed (Ho) and expected (He) heterozygosity were calculated using Arlequin vs 3.5.1.365. P-values for HWE were executed with 106 steps in the Markov chain and 106 dememorization steps. The Bonferroni adjustment (α = 0.05/15 = 0.00333 for 15 loci) was applied to the probability of HWE to minimize possible type I errors66. Heterozygote deficiency or excess as a cause of deviation from HWE was investigated using Genepop. Silent allele frequencies were estimated using a maximum likelihood approach as implemented in ML-NullFreq51.
Population structure
Population structure was evaluated using different approaches, summary statistics, analysis of molecular variance AMOVA, multivariate methods, and unsupervised model-based Bayesian clustering. Pairwise genetic distances (F ST ) and AMOVA were calculated with the program Arlequin vs 3.5.1.365. F ST P-values were estimated at a significance of 0.01 using 10,000 permutations and applied Bonferroni adjustment. AMOVA was run using a FST-like distance matrix at 10,000 permutations. Factorial correspondence analysis (FCA) was conducted using the program GENETIX vs 4.0567.
F ST , Hierarchical and non-hierarchical AMOVA, and FCA were used to test for heterogeneity over ethno-linguistic (Bantu Central K, R, and S and Khoisan Khoe-Kwadi, Kx’a and Tuu) and geographic subdivision (administrative districts). Non-hierarchical AMOVA was used to test for ethno-linguistic heterogeneity amongst the self-declared ethnolinguistic groups: (1) Central-K Bantu, (2) Central-R Bantu, (3) Central-S Bantu, (4) Khoe-Kwadi Khoisan, (5) Kx’a Khoisan, (6) Tuu Khoisan (This test was limited to n = 970 because there was no self-declared ethno-linguistic sub-language classifications for twenty donors); and geographic heterogeneity in the nine administrative districts: (1) Central, (2) Ghanzi, (3) Kgalagadi, (4) Kgatleng, (5) Kweneng, (6) Northeast, (7) Northwest, (8) Southern, (9) Southeast (n = 990). Hierarchical AMOVA grouping strategy tested for variation between the self-declared Bantu and Khoisan ethnolinguistic sub-population groups: (1) Bantu- Central-K + Central-R + Central-S; (2) Khoisan- Khoe-Kwadi + Kx’a + Tuu; and geographic variation between the north and south regions of the country: (1) North: Northwest + Ghanzi + Central + Northeast and (2) South: Kgalagadi + Kweneng + Kgatleng + Southern + Southeast.
Population structure was further evaluated using the unsupervised Bayesian clustering methods in STRUCTURE vs 2.3.468 and TESS vs 2.3.169. These two programs were used to take advantage of their different sensitivities to population structure and admixture. STRUCTURE is advantageous in that it is able to explore the number of populations in a dataset by optimizing Hardy-Weinberg equilibrium within putative groups, while as an addition TESS uses geographical information in assigning membership.
STRUCTURE was run using the parameters admixture and correlated allele frequencies model for a burn-in period of 1,000,000, followed by 100,000 iterations. Ten replicates were run for each K = 1 to K = 6. Summary reports were generated using the STRUCTURE HARVERSTER tool52, an ad hoc statistic-based approach which estimates the true number of populations K by estimating ΔK. Individual ancestral components were visualized using CLUMPAK (http://clumpak.tau.ac.il/index.html).
The Bayesian clustering algorithm in TESS was applied using geographical information as an additional parameter in the model. Initially TESS was run with the no-admixture model to estimate the upper bound on the number of distinct genetic clusters as recommended in the user manual. The spatial interaction parameter (ψ) was set to 0.6 as recommended69. The model was run for 1,000,000 iterations with a burn-in period of 100,000 iterations for K = 2 to K = 10 with ten replicates for each k. The ideal number of clusters (Kmax) was chosen based on when the deviance information criterion (DIC) values reached a plateau (as recommended in the user manual). Using the resulting Kmax, 10 replicates were run using the conditional auto-regressive (CAR) admixture model using the same parameters mentioned above. The average of each individual’s proportions of ancestral components was calculated using the program CLUMPP70 over the 10 replicates.
The CAR model uses a hidden regression approach which allows the possibility to display posterior predictive maps of admixture coefficients71. These maps provide useful information in addition to the standard unidimensional bar chart representation showing the predictions of admixture proportions for individuals at their geographic locations. The R (http://www.R-project.org) 3.0.372 package spatial 7.3.973 and maps 2.3.974 were used to map the extent of genetic clusters and identify barriers between clusters using ordinary kriging surface interpolation of admixture proportions. Each of these maps (equal to the number of genetic clusters) extrapolates the admixture proportions (proportion of genotype belonging to that particular cluster) across the study area. Areas with low values in the combined map were considered boundary regions between genetic clusters.
Assignment and Ancestry
Using the results of STRUCTURE at K = 2, three groups were identified according to whether their ancestral proportions at a cut-off of 0.7 for ancestral membership proportions for either the Bantu or Khoisan ethno-linguistic groups following criteria similar to Phillips et al.61 and Ristow et al.43. Group (1) consisted of individuals who had Bantu ethno-linguistic population group proportions ≥0.7, Group (2) individuals with Khoisan ethno-linguistic population group proportions ≥0.7, and Group (3) individuals whose membership probability proportions were below 0.7 for either the Bantu or Khoisan ethno-linguistic group and therefore admixed. Using proportions of ancestral components, the ability of Identifiler to determine ancestry was evaluated. Those individuals with a K ≥ 0.7 in a group that corresponded to their self-declared ethno-linguistic population group (Bantu or Khoisan) were “correctly assigned”. “Wrongly assigned” meant individuals whose self-declared ethno-linguistic population group showed ancestral proportion ≤0.7. Those whose ancestral components were below 0.7 for both Bantu and Khoisan were considered “admixed”.
The efficiency of assignment of the Identifiler markers was also evaluated using the program WHICHRUN vs 4.175. The genotypes of Group (1)-Bantu and Group (2)-Khoisan were used as a baseline (training set) in WHICHRUN using the critical populations method to determine the log likelihood (log(L)) of the population probabilities for all 990 samples used in the study. The individual genotypes probability ratio limit of 3 (log103 = 0.477) was chosen for population assignment to exclude false positives. An individual was considered “correctly assigned” when assigned to the self-declared population group. An individual was considered “wrongly assigned” when assignment corresponded to other than the self-declared population group. The proportion of wrongly assigned individuals is the error rate. Those individuals that were not assigned to a group were classified as “not assigned” and therefore considered admixed.
The ability of the markers in the inference of ancestry was evaluated in Group (1)-Bantu and Groups (2)-Khoisan using the loci informativeness for assignment (I n ) implemented in the program Infocalc76. I n ranges from zero (no information) to the natural logarithm of the number of populations (maximum information). The highest theoretically attainable I n per locus for this study was 0.693 (log102).
Forensic parameters
Standard summary statistics estimating forensic parameters77: Power of Exclusion (PE = h2(1–2hH2) where h and H are the number of heterozygotes and homozygotes respectively, and the Combined \(P{E}_{i=1}^{n}\) = 1 − π(1 − PEi) were PE i is the specific exclusion probability of the ith genetic marker and π(1 − PEi) means (1 − PE1) × (1 − PE2) × (1 − PE3) × … × (1 − PEn) from locus i = 1 to the nth locus, Match Probability (MP = \(\sum _{i=a}^{n}\,\sum _{j\ge 1}^{n}{{\rm{P}}}_{{{\rm{ij}}}^{2}}\) where i and j represent the frequencies of all possible alleles a through n. Pij represents the frequencies of all possible genotypes), the combined PM for more than one locus is the product of the individual PM at each locus assuming that they are not linked, Power of Discrimination (PD = 1 − MP), and Typical Paternity Index \(({\rm{TPI}}=\frac{H+h}{2H})\) were calculated using an Excel spreadsheet. Polymorphic information content (PIC) was calculated using Cervus vs 3.0778.
Data accessibility
All data used in this study has been deposited into the European Genome-Phenome Archive (EGA) (http://www.ebi.ac.uk/ega/), which is hosted by the European Bioinformatics Institute (EBI), under accession number EGAS00001002380.
References
Greenberg, J. H. The languages of Africa, (Indiana Univ., 1963).
Gordon, R. G. & Grimes, B. F. Ethnologue: Languages of the world, (SIL international Dallas, TX, 2005).
Barnard, A. Hunters and herders of southern Africa: a comparative ethnography of the Khoisan peoples, (Cambridge University Press, 1992).
Heine, B. & Honken, H. The Kx’a family: A new Khoisan genealogy. Journal of Asian and African Studies 79, 5–36 (2010).
Guldemann, T. A linguist’s view: Khoe-Kwadi speakers as the earliest food-producers of southern Africa. Southern African Humanities 20, 93–132 (2008).
Güldemann, T. Tuu as a language family. In Studies in Tuu (Southern Khoisan) 11–30 (Institut für Afrikanistik, Universität Leipzig, 2005).
Tlou, T. & Campbell, A. C. History of Botswana, (Macmillan Botswana, 1997).
Phillipson, D. W. African archaeology, (Cambridge University Press, 2005).
Lane, P., Reid, A. & Segobye, A. Ditswa mmung, (Pula Press and Botswana Society, 1998).
Potten, D. Aspects of the recent history of Ngamiland. Botswana Notes and Records, 63–86 (1976).
Marks, S. J. et al. Static and moving frontiers: the genetic landscape of Southern African Bantu-speaking populations. Molecular biology and evolution, msu263 (2014).
Pickrell, J. K. et al. The genetic prehistory of southern Africa. Nature communications 3, 1143 (2012).
Schlebusch, C. M., Lombard, M. & Soodyall, H. MtDNA control region variation affirms diversity and deep sub-structure in populations from southern Africa. BMC evolutionary biology 13, 56 (2013).
Petersen, D. C. et al. Complex patterns of genomic admixture within southern Africa. PLoS Genet 9, e1003309 (2013).
Batini, C. et al. Signatures of the preagricultural peopling processes in sub-Saharan Africa as revealed by the phylogeography of early Y chromosome lineages. Molecular biology and evolution 28, 2603–2613 (2011).
Beleza, S., Gusmao, L., Amorim, A., Carracedo, A. & Salas, A. The genetic legacy of western Bantu migrations. Human genetics 117, 366–375 (2005).
de Filippo, C. et al. Y-chromosomal variation in sub-Saharan Africa: insights into the history of Niger-Congo groups. Molecular biology and evolution 28, 1255–1269 (2011).
Diamond, J. & Bellwood, P. Farmers and their languages: the first expansions. Science 300, 597–603 (2003).
Barbieri, C., Butthof, A., Bostoen, K. & Pakendorf, B. Genetic perspectives on the origin of clicks in Bantu languages from southwestern Zambia. European journal of human genetics 21, 430–436 (2013).
Barbieri, C. et al. Migration and interaction in a contact zone: mtDNA variation among Bantu-speakers in southern Africa. PloS one 9, e99117 (2014).
Güldemann, T. & Stoneking, M. A historical appraisal of clicks: a linguistic and genetic population perspective. Annual Review of Anthropology 37, 93–109 (2008).
Bostoen, K. & Sands, B. Clicks in south-western Bantu languages: contact-induced vs. language-internal lexical change. In Proceedings of the 6th World Congress of African Linguistics Cologne 129–140 (2009).
Nyati-Ramahobo, L. Minority tribes in Botswana: The politics of recognition, (Minority Rights Group International London, 2008).
Tishkoff, S. A. et al. The genetic structure and history of Africans and African Americans. Science 324, 1035–1044 (2009).
Barbieri, C. et al. Unraveling the complex maternal history of Southern African Khoisan populations. American journal of physical anthropology 153, 435–448 (2014).
Mpoloka, S., Kgotlele, T. & Wally, A. Determination of allele frequencies in nine short tandem repeat loci of five human sub-populations in Botswana. African Journal of Biotechnology 7 (2008).
Tau, T., Davison, S. & D’Amato, M. E. Polymorphisms at 17 Y-STR loci in Botswana populations. Forensic Science International: Genetics 17, 47–52 (2015).
Tofanelli, S. et al. Variation at 16 STR loci in Rwandans (Hutu) and implications on profile frequency estimation in Bantu-speakers. International journal of legal medicine 117, 121–126 (2003).
Alves, C. n., Gusmão, L., Damasceno, A., Soares, B. & Amorim, A. Contribution for an African autosomic STR database (AmpF/STR Identifiler and Powerplex 16 System) and a report on genotypic variations. Forensic science international 139, 201–205 (2004).
Beleza, S. et al. 17 STR data (AmpF/STR identifiler and powerplex 16 system) from Cabinda (Angola). Forensic science international 141, 193–196 (2004).
Alves, C. n. et al. STR allelic frequencies for an African population sample (Equatorial Guinea) using AmpFlSTR Identifiler and Powerplex 16 kits. Forensic science international 148, 239–242 (2005).
Butler, J. M. Genetics and genomics of core short tandem repeat loci used in human identity testing. J Forensic Sci 51, 253–65 (2006).
Forward, B. W., Eastman, M. W., Nyambo, T. B. & Ballard, R. E. AMPFlSTR® Identifiler™ STR Allele Frequencies in Tanzania, Africa. Journal of forensic sciences 53, 245–247 (2008).
Muro, T. et al. Allele frequencies for 15 STR loci in Ovambo population using AmpFlSTR® Identifiler Kit. Legal Medicine 10, 157–159 (2008).
Gomes, V. et al. Population data defined by 15 autosomal STR loci in Karamoja population (Uganda) using AmpF/STR Identifiler kit. Forensic Science International: Genetics 3, e55–e58 (2009).
Tillmar, A. O., Bäckström, G. & Montelius, K. Genetic variation of 15 autosomal STR loci in a Somali population. Forensic Science International: Genetics 4, e19–e20 (2009).
Melo, M. M. et al. Genetic study of 15 STRs loci of Identifiler system in Angola population. Forensic Science International: Genetics 4, e153–e157 (2010).
Babiker, H., Schlebusch, C. M., Hassan, H. Y. & Jakobsson, M. Genetic variation and population structure of Sudanese populations as indicated by 15 Identifiler sequence-tagged repeat (STR) loci. Investig Genet 2, 12 (2011).
Schlebusch, C. M., Soodyall, H. & Jakobsson, M. Genetic variation of 15 autosomal STR loci in various populations from southern Africa. Forensic Science International: Genetics 6, e20–e21 (2012).
Lucassen, A., Ehlers, K., Grobler, P. J. & Shezi, A. L. Allele frequency data of 15 autosomal STR loci in four major population groups of South Africa. International journal of legal medicine 128, 275–276 (2014).
Nabwowe, J., Kirya, M., Okello, E. & Nanteza, A. Allele Frequency of 15 Short Tandem Repeats (Strs) in a Buganda Population (Central Uganda): Forensic Utility and Parentage Testing. Journal of Forensic Research 2014 (2014).
Ristow, P., Davison, S. & D’Amato, M. Implementing genotypic AmpFlSTR® Identifiler® Plus profiles to infer population groups. Forensic Science International: Genetics Supplement Series 5, e553–e554 (2015).
Ristow, P. G. & Cloete, K. W. GlobalFiler® Express DNA amplification kit in South Africa: Extracting the past from the present. Forensic Science International: Genetics (2016).
Butler, J. M. Advanced topics in forensic DNA typing: interpretation, (Academic Press, 2014).
Clayton, T. M., Guest, J. L., Urquhart, A. J. & Gill, P. D. A genetic basis for anomalous band patterns encountered during DNA STR profiling. Journal of forensic sciences 49, 1207–1214 (2004).
Lane, A. The nature of tri-allelic TPOX genotypes in African populations. Forensic Science International: Genetics 2, 134–137 (2008).
Picanço, J. B. et al. Identification of the third/extra allele for forensic application in cases with TPOX tri-allelic pattern. Forensic Science International: Genetics 16, 88–93 (2015).
Dakin, E. & Avise, J. Microsatellite null alleles in parentage analysis. Heredity 93, 504–509 (2004).
Amorim, A. & Carneiro, J. The impact of silent alleles in kinship probability calculations. Forensic Science International: Genetics Supplement Series 1, 638–639 (2008).
Lane, A. B. STR null alleles complicate parentage testing in South Africa. SAMJ: South African Medical Journal 103, 1004–1008 (2013).
Kalinowski, S. T. & Taper, M. L. Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conservation Genetics 7, 991–995 (2006).
Earl, D. A. Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conservation genetics resources 4, 359–361 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular ecology 14, 2611–2620 (2005).
Lowe, A. L., Urquhart, A., Foreman, L. A. & Evett, I. W. Inferring ethnic origin by means of an STR profile. Forensic Science International 119, 17–22 (2001).
Brenner, C. H. Some mathematical problems in the DNA identification of victims in the 2004 tsunami and similar mass fatalities. Forensic science international 157, 172–180 (2006).
Phillips, C. et al. Ancestry analysis in the 11-M Madrid bomb attack investigation. PLoS One 4, e6583 (2009).
Fosella, X. et al. Assigning individuals to ethnic groups based on 13 STR loci. In International Congress Series Vol. 1261, 59–61 (Elsevier, 2004).
Steele, C. D. & Balding, D. J. Choice of population database for forensic DNA profile analysis. Science & Justice 54, 487–493 (2014).
Phillips, C. Forensic genetic analysis of bio-geographical ancestry. Forensic Science International: Genetics (2015).
Graydon, M., Cholette, F. & Ng, L.-K. Inferring ethnicity using 15 autosomal STR loci—Comparisons among populations of similar and distinctly different physical traits. Forensic Science International: Genetics 3, 251–254 (2009).
Phillips, C. et al. Analysis of global variability in 15 established and 5 new European Standard Set (ESS) STRs using the CEPH human genome diversity panel. Forensic Science International: Genetics 5, 155–169 (2011).
Pereira, L. et al. PopAffiliator: online calculator for individual affiliation to a major population group based on 17 autosomal short tandem repeat genotype profile. International journal of legal medicine 125, 629–636 (2011).
Algee-Hewitt, B. F., Edge, M. D., Kim, J., Li, J. Z. & Rosenberg, N. A. Individual Identifiability Predicts Population Identifiability in Forensic Microsatellite Markers. Current Biology 26, 935–942 (2016).
Rousset, F. genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Molecular Ecology Resources 8, 103–106 (2008).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular ecology resources 10, 564–567 (2010).
Hochberg, Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika 75, 800–802 (1988).
Belkir, K., Borsa, P., Goudet, J., Chikhi, L. & Bonhomme, F. Genetix, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome et Populations, CNRS UPR 9060. Université de Montpellier II, Montpellier, France (1999).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Chen, C., Durand, E., Forbes, F. & François, O. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Molecular Ecology Notes 7, 747–756 (2007).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Durand, E., Jay, F., Gaggiotti, O. E. & François, O. Spatial inference of admixture proportions and secondary contact zones. Molecular Biology and Evolution 26, 1963–1973 (2009).
R: A language and environment for Statistical Computing, R Core Team, R Foundation for Statistical Computing, Vienna, Austria http://www.R-project.org (2014).
Ripley, B. Spatial: Functions for Kriging and Point Pattern Analysis 7.3. (R Documentation, 2011).
Brownrigg, R., Minka, T., Becker, R. & Wilks, A. Maps: draw geographical maps. R package version, 2.1–6 (2011).
Banks, M. & Eichert, W. Whichrun (version 3.2): a computer program for population assignment of individuals based on multilocus genotype data. Journal of Heredity 91, 87–89 (2000).
Rosenberg, N. A., Li, L. M., Ward, R. & Pritchard, J. K. Informativeness of genetic markers for inference of ancestry. The American Journal of Human Genetics 73, 1402–1422 (2003).
Evett, I. W. & Weir, B. S. Interpreting DNA evidence: statistical genetics for forensic scientists, (Sinauer, 1998).
Kalinowski, S. T., Taper, M. L. & Marshall, T. C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Molecular ecology 16, 1099–1106 (2007).
R: A language and environment for Statistical Computing, R Core Team, R Foundation for Statistical Computing, Vienna, Austria http://www.R-project.org (2016).
Hadley, W. ggplot2: Elegant graphics for data analysis https://cran.r-project.org/web/packages/ggmap/citation.html (2009).
Kahle, D. & Wickham, H. ggmap: Spatial Visualization with ggplot2. R Journal 5 https://cran.r-project.org/web/packages/ggplot2/citation.html (2013).
Acknowledgements
Many thanks to Peter Ristow for all his help with the graphics.
Author information
Authors and Affiliations
Contributions
Research was conceptualized by A.W. and S.W.M. T.P.F., G.L.N., A.W., and S.W.M. collected the samples. M.E.D. and T.T. designed the research. T.P.F. genotyped the samples. T.T. performed the statistical analysis and wrote the manuscript with supervision of M.E.D. All authors reviewed, corrected and accepted the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tau, T., Wally, A., Fanie, T. et al. Genetic variation and population structure of Botswana populations as identified with AmpFLSTR Identifiler short tandem repeat (STR) loci. Sci Rep 7, 6768 (2017). https://doi.org/10.1038/s41598-017-06365-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06365-y
- Springer Nature Limited
This article is cited by
-
Forensic autosomal and gonosomal short tandem repeat marker reference database for populations in Burkina Faso
Scientific Reports (2024)
-
Genetic analysis based on 15 autosomal short tandem repeats (STRs) in the Chaouia population, western center Morocco, and genetic relationships with worldwide populations
Molecular Genetics and Genomics (2023)
-
Allele frequency data for 23 aSTR for different ethnic groups from Republic of Zimbabwe
International Journal of Legal Medicine (2021)