
Abstract
The local electronic and atomic structures of the high-quality single crystal of SrFeO3-δ (δ~0.19) were studied using temperature-dependent x-ray absorption and valence-band photoemission spectroscopy (VB-PES) to investigate the origin of anisotropic resistivity in the ab-plane and along the c-axis close to the region of thermal hysteresis (near temperature for susceptibility maximum, Tm~78 K). All experiments herein were conducted during warming and cooling processes. The Fe L 3,2-edge X-ray linear dichroism results show that during cooling from room temperature to below the transition temperature, the unoccupied Fe 3d e g states remain in persistently out-of-plane 3d 3z 2 -r 2 orbitals. In contrast, in the warming process below the transition temperature, they change from 3d 3z 2 -r 2 to in-plane 3d x 2 -y 2 orbitals. The nearest-neighbor (NN) Fe-O bond lengths also exhibit anisotropic behavior in the ab-plane and along the c-axis below Tm. The anisotropic NN Fe-O bond lengths and Debye-Waller factors stabilize the in-plane Fe 3d x 2 -y 2 and out-of-plane 3d 3z 2 -r 2 orbitals during warming and cooling, respectively. Additionally, a VB-PES study further confirms that a relative band gap opens at low temperature in both the ab-plane and along the c-axis, providing the clear evidence of the charge-density-wave nature of SrFeO3-δ (δ~0.19) single crystal.
Similar content being viewed by others

Introduction
Recently, SrFeO3-δ based colossal magnetoresistance (CMR) materials have attracted much interest owing to their potential applications in the next generation of magnetic data storage read heads, and because of the research goal of elucidating the microscopic origin of CMR1,2,3,4. Extensive research on these CMR materials and associated electron structures that has been performed for the last decade is still ongoing5,6,7,8,9,10.
The structural, magnetic and transport properties of SrFeO3-δ vary dramatically with oxygen content and the valence state of Fe7, 11,12,13. The known phases of SrFeO3-δ include stoichiometric SrFeO3 (δ = 0), which has a cubic perovskite structure with a valence state of Fe4+, and oxygen-deficient phases with tetragonal (δ = 0.125), orthorhombic (δ = 0.25) and brownmillerite-type (δ = 0.50) crystal structures1, 2. Certain amount of oxygen result in the coexistence of these phases. The tetragonal phase comprises FeO6 octahedra, distorted/tilted FeO6 octahedra and square pyramidal FeO5 2. The orthorhombic phase reportedly includes distorted FeO6 octahedra and square pyramidal FeO5, in which Fe has valence of 3+ and 4+, respectively. The brownmillerite-type phase has tetrahedrally and octahedrally coordinated Fe sites with an Fe valence state of 3+2. In these oxygen-deficient SrFeO3-δ systems, giant negative magnetoresistance has been observed in the tetragonal phase around a coincident charge-spin ordering transition temperature1, 2. This transition exhibits thermal hysteresis of resistivity and magnetization, suggesting a first-order phase transition. The thermal hysteresis around the transition temperature (close to the temperature for susceptibility maximum, Tm) has been attributed to the coexistence of antiferromagnetic and paramagnetic phases5, 6. Further, a change in the lattice structure that originates from the charge ordering (CO) transition has been observed in Raman, far-infrared ellipsometric and neutron diffraction studies1, 2, 14. In transition metal oxides, CO-associated lattice distortion, which depends on the electron-lattice coupling strength, can cause orbital ordering (OO) so CO has been found to coexist with OO15,16,17,18. Bao et al. presented evidence of a CO-induced structural phase transition in single crystalline (Bi,Ca)MnO3 15. Chi et al. also observed strong enhancement of orthorhombic distortion owing by CO, which allows OO to be established in single crystals of Pr0.5Ca1.5MnO4 17. Similar observations have been made concerning other CO transitions, specifically, the charge-disproportionation of Fe ion and/or the formation of a charge-density-wave (CDW) in La1−xSrxFeO3 3, 19. However, in (La2−2xSr1+2x)Mn2O7, competing local lattice distortion and the hopping of charge carriers generate comparable amounts of Mn out-of-plane 3d 3z 2 -r 2 and in-plane 3d x 2 -y 2 orbitals in the metallic phase16. Therefore, the electrical and magnetic properties of (La2−2xSr1+2x)Mn2O7 depend on not only the out-of-plane but also the in-plane Mn 3d states. Clearly, these orbital effects influence the electronic properties of the sample, resulting in peculiar behavior that is associated with the resistivity-related phenomena20,21,22. Chen et al. used a theoretical model of iron pnictides to show that unequal d xz and d yz orbital populations can result in anisotropic resistivity23. They further noted that such preferable orbital states can be identified by x-ray linear dichroism (XLD)23. However, the complex interplay between the mechanisms that drive OO and cause these orbital effects remains unresolved24,25,26. As discussed above, the tetragonal phase of SrFeO3-δ undergoes a first-order spin-charge ordering transition that is accompanied by a change in lattice structure, which exhibits exotic electrical and magnetic properties1. Most spectroscopic investigations of SrFeO3-δ-based materials have involved varying the oxygen content and substitution at Sr or Fe sites; very few experimental and theoretical studies that involve the metal-to-insulator (MI)/semiconductor transition in SrFeO3-δ 3, 4, 19. Using resonant x-ray scattering (RXS), Martin et al. elucidated the CDW nature of the CO transition (MI/semiconductor transition) in La-substituted SrFeO3 19. However, their studies did not provide evidence of band gap opening in the formation of CDW19.
In this context, an investigation of the local electronic and atomic structures of tetragonal SrFeO3-δ by x-ray polarization (E)-dependent absorption and valence-band photoemission spectroscopy (VB-PES), can provide insight into the preferred orbital of Fe 3d states, lattice distortion that is related to the anisotropy of resistivity in the thermal hysteresis region, and band gap opening as a function of temperature. Therefore, a detailed study of single crystalline SrFeO3-δ (δ~0.19) is conducted herein using x-ray diffraction, magnetization, resistivity, x-ray absorption near-edge spectroscopy (XANES), XLD, extended x-ray absorption fine structure (EXAFS) and VB-PES. All experiments were carried out during warming and cooling runs with normal incidence (E//ab-plane, θ = 0°) and incidence at a glancing angle (near E//c-axis, θ = 70°).
Results and Discussion
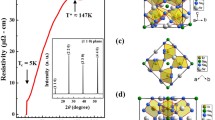
Figure 1(a) presents Lebail fits27,28,29 of the X-ray powder diffraction (XRPD) of SrFeO3-δ sample at room temperature based on the tetragonal I4/mmm space group model12. The refined lattice parameters are a = b = 10.9320(5) Å and c = 7.7012(5) Å. The insets in Fig. 1(a) magnify some of the Bragg reflections. The singlet nature of the (111)pc peak and the doublet nature of the (200)pc and (220)pc (pc = pseudocubic) peaks are characteristic of the tetragonal phase. Figure 1(b) schematically depicts the crystal structure of the tetragonal SrFeO3-δ, which includes Fe sites of three types pyramidal, [Fe(1), Fig. 1(c)], distorted/tilted octahedral, [Fe(2), Fig. 1(d)] and octahedral [Fe(3), Fig. 1(e)] with valence states of Fe4+, Fe3.5+and Fe4+, respectively on the basis of the Mössbauer spectroscopy results that were obtained by Lebon et al.1 and the bond valence calculations that were made by Hodges et al.12.

(a) Observed (red dots), calculated (black line) and difference (bottom green line) patterns of SrFeO2.81, obtained using Lebail refinement of synchrotron X-ray powder diffraction data. Vertical check marks above difference profiles indicate Bragg reflections. Insets magnify selected pseudocubic reflections, (b) crystalline structure of SrFeO2.81; valence Fe4+ is attributed to Fe(1), Fe(3) and Fe3.5+ is attributed to Fe(2), based on previous work1, (c) FeO5 square pyramidal, (d) FeO6 distorted octahedra, and (e) FeO6 octahedra.
Figure 2 plots the temperature-dependence of resistivity in the ab-plane and along the c-axis. The top right-inset plots the magnetic susceptibility (χ) of the crystal, which was also measured along the c-axis in a magnetic field of 1 Tesla in the zero-field-cooled (ZFC) and field-cooled (FC) runs. The top left-inset shows the room-temperature x-ray diffraction profile of the (004) Bragg peak of a single crystal of SrFeO3-δ. The sharpness of the peak (full width at half maximum~0.12°) indicates the high-quality of the single crystal27. Resistivity and magnetization measurements presented in Fig. 2 are consistent with those reported by Lebon et al. for SrFeO3-δ (δ = 0.19) crystal and therefore reveals that the oxygen content of the single crystal sample is close to that of SrFeO2.81 1, 2. A sharp rise in the susceptibility close to the temperature for the susceptibility maximum (Tm~78 K, top right-inset) indicates a transition from the paramagnetic phase to the antiferromagnetic phase and is consistent with earlier studies1, 2, 8. The ZFC and FC curves exhibit thermal hysteresis, revealing the first-order nature of the phase transition. Thermal hysteresis (thermal hysteresis region, ΔT~20 K) in the temperature-dependence of susceptibility in SrFeO2.81 has been attributed to the coexistence of paramagnetic and antiferromagnetic phases5, 6. Additionally, a peak that is characteristic of the residual cubic phase has also been observed at 130 K (indicated by the bar herein) in the temperature-dependence of χ1, 2, 8. However, RXS has recently been performed by co-authors of this work, and their results suggested that the transition at ~130 K is related to the weak localization of the charge on Fe3.5+, which is also magnetically active and leads to an anomaly in the paramagnetic region of susceptibility13. It is evident from Fig. 2 that during the warming and cooling processes resistivity exhibits thermal hysteresis both in the ab-plane and along the c-axis. At room temperature (paramagnetic phase) and ~10 K (antiferromagnetic phase), SrFeO2.81 has a resistivity of ~4 and ~104 mΩ-cm, respectively, reflecting typical metallic and semiconducting behaviors. According to the crystal structure in Fig. 1(b), at room temperature, SrFeO2.81 has three Fe sites with two valence states Fe4+ and Fe3.5+. The low resistivity in the paramagnetic phase is attributed to the delocalized electronic system with a high electron density of the Fe3.5+ state and the low electron density in the Fe4+ state, as determined by Mössbauer spectroscopy and refined room temperature neutron powder diffraction1, 2, 12. Moreover, the resistivity of the SrFeO2.81 crystal, which is independent of the warming or cooling of the sample and the measurement path, begins to increase as the temperature falls below ~130 K and is consistent with the anomaly in the magnetization data in Fig. 2. The change in resistivity as the temperature declines is reportedly associated with the beginning of the CO transition and similar to that associated with a MI phase transition13. Another signature of the phase transition is observed below ~78 K. This transition is accompanied by a thermal hysteresis that is characteristic of a first-order phase transition. According to the phenomenological theory of phase transition across a first-order MI phase transition, as the temperature is reduced, the volume fraction of the insulating phase increases but the metallic phase does not disappear (According to the results of Mössbauer spectroscopy, the calculated fraction of Fe3.5+ in the paramagnetic/metallic phase is ~55%, decreasing to ~10% in the antiferromagnetic/insulating phase. Lebon et al.1 found that the residual ~10% is caused by an oxygen deficiency), which results in the coexistence of two electronic phases over a certain temperature range, giving rise to the hysteresis of electronic properties30, 31. A recent study by Lee et al.13 on SrFeO2.81 with majority tetragonal phase revealed that magnetic and charge-related degrees of freedom are coupled with each other and thermal hysteresis in the resistivity is associated with the commensurate-to-incommensurate CO transition (The delocalized Fe3.5+ state with fractional valence changes to localized Fe3+ and Fe4+ states upon the CO transition). Therefore, the paramagnetic-to-antiferromagnetic transition in SrFeO2.81 coincides with a commensurate-to-incommensurate CO transition13. As expected, the resistivity of a single crystal of SrFeO2.81, plotted in Fig. 2, in the ab-plane is always larger than that along the c-axis, because the a and b lattice parameters, as reported by Reehuis et al.14, are greater than c at all temperatures, possibly lessening the conductivity of electrons. However, the results herein reveal another interesting phenomenon, which is a large difference between the thermal hysteresis region in the ab-plane (thermal hysteresis region, ΔT~19 K) and that along the c-axis (ΔT~30 K). The anisotropy or direction-dependence of resistivity in the thermal hysteresis region of a single crystal of SrFeO2.81 is not mostly explained by the coexistence of antiferromagnetic and paramagnetic phases, by the incommensurate-to-commensurate CO transition5, 6, 13, or in terms of the charge-disproportionation or CDW state3, 19. Several theoretical and experimental studies of single crystals and thin films of various materials have established that the anisotropy of resistivity is associated with OO or with the preferential occupation of orbitals, is primarily controlled by doping or lattice distortion/strains21, 23, 26, 29,30,31,32,33. Accordingly, the anisotropy of resistivity both in the ab-plane and along the c-axis in SrFeO2.81 crystal in the thermal hysteresis region is closely related to the changes in electronic and lattice structures.

Temperature-dependence of resistivity of a single crystal of SrFeO2.81, measured in ab-plane and along c-axis. Top inset shows temperature-dependence of magnetic susceptibility (χ) measured along c-axis in ZFC and FC runs in a magnetic field of 1 Tesla, and bottom inset presents room-temperature x-ray diffraction profile showing (004) Bragg peak obtained in θ-scan.
Figure 3(a–d) present the temperature-dependent Fe K-edge XANES spectra of single crystalline SrFeO2.81, at two angles of incidence, θ = 0° and 70°, to the normal of the ab-plane during warming and cooling processes. Corresponding spectra of FeO, Fe3O4, and Fe2O3 powder samples were obtained at room temperature, θ = 0°, for reference. According to the dipole-transition selection law, these Fe K-edge XANES spectra are primarily associated with the Fe 1 s → 4p transition, and the intensity of the main feature is attributable to the density of the unoccupied Fe 4p states. The bottom panels in Fig. 3(a–d) present the derivative results, to elucidate the variation of the valence state of the Fe ion in SrFeO2.81 with temperature, based on the position of the threshold feature33. The energy threshold of the Fe K-edge feature of SrFeO2.81 (feature d) is at 7127.5 ± 0.3 eV for both θ = 0° and 70° in warming and cooling at all temperatures; this threshold is above those of FeO/Fe2+ (feature a), Fe3O4/Fe(8/3)+ (feature b), and Fe2O3/Fe3+ (feature c). From the maximum of the derivative of the XANES spectrum, Blasco et al.3 obtained an average valence state of +4 for Fe ions in the powder sample of SrFeO2.96 with an energy threshold at 7127.5 ± 0.2 eV, which is close to that of SrFeO2.81 as displayed in Fig. 3. However, in the absence of standard Fe K-edge XANES for the 4+ valence state, the average valence state of Fe in SrFeO2.81 is determined to be between Fe3+ and Fe4+ and close to Fe4+. Additionally, the observed average valence state of Fe in SrFeO2.81, as stated above, obtained on the basis of Mössbauer spectroscopic results and the bond valence calculations, is determined to be Fe4+, Fe3.5+ and Fe4+ at the three crystallographic sites, in the ratio 1:2:11, 2, 12. Importantly, the MI/CO transition of SrFeO2.83±0.01 and La1/3Sr2/3FeO3 has been attributed to charge-disproportionation of Fe3.5+ and Fe3.66+, respectively, and new models, based on a CDW19, have been proposed to explain the charge modulation in these compounds13, 19. However, as shown in Fig. 3(a–d), the energy of Fe K-edge and the line-shape of SrFeO2.81 do not vary much with temperature during warming or cooling. Clearly, the Fe K-edge XANES studies do not support the claim that charge disproportionation influences resistivity (or the MI/metal-to-semiconductor transition) below the transition temperature of SrFeO2.81.

(a–d) The temperature-dependent Fe K-edge XANES spectra of single crystal SrFeO2.81 measured at two different angles of incidence θ = 0° (with electric field E parallel to the ab-plane) and 70° (with electric field E nearly parallel to the c-axis) on warming and cooling process. Corresponding spectra were obtained for FeO, Fe3O4, and Fe2O3 powder samples at room temperature with angle θ = 0° for reference.
Figure 4(a) and (b) present the temperature-dependent Fe L 3,2-edge XANES and XLD (bottom) spectra with E parallel to the ab-plane (angle of incidence, θ = 0°) and the c-axis (angle of incidence, θ = 70°), respectively. The Fe L 3,2-edge XANES spectra include two features- an L 3-edge around 708 eV and an L 2-edge around 720 eV, that are separated by spin-orbital splitting. These spectra are primarily associated with the Fe 2p → 3d transition, and the intensity of the main feature is attributable to the density of the unoccupied Fe 3d-O 2p hybridized states. Notably, as displayed in Fig. 4(a) and (b), the intensity of the Fe L 3,2-edge XANES spectra is greatly suppressed when E is parallel to the ab-plane and almost parallel to the c-axis at 60 K during warming and cooling. The Mn3+ ion (d 4) in an octahedral oxygen environment is well known to exhibit a large static Jahn-Teller (JT) distortion, which has an important role in the CO and MI transition20, 21, 34. SrFeO2.81 has two valence states Fe3.5+ and Fe4+, of which Fe4+ (d 4) is isoelectronic with Mn3+ and is therefore expected to exhibit a JT effect34, 35. Bocquet et al.36 noted that d 4 is a high-spin state with three electrons’ filling: the t 2g orbital and the remaining e g electron itinerant, so Fe4+ may induce a strong JT distortion and further split the 3d e g state into 3d x 2 -y 2 and 3d 3z 2 -r 2 35, 36. Figure S1 in the Supplementary Information schematically depicts the experiment to elucidate the in-plane and out-of-plane Fe 3d-O 2p hybridization states. The hybridizations of Fe 3d x 2 -y 2-O 2p x,y (in-plane, θ = 0°) and Fe 3d 3z 2 -r 2-O 2p z (out-of-plane, θ = 70°) are probed with the electric field E parallel to the ab-plane and almost parallel to the c-axis, respectively. The XLD spectra (difference in XANES intensity between θ = 0° and θ = 70°) of the Fe L 3,2-edge provide information on the preferred orbital of the Fe 3d electron, which is determined by the anisotropic effect with temperatures. The bottom panel in Fig. 4(b) reveals that the sign of the XLD feature is negative during all cooling, suggesting that Fe e g electrons preferentially occupy the out-of-plane 3d 3z 2 -r 2 orbitals. However, in the warming process, the signs of the XLD spectra are reversed, being positive at 40 and 60 K, suggesting that the Fe e g electrons preferentially occupied the in-plane 3d x 2 -y 2 orbitals in the thermal hysteresis. This behavior with respect to the preferential orbital can give rise to the anisotropy that is observed in the resistivity in the thermal hysteresis region, owing to the strong coupling between the lattice distortion and the electronic structures15,16,17,18. Several researchers have provided evidence of structural distortions of transition-metal oxides and their correlation with OO and CO15,16,17,18, 37, 38. The distortion of the unit cell is primarily responsible for the lowering of the energy of either out-of-plane e g orbitals or in-plane e g orbitals15,16,17,18. In single crystals of Pr0.5Ca1.5MnO4, an increase in orthorhombic distortion causes OO and changes the electronic properties17. However, in (La2−2xSr1+2x)Mn2O7, competition between local lattice distortion and the hopping of charge carriers is responsible for the comparable amounts of 3d 3z 2 -r 2 and 3d x 2 -y 2 orbitals16. Raman, far-infrared ellipsometric and neutron diffraction studies of tetragonal SrFeO3-δ have revealed a change in lattice structure as the temperature falls below Tm 1, 2, 14. Local lattice distortion in the SrFeO2.81 below Tm is evident in the Fe K-edge EXAFS results. Tetragonal SrFeO3-δ has three Fe sites, in pyramidal, distorted/tilted octahedral and octahedral networks of oxygen around Fe ions. The distortion of the oxygen octahedral/pyramidal networks by a change in the nearest-neighbor (NN) Fe-O bond length and the hybridization of Fe 3d-O 2p can stabilize different out-of-plane and in-plane Fe 3d e g orbitals, as evidenced by the temperature-dependent XLD spectra of SrFeO2.81 in Fig. 415,16,17,18. Figure S2(a–d) in the Supplementary Information plot the temperature-dependent Fourier transform (FT) Fe K-edge EXAFS spectra of SrFeO2.81 at θ = 0° (E//ab-plane) and θ = 70° (E//c-axis). The insets present corresponding EXAFS k 2χ data. The Fe K-edge EXAFS spectra include main FT features (A, B and C) that correspond to the NN bond lengths of Fe-O, Fe-Sr and Fe-Fe, respectively, in the SrFeO2.81 crystal3, 39, 40. To obtain detailed information about the temperature dependence of the local structure around the Fe atoms, Fig. 5 presents a magnified view of feature A (corresponding to the NN Fe-O bond length). The figure reveals that the intensity of FT feature is minimal at 300 K for both E//ab-plane and E//c-axis in both warming and cooling processes. However, in Figure S2, the intensities of FT features B and C (corresponding to the NN Fe-Sr and Fe-Fe bond lengths, respectively) follow the typical thermal trend: as the temperature increases, the intensity of each FT feature decreases, suggesting the importance of Fe-O bond lengths in the local distortions of the SrFeO2.81 crystal. Evidently, as the temperature is increased from 40 K to 80 K, the intensity of the FT feature increases, before decreasing with a further increasing in temperature. These changes are marked in the warming and cooling cycles for the E//ab-plane but also exist for E//c-axis. The intensity of the FT feature is determined by the coordination number, N and the DW factor σ2 41, 42. Figure S2(a–d) and Fig. 5(a–d) plot the EXAFS measurements in the ab-plane (E//ab-plane, θ = 0°) and along the c-axis (E//c-axis, θ = 70°): Normally, N does not vary with temperature, so a change in σ2 causes the intensity of the FT features to vary with temperature. The DW factor σ2 is composed of two components, σ2 stat and σ2(T)vib, which are associated with static disorder and thermal vibrations, respectively42. Component σ2 stat is related to the atomic structure and is unrelated to temperature, while the σ2(T)vib is associated with the lattice vibrations, which commonly become smaller as the temperature decreases, according to the Einstein or Debye model28, 41, 42. As expected, at high temperature (above Tm), reducing the temperature increases the intensity of the FT feature of the metallic/paramagnetic phase in the SrFeO2.81 crystal, owing to the key σ2(T)vib factor, but below Tm, the intensity of the FT feature decreases as the temperature declines. These anomalous results clearly reveal that σ2 stat dominates the FT intensity below Tm, suggesting that static disorders or JT distortions that are caused by Fe 3d electrons have a stronger effect than the influence of temperature. The large static distortions of the octahedral/pyramidal oxygen network around the Fe ions below Tm can be understood as being statically contributed to static DW factors that strongly influence the FT feature of Fe-O bonds in the SrFeO2.81.

(a) and (b) Temperature-dependence of normalized Fe L 3,2-edge XANES spectra of single crystal of SrFeO2.81 at two angles of incidence θ = 0° and 70° during warming and cooling. Bottom panels show corresponding XLD spectra.

Temperature-dependence of the main FT feature A (corresponding to the NN Fe-O bond distance) of Fe K-edge EXAFS for (a,b) E//ab-plane and (c,d) E//c-axis in the warming and cooling process.
To discuss quantitatively the local atomic structure of a single crystal of SrFeO2.81, fitting results for the NN Fe-O bond length, N and the DW factors are obtained using Artemis software41, 42 and presented in Table 1 of the Supplementary Information and Fig. 6. The DW factors and NN Fe-O bond lengths are plotted in Fig. 6 and exhibit anisotropic behavior in the ab-plane and along the c-axis. Generally, the DW factors that are related to atomic structure should decrease upon cooling of the sample as the thermal vibrations become less intense. However, DW factors in the ab-plane abruptly increase when the temperature declines below Tm. This sudden increase below the transition temperature [Fig. 6(a)] with thermal hysteresis indicates that the crystal structure of SrFeO2.81 exhibits much greater static disorder below Tm than above Tm, and this phenomenon of large static disorder dominates the thermal effect. This fact can be understood with reference to soft phonon mode behavior43, 44, which is related to a decrease or breaking of the crystal symmetry in the ab-plane of SrFeO2.81. Soft phonons are typically associated with phase transitions in crystals that can have more than one distinguishable lattice. The off-centering of the Fe ions or an order-disorder phase transition between dynamic and static distortions of Fe ions, in which interaction (that is phonon-mediated) between off-centered Fe and oxygen atoms drives phonon softening in the FeO6/FeO5 networks. Also, as shown in Fig. 6(a), DW factors along the c-axis are nearly constant as the temperature is reduced, indicating that the thermal effect compete with static disorder along the c-axis. The unusually high DW factors in the ab-plane and the thermal hysteresis suggest that the local structural ordering of SrFeO2.81 differs between the ab-plane and the c-axis in both warming and cooling processes. The significant changes of DW factors in the ab-plane in comparison to c-axis are consistent with Reehuis et al.14. In ref. 14, using neutron diffraction studies prominent changes in the bond lengths in ab-plane below transition temperature have been reported and it is anticipated to be due to orbital polarization. However, the greater value of DW factors in the ab-plane during cooling than warming below Tm is not currently explainable. We speculate that this difference may be related to the difference between the out-of-plane and in-plane Fe 3d orbitals, as observed in the temperature-dependent XLD spectra in Fig. 4. Therefore, a detailed theoretical calculation may provide a quantitative description of competing lattice, orbital and spin-related degrees of freedom in this system. Figure 6(b) presents the variation of the NN Fe-O bond lengths in the ab-plane and along the c-axis. As evidenced from Fig. 6(b), the NN Fe-O bond lengths exhibit hysteretic behavior below the transition temperature Tm; increase from 1.909 ± 0.005 to 1.933 ± 0.005 Å in the ab-plane, and increase from 1.900 ± 0.005 to 1.909 ± 0.005 Å along c-axis. Although the NN Fe-O bond lengths depend differently on temperature in the ab-plane and along the c-axis, the limitation on the resolution of the EXAFS analysis prevents precise determination of the Fe-O bond lengths. However, the anomalous variations of DW factors in the ab-plane and along the c-axis suggest that anisotropic distortion of the FeO6/FeO5 network and a change in XLD during warming and cooling processes are responsible for the anisotropy of the resistivity in the thermal hysteresis region. These results further demonstrate that the instabilities in the local Fe-O bond length/DW factors and preferred Fe 3d e g orbitals drive the MI/metal-to-semiconductor transition in SrFeO2.81, in a manner similar to the driving of the Peierls MI transition in VO2, which was elucidated by Budai et al. from first-principle calculations44. Their calculations also revealed that increased occupation of V 3d x 2 -y 2 orbitals induces the Peierls instability; lowers the total electronic energy, and opens the insulating band gap44. As discussed above, the XLD studies herein of the SrFeO2.81 crystal reveal similar phenomena. During the warming process below the transition temperature, the preferential occupation changes to in-plane Fe 3d x 2 -y 2 orbitals and these changes are associated with anisotropic distortion of the FeO6/FeO5 network during both warming and cooling.

Variation of (a) DW factors and (b) NN Fe-O bond lengths with temperature, obtained by fitting temperature-dependent Fe K-edge EXAFS for R from 1.15 to 1.96 Å with angle of incidence θ = 0°, and R from 1.04 to 1.77 Å with angle of incidence θ = 70°.
The above assertion may also be related to possible CDW behavior in SrFeO2.81 45, 46. As stated earlier, Lee et al.13 used RXS to provide evidence that satellite peaks are induced by charge modulation below the transition temperature in single crystalline tetragonal SrFeO2.81. As a consequence of an electron-phonon interaction, a CDW is associated with charge modulation and lattice distortion in SrFeO2.81 13, as in other CDW materials3, 19, 47. Lattice distortion is also evident from the anomalous DW factors/bond lengths below Tm for the single crystal of SrFeO2.81, as depicted in Fig. 6. Furthermore, a CDW is a charge/electronic modulation process that arises from the coupling of valence and conduction bands, opening a band gap at/near the Fermi level (E F)48, 49. Therefore, to further examine the band gap opening across a MI/metal-to-semiconducting transition in SrFeO2.81, VB-PES spectra with photon energy hυ = 58 eV and O K-edge XANES spectra of SrFeO2.81 were obtained during warming and cooling processes, and presented in Fig. 7(a–d) for E//ab-plane and E//c-axis. The VB-PES spectra exhibit three major features, b 1 , b 2 and b 3 . The line shape of each feature is similar to those in the experimental spectra of SrFeO3-δ, LaFeO3 and Sr-doped LaFeO3 50,51,52. According to the theoretical studies of SrFeO3-δ 50 and Sr-doped LaFeO3 51,52,53 at low photon energies (20 ≤ hυ ≤ 100 eV) the O 2p cross-section dominates the Fe 3d emission, so the region (from 0 to ~2 eV) at/below E F can be attributed to the O 2p and Fe 3d (e g) hybridized states, which are essential to have a critical role in the band gap opening in SrFeO2.81, as discussed below. The O K-edge XANES spectra of SrFeO2.81 reflect transitions from O 1 s to unoccupied 2p states. The empty O 2p states are hybridized with the 3d and 4sp bands of Fe with Sr 4d bands4, so the features that are indicated by a 1 , a 2 , a 3 and a 4 in Fig. 7(a–d) correspond to the hybridized O 2p-Fe 3d states that are subject to crystal-field splitting, forming t 2g↓, e g↑, e g↓ and e g↓ states, respectively, above/near the E F, as determined from earlier theoretical work4. The insets in Fig. 7(a–d) magnify VB-PES and O K-edge XANES spectra near E F, to elucidate the relative band gap opening (or energy separation) as a function of temperature. To calculate the band gap, the leading edges in both the VB-PES and O K-edge XANES spectra of SrFeO2.81 are extrapolated to the baselines to obtain valence-band maximum (E VBM) and conduction-band-minimum (E CBM)51, 54, respectively. At room temperature, the two extrapolated lines intersect each other, indicating the SrFeO2.81 sample has no band gap and exhibits metallic behavior, which is consistent with the resistivity measurement. The fact that the value of band gap at room temperature is zero in both directions, however, during both warming and cooling processes a semiconducting band gap opens up at/near the E F and the relative value of the band gap increases as the temperature decreases. At 40 K, these values are ~0.8 and 0.6 eV for E//ab-plane and ~0.7 and 0.6 eV for E//c-axis, during warming and cooling processes, respectively. These results also reveal that the band gaps are almost independent of direction and whether the process in question is warming or cooling. The above results provide further evidence of an abrupt increase in resistivity at ~130 K. However, CDW modulations and the relative band gaps do not exhibit the anisotropy close to the thermal hysteresis region (~78 K), indicating that they may not depend on direction in SrFeO2.81 crystal or they are outside the energy resolution limit of VB-PES (~18 meV), owing to the smallness of the relevant changes. The abrupt increase in the resistivity and relative band gap opening at low temperatures provide clear evidence of the CDW nature of single crystalline SrFeO2.81. Since resistivity is determined by the electron-phonon coupling, it depends on the lattice distortion. We therefore believe that the sudden increase of resistivity below ~130 K in both directions is associated with the formation of the CDW, this claim is consistent with the appearance of satellite peaks in the RXS studies of Lee et al.13. However, the anisotropic behavior of resistivity in the thermal hysteresis region (~78 K) arises from anisotropic DW factors/bond lengths, as evident from the EXAFS analysis, and further stabilizes the different in-plane and out-of-plane Fe 3d orbitals that are observed from XLD during warming and cooling processes.

(a–d) Normalized VB-PES and O K-edge XANES spectra of a single crystal SrFeO2.81 at two angles of incidence θ = 0° (E//ab-plane) and 70° (E//c-axis) during warming and cooling. VB-PES spectra are obtained at photon energy of 58 eV. Insets display linear fits to VB-PES and O K-edge XANES spectra at various temperatures and relative band gaps.
In summary, the local electronic and atomic structures of SrFeO2.81 were elucidated using temperature-dependent XANES and VB-PES techniques, to determine the origin of the anisotropy of resistivity in the ab-plane and along the c-axis in the thermal hysteresis region. The Fe L 3,2-edge XLD results reveal that, during cooling from room temperature to below Tm, the Fe 3d electrons preferentially occupy the out-of-plane Fe 3d 3z 2 -r 2 orbitals. However, during warming below Tm, they preferentially occupy the in-plane Fe 3d x 2 -y 2 orbitals. Local atomic structural analysis of the temperature-dependent Fe K-edge EXAFS of SrFeO2.81 reveals unusually large DW factors with thermal hysteresis below Tm. Additionally, NN Fe-O bond lengths exhibit anisotropy in the ab-plane. Experimental results suggest that the local atomic structural ordering in the ab-plane differs from that along the c-axis during both warming and cooling processes. This distinction stabilizes the difference between the forms of atomic structural ordering in the warming and cooling processes and is responsible for the anisotropy of resistivity below Tm in the thermal hysteresis region in the ab-plane and along the c-axis in a single crystal of SrFeO2.81. The abrupt increase in resistivity and evidence of relative band gap opening that were obtained from the O K-edge XANES and VB-PES experiment, along with the presence of satellite peaks in RXS spectra13, confirm the CDW nature of the SrFeO2.81 single crystal at low temperatures.
Methods
Sample preparation and characterization
High-quality single crystals of SrFeO2.81 were prepared using the floating zone method27. In ref. 13 reported by Lee et al., the estimated number of oxygen-content (2.875) in SrFeOx is based from their XRD measurements, however, they did not employ techniques such as thermogravimetric analysis of iodometric titration to determine the oxygen content. Meanwhile, the refined parameter of XRD cannot tell structure difference between SrFeO2.81 and SrFeO2.875 because of the insensitivity of X-ray to oxygen atom. Further, according to the refs 1 and 2, and introduction section of the present work, SrFeOx with oxygen-content x = 2.875 is reported to be pure tetragonal, however for x = 2.75, it is pure orthorhombic, so our sample (SrFeO2.81) with oxygen-content x = 2.81 lies in between these two and also XRD presented in Fig. 1 confirm the tetragonal nature of the sample. The magnetic susceptibility and electrical resistivity were measured using a superconducting quantum interference device and a physical property measurement system, respectively. XRPD patterns were obtained on an image plate at beamline-01C of the National Synchrotron Radiation Research Center (NSRRC) in Hsinchu, Taiwan, using X-ray with a wavelength of 0.728 Å (16 keV). Lebail refinements of XRPD patterns were performed using the Fullprof software package55,56,57. Fe L 3,2-edge XANES/XLD, K-edge XANES/EXAFS, O K-edge XANES and VB-PES (incident photon energy, hυ = 58 eV) experiments were conducted at the Dragon-11A, Wiggler-17C and U9-ARPES BL21B1 beamlines at the NSRRC. The measurements were made during warming and cooling at various temperatures to elucidate the local electronic and atomic structures of the SrFeO2.81 single crystal. Further, in the warming and cooling process, all the experiments have been carried out in the same conditions. In the warming process, sample is heated from the lowest temperature (30 K) to 40 K. At 40 K, sample temperature has been kept constant for ~30 mins and then data have been collected for two orientations of sample. This is repeated for all temperatures and during cooling process too. The temperature of the sample was controlled using a closed-cycle refrigerator with an accuracy of ±0.1 K. Local atomic structure analysis of EXAFS data was performed using the Artemis program. Artemis combines the multiple-scattering EXAFS computer program FEFF41 and the nonlinear least-squares-fitting computer program FEFFIT42.
References
Lebon, A. et al. Magnetism, charge order, and giant magnetoresistance in SrFeO3−δ single crystals. Phys. Rev. Lett. 92, 037202 (2004).
Adler, P. et al. Magnetoresistance effects in SrFeO3−δ: Dependence on phase composition and relation to magnetic and charge order. Phys. Rev. B 73, 094451 (2006).
Blasco, J. et al. Charge disproportionation in La1−xSrxFeO3 probed by diffraction and spectroscopic experiments. Phys. Rev. B 77, 054107 (2008).
Abbate, M. et al. X-ray absorption of the negative charge-transfer material SrFe1−xCoxO3. Phys. Rev. B 65, 165120 (2002).
Zhao, Y. M., Mahendiran, R., Nguyen, N., Raveau, B. & Yao, R. H. SrFeO2.95: A helical antiferromagnet with large magnetoresistance. Phys. Rev. B 64, 024414 (2001).
Zhao, Y. M. & Zhou, P. F. Metal–insulator transition in helical SrFeO3−δ antiferromagnet. J. Mag. Mag. Mats. 281, 214 (2004).
Tsujimoto, Y. et al. Infinite-layer iron oxide with a square-planar coordination. Nature 450, 1062 (2007).
Williams, G. V. M., Hemery, E. K. & McCann, D. Magnetic and transport properties of SrFeOx. Phys. Rev. B 79, 24412 (2009).
Seinberg, L. et al. Fe-site substitution effect on the structural and magnetic properties in SrFeO2. Inorg. Chem. 50, 3988 (2011).
Kienle, L. et al. Electron-beam induced in-situ transformations in SrFeO3−δ single crystals. J. Phys. Chem. Solids 68, 73 (2007).
Takeda, Y. et al. Phase relation in the oxygen nonstoichiometric system, SrFeOx (2.5 ≤ x ≤ 3.0). J. Solid State Chem. 63, 237 (1986).
Hodges, J. P. et al. Evolution of oxygen-vacancy ordered crystal structures in the perovskite series SrnFenO3n−1 (n = 2, 4, 8, and ∞), and the relationship to electronic and magnetic properties. J. Solid State Chem. 151, 190 (2000).
Lee, S. H. et al. Charge and spin coupling in magnetoresistive oxygen-vacancy strontium ferrate SrFeO3−δ. New J. Phys. 18, 093033 (2016).
Reehuis, M. et al. Neutron diffraction study of spin and charge ordering in SrFeO3−δ. Phys. Rev. B 85, 184109 (2012).
Bao, W., Axe, J. D., Chen, C. H. & Cheong, S.-W. Impact of charge ordering on magnetic correlations in perovskite (Bi,Ca)MnO3. Phys. Rev. Lett. 78, 543 (1997).
Park, J.-H., Kimura, T. & Tokura, Y. Competition between lattice distortion and charge dynamics for the charge carriers of double-layered manganites. Phys. Rev. B 58, R13330 (1998).
Chi, S. et al. Effect of antiferromagnetic spin correlations on lattice distortion and charge ordering in Pr0.5Ca1.5MnO4. Proc. Nat. Acad. Sci. USA 104, 10796 (2007).
Capogrosso, V. et al. Effects of charge-orbital order-disorder phenomena on the unoccupied electronic states in the single-layered half-doped Pr0.5Ca1.5MnO4. Phys. Rev. B 87, 155118 (2013).
Herrero-Martin, J., Subias, G., Garcia, J., Blasco, J. & Sánchez, M. C. Evidence for charge-density-wave nature of the charge-ordered phase in La1/3Sr2/3FeO3. Phys. Rev. B 79, 045121 (2009).
Moritome, Y., Asamitsu, A., Kuwahara, H. & Tokura, Y. Giant magnetoresistance of manganese oxides with a layered perovskite structure. Nature 380, 141 (1996).
Imada, M., Fujimori, A. & Tokura, Y. Metal-insulator transitions. Rev. Mod. Phys. 70, 1039 (1998).
Fang, Z., Solovyev, I. V. & Terakura, K. Phase diagram of tetragonal manganites. Phys. Rev. Lett. 84, 3169 (2000).
Chen, C.-C. et al. Orbital order and spontaneous orthorhombicity in iron pnictides. Phys. Rev. B 82, 100504(R) (2010).
Wang, X. F. et al. Anisotropy in the electrical resistivity and susceptibility of superconducting BaFe2As2 single crystals. Phys. Rev. Lett. 102, 117005 (2009).
Medvedeva, J. E., Anisimov, V., Mryasov, O. N. & Freeman, A. J. The role of Coulomb correlation in magnetic and transport properties of doped manganites: La0.5Sr0.5MnO3 and LaSr2Mn2O7. J. Phys.: Condens. Matter 14, 4533 (2002).
Liu, L. et al. In-plane electronic anisotropy in the antiferromagnetic orthorhombic phase of isovalent-substituted Ba(Fe1−xRux)2As2. Phys. Rev. B 92, 094503 (2015).
Maljuk, A., Strempfer, J., Ulrich, C., Lebon, A. & Lin, C. T. Growth and characterization of high-quality SrFeOx single crystals, J. Cryst. Growth 257, 427 (2003).
Piamonteze, C. et al. Short-range charge order in RNiO3 perovskites (R = Pr, Nd, Eu, Y) probed by x-ray-absorption spectroscopy. Phys. Rev. B 71, 012104 (2005).
Perroni, C. A. et al. Modeling of strain effects in manganite films. Phys. Rev. B 68, 224424 (2003).
Klein, J. et al. Transport anisotropy in biaxially strained La2/3Ca1/3MnO3 thin films. Phys. Rev. B 66, 052414 (2002).
Lu, Y. et al. Effect of strain and tetragonal lattice distortions in doped perovskite manganites. Phys. Rev. B 73, 184406 (2006).
Aruta, C. et al. Strain induced x-ray absorption linear dichroism in La0.7Sr0.3MnO3 thin films. Phys. Rev. B 73, 235121 (2006).
Asokan, K. et al. Effect of Co, Ni, and Cu substitution on the electronic structure of hexagonal YMnO3 studied by x-ray absorption spectroscopy. Appl. Phys. Lett. 95, 131901 (2009).
Kong, H. & Zhu, C. Ultrasonic anomalies in La1∕3Sr2∕3MO3 (M = Fe, Mn). Appl. Phys. Lett. 88, 041920 (2006).
Haas, O. et al. The Fe K-edge X-ray absorption characteristics of La1−xSrxFeO3−δ prepared by solid state reaction. Mat. Res. Bull. 44, 1397 (2009).
Bocquet, A. E. et al. Electronic structure of SrFe4+O3 and related Fe perovskite oxides. Phys. Rev. B 45, 1561 (1992).
Bari, R. A. Effects of short-range interactions on electron-charge ordering and lattice distortions in the localized state. Phys. Rev. B 3, 2662 (1971).
Senn, M. S., Wright, J. P. & Attfield, Paul. J. Charge order and three-site distortions in the Verwey structure of magnetite. Nature 481, 173 (2012).
Battle, P. D., Gibb, T. C. & Steel, A. T. An investigation of non-stoichiometry in the systems SrFeO3–y and SrCoO3–y by iron and cobalt K-edge extended x-ray absorption fine structure spectroscopy. J. Chem. Soc. Dalton Trans. 1, 83 (1988).
Piovano, A. et al. Time resolved in situ XAFS study of the electrochemical oxygen intercalation in SrFeO2.5 brownmillerite structure: comparison with the homologous SrCoO2.5 system. J. Phys. Chem. C 115, 1311 (2011).
Rehr, J. J., Mustre de Leon, J., Zabinsky, S. I. & Albers, R. C. Theoretical x-ray absorption fine structure standards. J. Am. Chem. Soc. 113, 5135 (1991).
Frenkel, A. I., Stern, E. A., Qian, M. & Newville, M. Multiple-scattering x-ray-absorption fine-structure analysis and thermal expansion of alkali halides. Phys. Rev. B 48, 12449 (1993).
Islam, Q. T. & Bunker, B. A. Ferroelectric transition in Pb1−xGexTe: Extended x-ray-absorption fine-structure investigation of the Ge and Pb Sites. Phys. Rev. Lett. 59, 2701 (1987).
Budai, J. D. et al. Metallization of vanadium dioxide driven by large phonon entropy. Nature 515, 535 (2015).
Grüner, G. The dynamics of charge-density waves. Rev. Mod. Phys. 60, 1129 (1988).
Gabovich, A. M., Voitenko, A. I., Annett, J. F. & Ausloos, M. Charge- and spin-density-wave superconductors. Supercond. Sci. Technol. 14, R1–R27 (2001).
Clerc, F. et al. Lattice-distortion-enhanced electron-phonon coupling and Fermi surface nesting in 1T−TaS2. Phys. Rev. B 74, 155114 (2006).
Cercellier, H. et al. Evidence for an excitonic insulator phase in 1T−TiSe2. Phys. Rev. Lett. 99, 146403 (2007).
Monney, C. et al. Temperature-dependent photoemission on 1T-TiSe2: Interpretation within the exciton condensate phase model. Phys. Rev. B 81, 155104 (2010).
Galakhov, V. R. et al. Valence band structure and x-ray spectra of oxygen-deficient ferrites SrFeOx. J. Phys. Chem. C 114, 5154 (2010).
Wadati, H. et al. Hole-doping-induced changes in the electronic structure of La1−xSrxFeO3: Soft x-ray photoemission and absorption study of epitaxial thin films. Phys. Rev. B 71, 035108 (2005).
Wadati, H. et al. Temperature-dependent soft x-ray photoemission and absorption studies of charge disproportionation in La1−xSrxFeO3. J. Phys. Soc. Jpn. 75, 054704 (2006).
Matsuno, J. et al. Photoemission and Hartree-Fock studies of oxygen-hole ordering in charge-disproportionated La1−xSrxFeO3. Phys. Rev. B 60, 4605 (1999).
Chuang, C.-H. et al. The effect of thermal reduction on the photoluminescence and electronic structures of graphene oxides. Sci. Rep. 4, 4525 (2014).
LeBail, A., Duroy, H. & Fourquet, J. L. Ab-initio structure determination of LiSbWO6 by x-ray powder diffraction. Mat. Res. Bull. 23, 447 (1988).
Rodriguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Physica B 192, 55 (1993).
Rodriguez-Carvajal, J. Laboratory, FULLPROF, a Rietveld and pattern matching and analysis program, Laboratoire Leon Brillouin, CEA-CNRS, France [http://www.ill.eu/sites/fullprof/], version July (2011).
Acknowledgements
The author (W.F.P.) would like to thank the National Science Council of Taiwan and Ministry of Science and Technology of Taiwan (MoST) for providing financial support for the research under project No. NSC 102-2112-M-032-007-MY3 and NSC 102-2632-M-032-001-MY3. R.S.S. is thankful to the Department of Science and Technology (DST), India for DST INSPIRE Faculty award (DST/INSPIRE/04/2015/002300).
Author information
Authors and Affiliations
Contributions
S.H.H., R.S.S. and W.F.P. designed the experiments having prior discussion with C.H.D. The SrFeO2.81 sample was synthesized by C.H.Y., C.H.D. and F.C.C. All measurements are performed by S.H.H., R.S.S., Y.F.W., Y.C.S., S.H.L., H.T.W., J.W.C., Y.Y.C., H.M.T., J.L.C., C.W.P., C.M.C., W.C.C., H.J.L. and J.F.L. The data analysis and manuscript writing are done by S.H.H., R.S.S. and W.F.P. All authors discussed the results and contributed to finalization of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hsieh, S.H., Solanki, R.S., Wang, Y.F. et al. Anisotropy in the thermal hysteresis of resistivity and charge density wave nature of single crystal SrFeO3-δ: X-ray absorption and photoemission studies. Sci Rep 7, 161 (2017). https://doi.org/10.1038/s41598-017-00247-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-00247-z
- Springer Nature Limited
This article is cited by
-
Simple analytical method for determining electrical resistivity and sheet resistance using the van der Pauw procedure
Scientific Reports (2020)
-
The effect of orbital-lattice coupling on the electrical resistivity of YBaCuFeO5 investigated by X-ray absorption
Scientific Reports (2019)
-
Anisotropy in the magnetic interaction and lattice-orbital coupling of single crystal Ni3TeO6
Scientific Reports (2018)