
Abstract
Achelura yunnanensis is a destructive pest of forests, causing substantial damage on tree growth and severe economic losses. Additionally, as a daytime-active moth, this species also holds important scientific value for investigating the genetic mechanisms governing day-night activity patterns of Lepidoptera. To facilitate effective pest control and deepen our understanding of the diurnal behavior’s genetic basis of moths, genomic data for this species are crucial. In this study, we present a chromosome-level reference genome of A. yunnanensis (368.15 Mb in 32 chromosomes; scaffold N50 = 12.61 Mb; BUSCO completeness = 98.0%). Genome annotation shows that the new assembly comprises 37.10% (136.55 Mb) repetitive elements, 1,828 non-coding RNAs, and 15,523 protein-coding genes. Genes involved in lipid metabolism and xenobiotics biodegradation and metabolism, such as cytochrome P450 families, experienced significant expansion in the A. yunnanensis genome. The chromosome-level genome of A. yunnanensis provides a valuable genomic resource for devising novel pest control strategies, and will also help to study the genetic mechanism of the shift of diurnal behavior in Lepidoptera.
Similar content being viewed by others
Background & Summary
Achelura yunnanensis is a notorious pest that feeds on the flowering cherry trees1,2, which are economically valuable ornamental plants3. During periods of high infestation, a single cherry tree can harbor up to hundreds of larvae, significantly impacting tree growth and resulting in substantial economic losses4. Currently, chemical pesticides still serve as the primary method for controlling A. yunnanensis outbreaks; however, these chemicals often lead to environmental pollution and pose food safety risks5,6. Moreover, previous studies have found that the expansion of the uridine diphosphate glycosyltransferases gene family in A. yunnanensis may be linked to its increased resistance to both plant metabolites and pesticides, exacerbating the challenges of chemical pest control7,8,9. Therefore, there is an urgent need to explore alternative biocontrol methods to achieve effective and environmentally safe pest management to this species. However, the lack of genomic resources has hindered the development of biocontrol strategies, including those based on specific molecular targets.
Taxonomically, A. yunnanensis belongs to the Zygaenidae family, a diverse group of moths distributed throughout the world1,10. Unlike most nocturnal moths, most Zygaenidae species are diurnal and have eye-catching aposematic colors and patterns on their wings to warn off daytime predators11,12. Although diurnality is widespread in Lepidoptera (moths and butterflies) and has independently evolved many times, the molecular mechanisms underlying this behavior remain poorly understood13. A recent study based on transcriptome data by Akiyama et al.14 suggested that the parallel evolution of opsins may contribute to the diurnal adaptation of certain day-flying species within the hawkmoth family (Lepidoptera, Sphingidae)14. Undoubtedly, to fully understand the genetic mechanism behind the shift of day-night activity of Lepidoptera, we need to study the genome data across various lepidopteran taxa, including Zygaenidae. However, genomic resources for Zygaenidae are extremely limited. Before this study, of the four subfamilies of Zygaenidae, only the Zygaeninae subfamily has one species been sequenced, and the subfamilies Chalcosiinae, Callizygaeninae, and Procridinae all lack of genomic data15. This scarcity of genomic resources has impeded further exploration of the genetic basis underlying diurnality in this moth family.
In this study, we present a chromosome-level genome of A. yunnanensis, a representative species of the Chalcosiinae subfamily of Zygaenidae. The final genome assembly was 368.15 Mb, with contig N50 and scaffold N50 values of 12.20 Mb and 12.61 Mb, respectively, indicating a high level of completeness and contiguity (Table 1). Comparative genomic analysis revealed a significant expansion of gene families associated with lipid catabolism and xenobiotic biodegradation and metabolism in A. yunnanensis, which may contribute to the species’ remarkable adaptability, including its broad host range and its ability to degrade toxic compounds from both plants and the environment. Overall, this genome assembly serves as a valuable resource for future endeavors in the integrated pest management of A. yunnanensis and has the potential to uncover the genetic mechanisms governing day-night activity patterns in Lepidoptera through comparative genomics studies.
Methods
Sample collection, library construction and sequencing
Two fifth-instar larvae samples of A. yunnanensis were collected in September 2023 from cherry trees located at Yunnan University, Kunming, Yunnan Province, China. Guts were removed from each larvae sample to reduce gut microbe contamination, then each sample was cleaned with phosphate-buffered saline (PBS) buffer twice.
DNA and RNA were extracted from one larvae sample using the TIANGEN Blood & Tissue Kit (Tiangen, Beijing, China) and the TRIzol Reagent Kit (Invitrogen, USA), respectively. The quality and concentration of nucleic acid were assessed using a Qubit 3.0 Fluorometer (Life Technologies, CA, USA) and 1.0% TBE agarose gel electrophoresis. For short-read genomic sequencing, DNA sequencing libraries were constructed according to the TruSeq DNA Sample Preparation Guide (Illumina, USA) and sequenced on the Illumina NovaSeq. 6000 platform. For PacBio HiFi sequencing, circular consensus sequencing (CCS) libraries were constructed using the Pacific Biosciences SMRT bell Express Template Prep Kit 2.0, and sequenced on the PacBio Sequel II System with HiFi mode. For Hi-C sequencing, the Hi-C libraries were prepared from the other larvae sample according to the standard procedure with minor modifications16 and sequenced on the Illumina NovaSeq. 6000 platform. For transcriptome sequencing, RNA-seq libraries were constructed using the Illumina TruSeq Stranded mRNA Library Prep Kit (Illumina, USA) and sequenced on the Illumina NovaSeq6000 platform. After filtering the low-quality reads and trimming adaptor sequences from the raw data using fastp (v0.23.2)17, we obtained a total of 57.00 Gb Illumina short-reads (~155-fold coverage), 26.12 Gb PacBio HiFi long-reads (~71-fold coverage), 55.94 Gb Hi-C reads (~152-fold coverage) and 6.5 Gb RNA-seq data (Supplementary Table 1).
Genome survey and de novo assembly
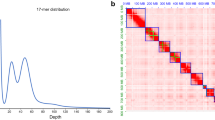
The 57.00 Gb Illumina short-reads were used for genome survey to estimate genome characteristics such as genome size, repetitive sequence content, and heterozygosity. K-mer frequencies were assessed using jellyfish (v2.3.0)18 with a length set to 17 k-mer and were then used to conduct a genome survey using GenomeScope (v1.0)19. As a result, the estimated genome size was approximately 320.47 Mb, with a heterozygosity rate of 1.43% and a repetitive sequence content of 35.64% (Fig. 1a; Table 2).

(a) Result of the 17-mer frequency distribution analysis for the Achelura yunnanensis genome. (b) Hi-C interactions heatmap for the A. yunnanensis genome. The color gradient bars on the right side of the map represent chromosome interaction strengths, which range from yellow (low) to red (high). It shows that intra-chromosome (red blocks on the diagonal line) interactions are stronger than inter-chromosome.
The PacBio HiFi long-read (quality value > = 20) data were de novo assembled into a draft genome (comprising dozens of contigs) using Hifiasm (v0.19.6)20 with the default parameters. To generate a chromosome-scale genome assembly of A. yunnanensis, Hi-C reads were mapped to the draft genome with the BWA mem algorithm21. Based on the quality-controlled Hi-C read alignments, a contact matrix was generated using Juicer (v1.6.2)22 with default parameters. 3D-DNA (v190716)23 was then employed to correct misjoins, and order and orientate the contigs, resulting in most of the contigs being anchored to the pseudochromosomes. And JuiceBox (v2.17.0)24 was used to visualize the Hi-C interactions between contigs, and manually correct any misjoins, translocations, and inversions. For contigs that could not be anchored to the chromosomes, BLASTN (v2.15.0)25 was used to search them against the Nucleotide Sequence Database (NT). Contigs that hit non-metazoan targets were viewed as contamination and discarded. Next, genome integrity was assessed by BUSCO (v5.4.3)26 based on the Lepidoptera_odb10 database (n = 5,286 single-copy orthologues). To calculate the mapping rate and identify sex chromosomes27, we mapped Illumina short-reads to the genome assembly using BWA (v0.7.17)21. The mapping rate and sequencing depth for each chromosome were then calculated by using QualiMap (v.2.3)28. Chromosomes with half the sequencing depth were identified as sex chromosomes. As a result, the initial assembly based on PacBio HiFi long-reads yielded a draft genome of 375.40 Mb, comprising 96 contigs with an N50 size of 12.20 Mb. These contigs were anchored to 32 chromosomes by the Hi-C data (Fig. 1b). Chr1 and Chr32 were identified to be the sex chromosomes. After removal of the contaminating contigs and mitochondria sequences, the resulting chromosome-level genome was 368.15 Mb in length with a scaffold N50 of 12.61 Mb and a GC content of 35.15% (Table 1; Fig. 2). Quality evaluation of the genome assembly showed that a total of 99.02% of the Illumina short-reads were properly mapped to it. Furthermore, a BUSCO assessment indicated that 98.0% of the target orthologous genes could be identified in complete form from the genome assembly (Supplementary Table 2). Together, these evaluations suggest a remarkably high level of completeness, contiguity, and accuracy of the genome assembly of A. yunnanensis.

Circos plot of the Achelura yunnanensis genomic features. The tracks from inside to outside: (A) DNA TE abundance; (B) LINE abundance; (C) GC content; (D) Gene density; (E) Pseudo-chromosomes. Window size = 100 kb.
Repetitive element and noncoding RNA annotation
To annotate repeat elements in the A. yunnanensis genome, we firstly de novo constructed a repeat library from the genome using the integrated results from three embedded programs (RECON (v1.0.8)29, RepeatScout (v1.0.6)30 and TRF (v4.09)31) in RepeatModeler (v2.0.3)32. This library was then merged with the known repeat element database, including the Insecta set of Repbase-2018102633 and Dfam 3.734, to form a custom library. Based on this custom library, RepeatMasker (v4.1.5)35 was used to identify and mask repetitive regions in the genome assembly with xsmall parameter. In total, 136.55 Mb of repeat sequences were identified, which accounted for 37.10% of the genome assembly. Among these repeat elements, long interspersed elements (LINEs) represented the most abundant class of repeats, constituting 52.48 Mb (14.26% of the whole genome). Additionally, DNA transposons, short interspersed nuclear elements (SINEs), and long terminal repeats (LTR) account for 6.43%, 2.47%, and 1.56% of the whole genome, respectively (Table 3).
For the discovery of transfer RNAs (tRNAs), tRNAscan-SE (v2.0.12)36 was applied with eukaryotic parameters according to the characteristics of tRNA. The identification of ribosomal RNAs (rRNAs) and its various copies was performed using Barrnap (https://github.com/tseemann/barrnap). Other genomic noncoding RNAs (ncRNAs), such as small nuclear RNAs (snRNAs) and microRNAs (miRNAs), were identified through comparison with the Rfam37 database (release 14.10) using Infernal (v1.1.5)38. Totally, 1828 ncRNAs were identified in the A. yunnanensis genome, including 483 rRNAs, 66 miRNAs, 72 snRNAs, 1099 tRNAs, and 108 other ncRNAs, respectively (Table 4).
Protein-coding gene prediction and function annotation
Protein-coding gene structure predictions were conducted by combining evidence from the transcriptome-based, ab initio, and homology-based predictions. For transcriptome-based prediction, RNA-seq data from the larvae body and the adult sex pheromone glands39 were aligned with the genome to produce BAM alignments by HISAT (v2.2.1)40 and Samtools (v1.19)41. The RNA-seq alignments were used to perform genome-guided assembly by StringTie (v2.2.1)42, and the likely open reading frames within the transcripts were identified with TransDecoder (v5.5.0)43. For the ab initio predictions, BRAKER (v3.0.7)44 was adopted, which automatically trained the predictors Augustus (v3.4.0)45 and GeneMark-ETP (v4.72)46 using the Arthropoda reference proteins database from OrthoDB10 (v10)47 and the RNA-seq alignments mentioned above. SNAP (v2006-07-28)48 was also used for ab initio gene prediction, where the B.mori.hmm was selected as the training set. For the homology-based prediction, we downloaded the reference gene sets of six related species from Ensembl and NCBI databases, namely Bombyx mori49, Colias croceus50, Helicoverpa armigera51, Spodoptera frugiperda52, Vanessa cardui53 and Zygaena filipendulae54, to generate a homology-based gene set (Supplementary Table 3). GeMoMa (v1.9)55, GenomeThreader (v1.7.3)56, and Miniport (v0.12)57 were used to align the homology-based gene set to the genome and predict the gene structure information. Finally, EVidenceModeler (v2.1.0)58 was used to integrate the gene-predicted results from the three methods and generate a consensus gene set. As a result, a total of 15,523 protein-coding genes were predicted from the A. yunnanensis genome, with an average gene length of 770,1.7 bp. These genes have an average of 6.1 exons per gene, with an average exon length of 235.5 bp, and an average of 5.1 introns per gene, with an average intron length of 1230.2 bp (Table 5). The completeness of the predicted protein gene sequences was 97.3% (96.3% single-copied genes and 1.0% duplicated genes) as assessed by BUSCO (v5.4.3)26 search based on the Lepidoptera_odb10 database (n = 5,286 single-copy orthologues) (Table 5).
To add functional annotation to the predicted protein-coding genes, we searched the predicted genes against the UniProtKB database (SwissPro and TrEMBL) and the nonredundant protein sequence database (NR) using the high-sensitivity mode of Diamond (v2.1.8)59. We further employed eggNOG-mapper (v2.1.12)60 to search the eggNOG (v5.0)61 database. In addition, InterProScan (v5.59–91.0)62 was used to assign Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome pathway annotations to the predicted genes, and to identify protein domains among the predicted genes. A total of 15,116 (97.38%) protein-coding genes obtained a final prediction of gene functions following the above steps (Table 6). The final physical characteristics of the genome assembly features were visualized using Circos (v 0.69-8)63 (Fig. 2).
Data Records
The raw sequencing data of A. yunnanensis reported in this paper have been submitted to the NCBI with Bioproject ID PRJNA1115809. Illumina, PacBio, Hi-C, and transcriptome raw data have been deposited in the NCBI Sequence Read Archive with accession numbers SRR29152278-SRR2915228164,65,66,67. The final assembled genome has been submitted to the Genome database of NCBI with accession numbers GCA_041274885.168. The annotation file is available in figshare2596283569.
Technical Validation
Evaluation of the genome assembly
Three independent methods were used to assess the completeness, contiguity, and accuracy of the A. yunnanensis genome assembly. Firstly, the initial assembly contained a total of 96 contigs, with a contig N50 size of 12.20 Mb and the longest contig of 17.44 Mb. After the Hi-C data was added, the chromosome-level assembly was characterized by a scaffold N50 size of 12.61 Mb and the longest scaffold of 17.44 Mb, which indicates high continuity of the genome assembly. Secondly, the genome assembly displayed a BUSCO completeness of 98.0% (97.4% single-copied genes and 0.6% duplicated genes) based on the Lepidoptera_odb10 database. Finally, to verify the accuracy of the genome assembly, we calculated mapping rates by aligning clean Illumina data to the genome assembly. As a result, 99.02% of the Illumina reads aligned with the genome assembly. Overall, these assessments reflect the high quality and accuracy of the chromosome-level assemblies.
Genomic synteny analysis
Genome synteny analysis of A. yunnanensis and another Zygaenidae species, Zygaena filipendulae54, was conducted using MCScanX70 to identify the Z and W chromosomes and evaluate the accuracy of the genome assembly. A high degree of collinearity was observed between our assembly and the Z. filipendulae genome (Fig. 3). All chromosomes in our assembled genome, except for the W chromosome, exhibited strong collinearity with those of Z. filipendulae. The lack of linear correlation between the W chromosomes is likely due to the fact that MCScanX synteny analysis relies on the collinear analysis of coding genes, while the W chromosome contains few coding genes. Additionally, we identified a chromosomal fusion and fission event between the two genomes, with chromosome 21 of Z. filipendulae being syntenic to chromosomes 28 and 29 of A. yunnanensis. Apart from the W chromosome and that chromosomal fusion-fission event, all chromosomes in the assembled genome demonstrated one-to-one collinearity with those of Z. filipendulae, highlighting the accuracy of our genome assembly.

Chromosome-level genomic synteny between Achelura yunnanensis and another Zygaenidae species, Zygaena filipendulae. Gray lines indicate conserved syntenic blocks between the two genomes.
Phylogenetic analysis
To determine the phylogenetic position of A. yunnanensis, we performed a phylogenomic analysis based on 4,316 single-copy protein-coding genes collected from the genomes of A. yunnanensis and 14 Lepidopteran species49,54,71,72,73,74,75,76,77,78,79,80,81,82 (Supplementary Table 4). Orthologous sequences of single-copy protein-coding genes among all species were determined using OrthoFinder (v2.5.4)83. Protein sequence alignments for each gene were built using MAFFT (v7.505)84 and poorly aligned regions were removed using Gblocks (v0.91b)85 with default settings. Phylogenetic tree was constructed from the concatenated supermatrix using FastTree (v2.1.11)86 under the JTTCAT model. Based on the phylogenetic tree, r8s (v1.81)87 was used to estimate the divergence times among taxa. To calibrate the timetree, the divergence time between E. monodactyla and B. mori was fixed at 98 million years ago (Mya) according to the documented divergence time available in the TimeTree database88. Our phylogenetic tree (Fig. 4a; rooted with Plutella xylostella82) showed that Zygaenidae, which A. yunnanensis and Z. filipendulae belong to, is the sister group of Limacodidae, and the divergence between the two families took place at approximately 70.10 Mya. Within the family Zygaenidae, the divergence time between A. yunnanensis and its European relative Z. filipendulae was estimated to be 58.11 Mya.

Gene family evolution in Achelura yunnanensis. (a) The numbers of the expanded gene families (red) and contracted gene families (blue) are shown to the right of each branch. The pie charts represent the proportions of gene family expansions (red) and contractions (blue). (b) GO enrichment analysis on the expanded gene families of A. yunnanensis. The top 20 most significant GO categories were included (p < 0.05). (c) KEGG pathways enrichment analysis on the expanded gene families of A. yunnanensis. The graph depicts the most highly enriched pathways.
Gene family expansion and contraction
To investigate genome-wide changes associated with adaptation in the A. yunnanensis genome, we performed an analysis of gene family expansion and contraction across 15 Lepidopteran species using CAFÉ (v5.0)89 with a p-value threshold < 0.05 as the cut-off. Subsequently, we used the R package clusterProfiler (v4.10.0)90 to conduct GO and KEGG enrichment analyses on the significantly expanded gene families (p < 0.05).
We identified 531 and 467 gene families that had expanded and contracted in A. yunnanensis, respectively (Fig. 4a; the detailed analysis results of the expanded and contracted gene families of A. yunnanensis are given in Supplementary Tables 5, 6). GO enrichment analysis (Fig. 4b) showed that the expanded genes were significantly enriched in catabolic processes, such as glycosphingolipid catabolic process (GO:0046479, P = 3.72 × 10−30) and lipid catabolic process (GO:0016042, P = 3.74 × 10−22). KEGG pathway enrichment analysis (Fig. 4c) suggested that the expanded genes were significantly involved in lipid metabolism and xenobiotics biodegradation and metabolism, such as the metabolism of xenobiotics by cytochrome P450 pathway (ko00980, P = 5.39 × 10−17).
A. yunnanensis larvae feed on various plant species of the Rosaceae family9. The expansion of the catabolism-related genes may contribute to their ability to feed on a wide range of plants, enhancing their adaptability as pests. Additionally, detoxification-related genes are crucial for herbivorous insects to neutralize toxic chemicals from their host plants or the environment. Several expanded gene families in the A. yunnanensis genome were significantly enriched with xenobiotics detoxification systems, further increasing their adaptability and complicating pest control efforts. Therefore, understanding the functions of the genes within these expanded gene families may help in developing novel pest management strategies.
Code availability
No specific script was used in this work. All software and bioinformatic analyses were performed according to the manuals and protocols of the corresponding bioinformatic software. If manually adjusted parameters were used, the software version and method used are described in the Methods.
References
Owada, M., Horie, K. & Dayong, X. Three new chalcosiine moths of the genus Achelura (Lepidoptera, Zygaenidae) from the northern Indo-Chinese Peninsula and southwestern China. Tinea 16, 20–28 (1999).
Tian, M., Rong, C., Bai, B., Yang, Z. & Liu, P. Biological characteristics and occurrence patterns of Achelura yunnanensis (Lepidoptera: Zygaenidae) in Yunnan Province. Plant Prot. 44, 191–194 (2018).
Yi, X. G. et al. The genome of Chinese flowering cherry (Cerasus serrulata) provides new insights into Cerasus species. Hortic. Res. 7, 165 (2020).
Rong, C. et al. Spatial distribution patterns and sampling techniques of the Achelura yunnanensis. J. West China For. Sci. 44, 72–76 (2015).
Kim, K. H., Kabir, E. & Jahan, S. A. Exposure to pesticides and the associated human health effects. Sci. Total Environ. 575, 525–535 (2017).
Rong, C. et al. Toxicity test of 5 insecticides against Achelura yunnanensis. J. West China For. Sci. 45, 142–144 (2016).
Ahn, S. J., Chertemps, T., Maïbèche, M., Marygold, S. J. & Van Leeuwen, T. Editorial: invertebrate UDP-glycosyltransferases: nomenclature, diversity and functions. Front. Physiol. 12, 748290 (2021).
Després, L., David, J. P. & Gallet, C. The evolutionary ecology of insect resistance to plant chemicals. Trends Ecol. Evol. 22, 298–307 (2007).
Xiao, H. Y., Chen, D. L., Lu, T. T., Yao, Y. J. & Liu, N. Y. The UDP-glycosyltransferase gene family in Achelura yunnanensis (Lepidoptera: Zygaenidae): identification, phylogeny, and diverse expression patterns. Diversity 14, 407 (2022).
Epstein, M. E., Geertsema, H., Naumann, C. M. & Tarmann, G. M. Handbook Of Zoology. (De Gruyter, 1999).
Yen, S., Robinson, G. S. & Quicke, D. L. J. Phylogeny, systematics and evolution of mimetic wing patterns of Eterusia moths (Lepidoptera, Zygaenidae, Chalcosiinae). Syst. Entomol. 30, 358–397 (2005).
Kawahara, A. Y. et al. Diel behavior in moths and butterflies: a synthesis of data illuminates the evolution of temporal activity. Org. Divers. Evol. 18, 13–27 (2018).
Seifert, C. L., Strutzenberger, P. & Fiedler, K. Are day-flying moths more specialized in larval dietary breadth?—a test of the ‘Salient Aroma Hypothesis’ in a predominantly nocturnal clade. Evolution 78, 1174–1182 (2024).
Akiyama, T., Uchiyama, H., Yajima, S., Arikawa, K. & Terai, Y. Parallel evolution of opsin visual pigments in hawkmoths by tuning of spectral sensitivities during transition from a nocturnal to a diurnal ecology. J. Exp. Biol. 225, jeb244541 (2022).
Niehuis, O., Yen, S. H., Naumann, C. M. & Misof, B. Higher phylogeny of zygaenid moths (Insecta: Lepidoptera) inferred from nuclear and mitochondrial sequence data and the evolution of larval cuticular cavities for chemical defence. Mol. Phylogenet. Evol. 39, 812–829 (2006).
Rao, S. S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinforma. Oxf. Engl. 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Li, H. & Durbin, R. Fast and accurate long-read alignment with burrows–wheeler transform. Bioinformatics 26, 589–595 (2010).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Mount, D. W. Using the basic local alignment search tool (BLAST). Cold Spring Harb. Protoc. 6 (2007).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Mongue, A. J., Nguyen, P., Voleníková, A. & Walters, J. R. Neo-sex chromosomes in the monarch butterfly. Danaus plexippus. G3 7, 3281–3294 (2017).
Okonechnikov, K., Conesa, A. & García Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinforma. Oxf. Engl. 32, 292–294 (2016).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinforma. Oxf. Engl. 21, 351–358 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–89 (2016).
Tarailo Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. 4, 4.10.1–4.10.14 (2009).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Griffiths Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR15959226 (2021).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinforma. Oxf. Engl. 25, 2078–2079 (2009).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Gabriel, L. et al. BRAKER3: fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. Preprint at https://www.biorxiv.org/content/10.1101/2023.06.10.544449v4 (2024).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–312 (2004).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-ETP: automatic gene finding in eukaryotic genomes in consistency with extrinsic data. Preprint at https://www.biorxiv.org/content/10.1101/2023.01.13.524024v5 (2024).
Kriventseva, E. V. et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811 (2019).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_014905235.2 (2020).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_905220415.1 (2021).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_023701775.1 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_023101765.3 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_905220365.1 (2021).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_907165275.2 (2021).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inf. Softw. Technol. 47, 965–978 (2005).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014 (2023).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Huerta Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122 (2017).
Huerta Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29152278 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29152279 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29152280 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR29152281 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_041274885.1 (2024).
Fang, R. Z. The genome and the genome annotation of the Achelura yunnanensis. figshare https://doi.org/10.6084/m9.figshare.25962835 (2024).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40 (2012).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_946406115.1 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_905404145.2 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_949316135.1 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_033807575.1 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_916618145.1 (2021).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_907165245.1 (2021).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_022581195.1 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_023078275.1 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_024362695.1 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_905475465.2 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_027563975.2 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_932276165.1 (2024).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS One 5, e9490 (2010).
Sanderson, M. J. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinforma. Oxf. Engl. 19, 301–302 (2003).
Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819 (2017).
Mendes, F. K., Vanderpool, D., Fulton, B. & Hahn, M. W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 36, 5516–5518 (2021).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innov. Camb. Mass 2, 100141 (2021).
Acknowledgements
We would like to thank Dr. ShaoJi Hu for help in collecting A. yunnanensis larvae samples. This work was supported by the National Natural Science Foundation of China (No. 32370477, 32370541), and the Natural Science Foundation of Guangdong Province Outstanding Youth Team Project (No. 2023B1515040002).
Author information
Authors and Affiliations
Contributions
Z.P. and L.D. conceived and designed the study. T.X. and F.R.Z. generated the genome data. F.R.Z. performed the genomic analyses. T.X. and F.R.Z. drafted the manuscript. Z.P. and L.D. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41597_2024_3849_MOESM5_ESM.xlsx
Supplementary Table 5 The expanded gene families of Achelura yunnanensis and their corresponding numbers of these expanded gene families in the 15 lepidopteran genomes
41597_2024_3849_MOESM6_ESM.xlsx
Supplementary Table 6 The annotation of the contracted gene families of Achelura yunnanensis and their corresponding numbers of these contracted gene families in the 15 lepidopteran genomes
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fang, R., Tian, X., Liang, D. et al. Chromosome-level genome assembly of the forest pest Achelura yunnanensis (Lepidoptera: Zygaenidae). Sci Data 11, 995 (2024). https://doi.org/10.1038/s41597-024-03849-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03849-1
- Springer Nature Limited