
Abstract
The two-spotted spider mite, Tetranychus urticae Koch (Acari: Tetranychidae), is a notorious pest in agriculture that has developed resistance to almost all chemical types used for its control. Here, we assembled a chromosome-level genome for the TSSM using Illumina, Nanopore, and Hi-C sequencing technologies. The assembled contigs had a total length of 103.94 Mb with an N50 of 3.46 Mb, with 87.7 Mb of 34 contigs anchored to three chromosomes. The chromosome-level genome assembly had a BUSCO completeness of 94.8%. We identified 15,604 protein-coding genes, with 11,435 genes that could be functionally annotated. The high-quality genome provides invaluable resources for the genetic and evolutionary study of TSSM.
Similar content being viewed by others


Background & Summary
The two-spotted spider mite (TSSM), Tetranychus urticae Koch (Acari: Tetranychidae), is a notorious agricultural pest, with over 1,100 documented host plants1. It causes damage to a wide variety of vegetables, fruit trees, and flowers worldwide. Despite numerous control methods developed to control TSSM, it remains one of the major challenges to mitigating the damage of the TSSM in fields2,3,4,5. The TSSM has a high potential to adapt to environmental changes6,7. It has developed resistance to almost all types of pesticide used to its control8. A reference genome is essential for understanding the ecology and genetics of adaptation as well as for developing new control methods of TSSM. A TSSM genome was determined using Sanger sequencing, which is one of the early reported pest genomes7. The assembly has a size of 89.6 Mb with 640 scaffolds7. It has been widely used and significantly enhanced the studies of TSSM, especially in the fields of pesticide resistance, adaptation to host plants, and environmental changes9,10,11,12,13,14. To improve the continuity of the TSSM genome and correct misassembled scaffolds, Wybouw, et al.15 assembled the Sanger sequences into three pseudochromosomes by using population allele frequency data and de novo assemblies of seven strains from Illumina data. The number of chromosomes is consistent with previous cytological work16,17.This chromosome-level genome resolves discontinuities of allele frequencies and facilitates the genome-wide scanning of genes and mutations underlying the evolutionary adaptation of TSSM15,18,19.
In this study, we assembled a chromosome-level genome for the TSSM using a combination of Nanopore long-read and Illumina short-read sequencing, Hi-C technology, and RNA-sequencing (RNA-seq). We yielded a nuclear genome assembly of 87.7 Mb, with an N50 of 29.6 Mb and BUSCO (Benchmarking Universal Single-Copy Ortholog) completeness of 93.4%. This high-quality genome will provide invaluable resources for the study of the TSSM and its relative issues.
Methods
Materials and sequencing
The TSSM strain used for sequencing was collected from Xiaoshan City of Zhejiang province. To decrease the effect of heterozygosity on subsequent analysis, a lab population was reared on French bean Phaseolus vulgaris from a small population (about 200 individuals) for continuous generations (about 20 generations) before sequencing, under 25 ± 1 °C, 60 ± 5% relative humidity and L16: D8 photoperiod. Approximately 200 individuals were used for Illumina, 2000 for NanoPore, and 3000 for Hi-C proximity ligation library construction. About 200 larvae and adults were used for transcriptome sequencing for each of the three libraries. Genomic DNA was extracted using the DNeasy tissue kit (Qiagen, Hilden, Germany) for Illumina library construction and the MagAttract HMW DNA kit (Qiagen, Hilden, Germany) for NanoPore library construction. For the Hi-C library, the genome was digested by the restriction enzyme DpnII, and fragments were then sheared into ~400 bp. The Hi-C library was sequenced using the DNBSEQ-T7 platform. RNA-seq libraries were prepared using VAHTSTM mRNA-seq V2 Library Prep Kit (Vazyme, Nanjing, China) and sequenced on the Illumina NovaSeq platform. Sequencing data generated from each library are provide in Table 1.
Genome survey
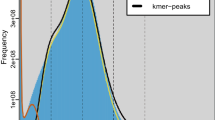
Genome survey was performed using a k-mer based method. The k-mer coverage was counted from Illumina short reads using Jellyfish version 2.2.1020 with k-mers of 17, 21, 25, and 31. Genome size, heterozygosity, and duplication rate were estimated using GenomeScope version 2.021. The estimated size of the TSSM genome rangs from 87.25 Mb to 88.05 Mb, with a heterozygosity rate of 0.60% to 0.64%, and a duplication rate of 3.25% to 4.41% (Fig. 1a–d).

Genome survey and assembly of the two-spotted spider mite (TSSM) Tetranychus urticae. Genome size, heterozygosity and rate of duplication were estimated using Genomescope when k-mer = 17 (a), 21 (b), 25 (c), and 31 (d). (e) The genome-wide all-by-all Hi-C matrix of TSSM. Three linkage groups were identified based on Hi-C contacts, indicated by blue boxes. Sequences anchored on chromosomes are shown in the plot. (f) Synteny blocks between our new assembly and two previously published genome assemblies of TSSM.
Genome assembly
Nanopore long-reads were corrected and assembled using Nextdenovo22 with default parameters. In order to remove possible secondary alleles, the assembled contigs were filtered using the pipeline Purge Haplotigs23, which produced 177 contigs with a total length of 103.35 Mb and a contig N50 of 3.46 Mb. Raw Illumina whole-genome short-reads were used to polish the long-read contig-level assembly using Pilon v1.2224. Hi-C Illumina short-reads were used to assemble contigs into a chromosome-level genome using Juicer v1.525 and 3D-DNA26. The final assembly contains three chromosomes composed of 34 contigs with a total length of 87.7 Mb (Fig. 1e). This newly assembled genome has greater continuity, with 33 gaps, compared to a previously reported pseudochromosome-level genome, which consisted of 42 scaffolds with over 800 gaps15,27.
Genome annotation
The repeat annotation was performed with RepeatModeler v2.0.428 and RepeatMasker v4.1.429 using a species-specific repeat library, a RepBase database, and a repeat element library for Arthropoda from the Dfam database. The protein-coding genes were annotated using RNA-seq-based, ab initio, and homolog-based methods in the MAKER v3.01.04 pipeline30. For the RNA-seq-based method, the RNA-seq reads of three libraries were mapped to our TSSM assembly with Hisat v2.2.031. The transcripts were then assembled using Stringtie v2.1.2. For ab initio annotation, SNAP v2013-02-1632 and Augustus v3.2.333 parameters were estimated or trained before using them to predict genes in MAKER30. The SNAP parameters were estimated from high-quality transcripts obtained by improvement and filtering using PASA v2.4.134. The gene model of Augustus was directly obtained from the above BUSCO analysis of the genome assembly. For the homolog-based method, we the used protein-coding genes of Drosophila melanogaster (dmel_r6.06) and the previously published genome of TSSM (Accession: GCF_000239435.1)7. Another homology-based method implemented in GeMoMa35 and transcript-based gene predictions utilized in the PASA pipeline v2.1.08734 were performed. Gene models from the three main sources were merged to produce consensus models by EvidenceModeler36. Finally, we identified 15,604 protein-coding genes, 11,232 of which were identical (>95%) to 10,725 protein sequences of the previous version15. Functions of the protein-coding genes were annotated using EggNOG-Mapper v2.1.737 against the database EggNOG v5.0.238, NR39, Swiss-Prot40, GO41, KEGG42, COG43 and PFAM44. In total, 11,435 genes could be functionally annotated. The gene count, Guanine-Cytosine(GC) content, and repeat sequence content were calculated in 100Kbp non-overlapping sliding windows using Bedtools v2.3045 and displayed in a Circos plot by TBtools v2.09346 (Fig. 2a).

Circos plot of GC content, gene count, and repeat content of Tetranychus urticae genome.
Data Records
Illumina short-reads, Nanopore, Hi-C raw reads for T. urticae genome sequencing and Illumina transcriptome data can be accessed in the NCBI Sequence Read Archive under project accession number PRJNA78838547, with accession numbers SRR2800046548, SRR2800045748, SRR2800006648 and SRR28000928- SRR2800093048, respectively. The finally assembled genome has been deposited in the NCBI with an accession number of JALDPR010000001-JALDPR01000005149. The genome assembly and annotation files are available in Figshare (https://doi.org/10.6084/m9.figshare.25241794)50.
Technical Validation
Completeness of the genome assembly was up to 94.8% (90.8% single-copied genes, 4.0% duplicated genes, 0.5% fragmented, and 4.7% missing genes) as assessed using BUSCO v3.0.251 with the ‘arachnida_odb10’ database (n = 2934), similar to the previously assembled pseudochromosome-level genome (95.0% completeness with 90.8% single-copied genes, 4.2% duplicated genes, 0.3% fragmented, and 4.7% missing genes). The completeness for annotated gene set was 93.4% (87.1 single-copied genes, 6.3% duplicated genes, 0.7% fragmented, and 5.9% missing genes). Synteny between our assembly and a previously published assembly (the London strain, Assembly accession: GCF_000239435.1) as well as the chromosome-level reassembly7,15 was analyzed using MCSCAN52. This genome showed high synteny to a previously assembled scaffold-level and pseudochromosome-level genome (Fig. 1e). As noted by previous studies15,27, errors on scaffolds 1, 2, 4, and 8 of the Sanger assembly were resolved by our new assembly.
Code availability
No custom scripts or code were used in this study.
References
Gerson, U. & Weintraub, P. G. Mites (Acari) as a factor in greenhouse management. Annu. Rev. Entomol. 57, 229–247, https://doi.org/10.1146/annurev-ento-120710-100639 (2012).
Reichert, M. B., Schneider, J. R., Wurlitzer, W. B. & Ferla, N. J. Impacts of cultivar and management practices on the diversity and population dynamics of mites in soybean crops. Exp. Appl. Acarol. 92, 41–59, https://doi.org/10.1007/s10493-023-00862-8 (2024).
Mérida-Torres, N. M., Cruz-López, L., Malo, E. A. & Cruz-Esteban, S. Attraction of the two-spotted spider mite, Tetranychus urticae (Acari: Tetranychidae), to healthy and damaged strawberry plants mediated by volatile cues. Exp. Appl. Acarol. 91, 413–427, https://doi.org/10.1007/s10493-023-00852-w (2023).
Gong, Y.-J. et al. Efficacy of carbon dioxide treatments for the control of the two-spotted spider mite, Tetranychus urticae, and treatment impact on plant seedlings. Exp. Appl. Acarol. 75, 143–153, https://doi.org/10.1007/s10493-018-0251-1 (2018).
Tanaka, M., Yase, J., Kanto, T. & Osakabe, M. Combined nighttime ultraviolet B irradiation and phytoseiid mite application provide optimal control of the spider mite Tetranychus urticae on greenhouse strawberry plants. Pest Manage. Sci. 80, 698–707, https://doi.org/10.1002/ps.7798 (2024).
Bajda, S. A., Wybouw, N., Nguyễn, V. H., Clercq, P. D. & Leeuwen, T. V. Adaptation of an arthropod predator to a challenging environment is associated with a loss of a genome‐wide plastic transcriptional response. Pest Manage. Sci., https://doi.org/10.1002/ps.7936 (2024).
Grbic, M. et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature 479, 487–492, https://doi.org/10.1038/nature10640 (2011).
Sparks, T. C. & Nauen, R. IRAC: Mode of action classification and insecticide resistance management. Pestic. Biochem. Physiol. 121, 122–128, https://doi.org/10.1016/j.pestbp.2014.11.014 (2015).
Vandenhole, M. et al. Contrasting roles of cytochrome P450s in amitraz and chlorfenapyr resistance in the crop pest Tetranychus urticae. Insect Biochem. Mol. Biol. 164, 104039, https://doi.org/10.1016/j.ibmb.2023.104039 (2024).
De Rouck, S., İnak, E., Dermauw, W. & Van Leeuwen, T. A review of the molecular mechanisms of acaricide resistance in mites and ticks. Insect Biochem. Mol. Biol. 159, 103981, https://doi.org/10.1016/j.ibmb.2023.103981 (2023).
Fotoukkiaii, S. M. et al. High-resolution genetic mapping reveals cis-regulatory and copy number variation in loci associated with cytochrome P450-mediated detoxification in a generalist arthropod pest. PLoS Genet. 17, e1009422, https://doi.org/10.1371/journal.pgen.1009422 (2021).
Rouck, S. D., Mocchetti, A., Dermauw, W. & Leeuwen, T. V. SYNCAS: Efficient CRISPR/Cas9 gene-editing in difficult to transform arthropods. Insect Biochem. Mol. Biol. 165, 104068, https://doi.org/10.1016/j.ibmb.2023.104068 (2024).
Shi, P. et al. Independently evolved and gene flow-accelerated pesticide resistance in two-spotted spider mites. Ecol. Evol. 9, 2206–2219, https://doi.org/10.1002/ece3.4916 (2019).
Bruinsma, K. et al. Host adaptation and specialization in Tetranychidae mites. Plant Physiol. 193, 2605–2621, https://doi.org/10.1093/plphys/kiad412 (2023).
Wybouw, N. et al. Long-term population studies uncover the genome structure and genetic basis of xenobiotic and host plant adaptation in the herbivore Tetranychus urticae. Genetics 211, 1409–1427, https://doi.org/10.1534/genetics.118.301803 (2019).
Helle, W. & Bolland, H. R. Karyotypes and sex-determination in spider mites (Tetranychidae). Genetica 38, 43–53, https://doi.org/10.1007/BF01507446 (1967).
Grbic, M. et al. Mity model: Tetranychus urticae, a candidate for chelicerate model organism. Bioessays 29, 489–496, https://doi.org/10.1002/bies.20564 (2007).
Ji, M. et al. A nuclear receptor HR96-related gene underlies large trans-driven differences in detoxification gene expression in a generalist herbivore. Nat. Commun. 14, 4990, https://doi.org/10.1038/s41467-023-40778-w (2023).
Sugimoto, N. et al. QTL mapping using microsatellite linkage reveals target-site mutations associated with high levels of resistance against three mitochondrial complex II inhibitors in Tetranychus urticae. Insect Biochem. Mol. Biol. 123, 103410, https://doi.org/10.1016/j.ibmb.2020.103410 (2020).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Hu, J. et al. An efficient error correction and accurate assembly tool for noisy long reads. bioRxiv, 2023.2003.2009.531669 https://doi.org/10.1101/2023.03.09.531669 (2023).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19, 460, https://doi.org/10.1186/s12859-018-2485-7 (2018).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963, https://doi.org/10.1371/journal.pone.0112963 (2014).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Bryon, A. et al. Disruption of a horizontally transferred phytoene desaturase abolishes carotenoid accumulation and diapause in Tetranychus urticae. Proc. Natl. Acad. Sci. 114, E5871–E5880, https://doi.org/10.1073/pnas.1706865114 (2017).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinformatics 25, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12, 491, https://doi.org/10.1186/1471-2105-12-491 (2011).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19(Suppl 2), ii215–225, https://doi.org/10.1093/bioinformatics/btg1080 (2003).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Keilwagen, J., Hartung, F. & Grau, J. in Gene prediction: Methods and protocols Vol. 1962 Methods in Molecular Biology (ed M. Kollmar) 161-177 (2019).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122, https://doi.org/10.1093/molbev/msx148 (2017).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314, https://doi.org/10.1093/nar/gky1085 (2019).
Deng, Y. Y. et al. Integrated nr database in protein annotation system and its localization. Computer Engineering 32, 71–72, https://doi.org/10.3969/j.issn.1000-3428.2006.05.026 (2006).
Consortium, T. U. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531, https://doi.org/10.1093/nar/gkac1052 (2022).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29, https://doi.org/10.1038/75556 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–462, https://doi.org/10.1093/nar/gkv1070 (2016).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36, https://doi.org/10.1093/nar/28.1.33 (2000).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–230, https://doi.org/10.1093/nar/gkt1223 (2014).
Quinlan, A. R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinformatics 47, 11.12.11–34, https://doi.org/10.1002/0471250953.bi1112s47 (2014).
Chen, C. et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 16, 1733–1742, https://doi.org/10.1016/j.molp.2023.09.010 (2023).
NCBI BioProject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA788385 (2021).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP490166 (2024).
Genbank https://identifiers.org/ncbi/insdc.gca:GCA_036877765.1 (2024).
Wei, S.-J. & Cao, L.-J. Chromosome-level genome and annotation of the two-spotted spider mite Tetranychus urticae. figshare. https://doi.org/10.6084/m9.figshare.25241794.v3 (2024).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49, https://doi.org/10.1093/nar/gkr1293 (2012).
Acknowledgements
This work was supported by National Key R&D Program of China (2023YFD1401200), Key Laboratory of Urban Agriculture (North China, Ministry of Agriculture and Rural Affairs of the People’s Republic of China), Key Laboratory of Environment Friendly Management on Fruit and Vegetable Pests in North China (Co-construction by Ministry of Agriculture and Rural Affairs of the People’s Republic of China and Province), and Program of Beijing Academy of Agriculture and Forestry Sciences (JKZX202208).
Author information
Authors and Affiliations
Contributions
S.J.W. designed the study. J.C.C. contributed to the materials. L.J.C., T.B.G., and F.Y. analyzed the data. L.J.C., and F.Y.Y. wrote the manuscript. S.J.W., J.X.L., and F.L.J. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, LJ., Guan, TB., Chen, JC. et al. Chromosome-level genome assembly of the two-spotted spider mite Tetranychus urticae. Sci Data 11, 798 (2024). https://doi.org/10.1038/s41597-024-03640-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03640-2
- Springer Nature Limited