
Abstract
Understanding the transcriptional regulatory characteristics throughout the embryogenesis of plant-parasitic nematodes is crucial for elucidating their developmental processes’ uniqueness. However, a challenge arises due to the lack of suitable technical methods for synchronizing the age of plant-parasitic nematodes embryo, it is difficult to collect detailed transcriptome data at each stage of embryonic development. Here, we recorded the 11 embryonic developmental time-points of endophytic nematode Meloidogyne incognita (isolated from Wuhan, China), Heterodera glycines (isolated from Wuhan, China), and Ditylenchus destructor (isolated from Jinan, China) species, and constructed transcriptome datasets of single embryos of these three species utilizing low-input smart-seq2 technology. The datasets encompassed 11 complete embryonic development stages, including Zygote, 2-cell, 4-cell, 8-cell, 24–44 cell, 64–78 cell, Comma, 1.5-fold, 2-fold, Moving, and L1, each stage generated four to five replicates, resulting in a total of 162 high-resolution transcriptome libraries. This high-resolution cross-species dataset serves as a crucial resource for comprehending the embryonic developmental properties of plant-parasitic nematodes and for identifying functional regulatory genes during embryogenesis.
Similar content being viewed by others

Background & Summary
Plant-parasitic nematodes are widely distributed in the ecosystem and have the ability to parasitize various tissues of thousands of plants, causing serious plant diseases and global economic agricultural crop losses1,2. The life history of plant-parasitic nematodes is completed within the infected host, where a zygotic cell undergoes a complex series of developmental processes to form a multicellular, functional embryo3,4. Crucial events in embryogenesis include reproductive system formation, digestive system determination, stylet formation and neuronal development5. These events involve rapid changes and complex regulatory mechanisms in molecular and cellular processes that are essential for the nematode’s parasitism6.
Previous research has established distinct variations in early embryonic cleavage patterns across nematode species, highlighting diverse and specific embryonic developmental characteristics that correlate with their phylogenetic positions7,8. Notable examples include the plasticity in embryogenesis pattern formation observed in Acrobeloides nanus9 and the postembryonic mouth dimorphism seen in Pristionchus pacificus10. Particularly striking peculiarities are observed within the Enoplea group, since members of clade 1B,such as Enoplus brevis11 and Pontonema vulgaris12, lack asymmetric cleavages during initial blastomere development, where a gut lineage was evident. Another example is Tobrilus diversipapillatus13 which displays a unique developmental feature of a prominent coeloblastula, a trait normally thought to be absent in nematodes. This diversity in embryonic development is further reflected in the considerable variability in the time taken by each species to reach equivalent developmental stages. For example, H. glycines embryos progress to the J2 stage in approximately six days14, while free-living C. elegans requires approximately three days to develop from zygote eggs into adults15. In contrast, M. incognita show a longer development time of up to 20 days16. The regulation of embryonic development time is species specific and is regulated by the complex interplay of genes involved in embryonic development. Despite their similar morphology and close genetic relationships, many nematode species exhibit distinct genome and developmental characteristics due to developmental drift or selection17. Comprehensive comparative analysis is essential to elucidate the differences between different nematode species.
Comparative transcriptome analysis across species during embryonic development is a valuable approach for elucidating gene expression patterns and understand the evolution of species with interspecific and intraspecific phenotypic variation18,19. For example, developmental disparities between C. elegans and C. briggase were analyzed by comparing the synchronized transition from embryo to adult stage and assessing the mRNA and protein levels changes throughout their life stages20. Another study has explored the relationship between embryonic development morphology and gene expression in five free-living nematode species, revealing both similarities and differences in gene expression that underline morphological differences of different species21. Moreover, more comprehensive studies spanning multiple nematode species have shed light on the evolutionary aspects of life cycle post-embryonic development. By comparing the developmental transcriptomes of eight nematode species at three developmental stages (embryo, larva and adult), analyzed the evolution of the life cycle after embryonic development22. All these studies focused on free-living nematodes species, while the current basic biology research on plant-parasitic nematodes is mainly focused on the parasitic stage, and the embryonic development is more about the description of morphology and development time, lacking data resources on its fine development stage. Thus, analyzing the commonality and specificity of gene expression at different stages of embryonic development across species can provide insight into the evolutionary relationships and courses among different nematode species. Additionally, understanding the embryonic transcriptome information can deepen our knowledge of various biological processes such as cell behavior, signal transduction and gene regulation within organisms, which holds value for understanding the adaptive evolution strategy of nematode defense against parasitic parasites23. To achieve this, comprehensive sampling and systematic study of the embryonic development process of different species are required to enhance our understanding of the embryogenesis behavior of plant-parasitic nematodes.
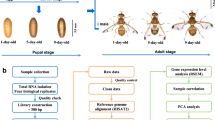
Here, the migratory endophytic potato tuber nematode (Ditylenchus destructor), the sedentary endophytic southern root-knot nematode (Meloidogyne incognita), and the sedentary endophytic soybean cyst nematode (Heterodera glycines) located on Clade 12 were selected (Fig. 1a). All of them are top-10 plant-parasitic nematodes known for causing significant damage to economic crops1. With well-establish laboratory culture, these nematodes can be easily isolation and purification using shallow plant method and sucrose gradient techniques24,25. Moreover, their high-quality reference genomes, comprehensive multi-omics functional gene annotations, and extensive basic research make them ideal candidates for dataset utilization and species development analysis. We utilized smart-seq226 (Fig. 2) protocol to conduct single-embryo transcriptomics in three plant-parasitic nematodes. This approach effectively overcame technical challenges related to eggs age synchronization in nematode culture and conducting low-input single egg RNA sequencing. A total of 162 individual embryo libraries across 11 developmental stages were constructed to fill the existing data gap in understanding the fine embryonic developmental stages of plant-parasitic nematodes. These datasets substantially enrich the omics resources available by providing insights into the global gene expression dynamics at each embryonic developmental stage of the three plant-parasitic nematodes, and aim to elucidate the features of cross-species embryonic development.

The embryogenesis characteristics of three plant-parasitic nematodes. (a) phylogenetic relationships between plant-parasitic nematodes and free-living nematodes species, (b) morphological characteristics of embryonic development during zygote to L1 stage.

The workflow of single-embryo RNA sequencing based on the smart-seq2 protocol. (a) Schematic flow chart of Smart-seq2 library construction, (b) the experimental principle of each step.
Methods
Experimental design
In the large-scale investigation of nematode populations, the embryonic development of different nematodes was observed to exhibit similar gene expression patterns, primarily dictated by morphological characteristics rather than development time27,28. This led to the understanding that different nematode species share commonalities in embryonic development, as indicated by the classical description of embryonic development in nematodes15. Consequently, we continuously recorded of the embryonic development phenotypes of D. destructor, M. incognita and H. glycines from zygote stage to L1 larvae, namely Zygote, 2-cell, 4-cell, 8-cell, 24–44 cell, 64–78 cell, Comma, 1.5-fold, 2-fold, Moving, and L1 (Fig. 1b). The information gathered from these developmental stages can be well match with data from other nematode species for further comparative analyses.
Nematode culture, collection and single embryo isolation
Nematode of the M. incognita strain, WHF4-1, were obtained from tobacco roots that had been infected for 35 days. The process of nematode isolation involved, firstly, cleaning and cutting the roots into pieces, followed by the release of the eggs through incubation in 10% sodium hypochlorite for 8 minutes. Subsequently, the mixture was transferred to a 500 μm filter and washed with tap water, followed by washing in a 0.01% Triton X-100 solution. Finally, the purified eggs were collected through centrifugation with 35% sucrose. Similarly, H. glycines (strain: Hg5#) nematodes were isolated from infected soybean roots after 35 days. The mature cysts were collected and mechanically disrupted to release the eggs, which were then sterilized with sodium hypochlorite and centrifuged with 35% sucrose to obtain purified eggs for further examination. D. destructor (strain: Dd1115) nematodes were isolated from sweet potato tubers after 28 days of infection. The process involved washing the infected potato pieces with sterile water, drying, and cutting them into a shallow dish. A screen of three layers of gauze was used to filter the sweet potato residue, and water was added until the residue was just submerged. The mixture was left at room temperature for 24 hours before collecting and crushing the lumps of sweet potatoes. The purified eggs were then obtained through centrifugation with a 35% sucrose solution.
Single-embryo isolation and library preparation
Initially, single embryos were transferred into pre-labelled PCR microtubes containing lysis buffer (18 μl 0.3% Triton-X100 + 2 μl RNase inhibitor) measuring 1.5 μl in volume. The low-input mRNA from each of the individual embryos was then subjected to reverse transcription into cDNA using a sensitive highly-multiplexed single-cell RNA-seq (Smart-seq2) protocol. The embryos samples were then carefully cut under a microscope with a scalpel and subjected to rapid centrifugation to initiate the cleavage reaction procedure at 65°C for 10 minutes, followed by an additional incubation at 85°C for 1 minute. Subsequently, the reaction system for the synthesis of the template strand of cDNA was configured, by adding 1 μl of oligo-(dT)30 primer and 1 μl of dNTPs mix to the reaction test tube, followed by incubation at 72°C for 30 seconds to facilitate the specific binding of the PolyA tail. Upon completion of the reaction, the test tube was promptly placed on ice to proceed to the next step. Finally, the synthesis of the first-strand and second-strand reverse transcribed and PCR amplified was carried out in accordance with the Smart-seq2 protocol. The cDNA amplification products were subsequently purified using VAHTS DNA clean beads (Vazyme, CHINA). Following this, 25 μl of beads was added to the reaction system and thoroughly mixed to initiate the purification process, and the mixture was incubated for 8 minutes at room temperature to allow the binding of cDNA to the magnetic beads. With the aid of a magnetic rack, the magnetic beads were separated from the liquid, and the supernatant was carefully removed to clarify the solution. Throughout the process, the PCR tube was kept on the magnetic holder. Subsequently, 200 µl of freshly prepared 80% ethanol was added, and the mixture was incubated for 30 seconds at room temperature. After careful removal, the rinse was repeated. The cleaned-up cDNA was then eluted with 17 µl of Elution Buffer supernatant, resulting in the recovery of a total of 15 µl sample, which was subsequently transferred to a new low-adsorption EP tube and stored at −20 °C.
The library construction process began with a 20 µl reaction, comprising 4 µl of 5 × TTBL, 5 µl of TTE Mix V5, and 5 ng of cDNA (TruePrep DNA Library Prep Kit V2 for Illumina #TD502, Vazyme, CHINA), followed by an incubation at 55°C for 10 minutes. Afterward, 5 µl of 5 × TS solution was added to halt the fragmentation reaction. The addition of index sequences to both ends of the fragmented sequence was carried out using the TruePrep Index Kit V2 for Illumina #TD202, Vazyme, CHINA, with a 10 µl 5 × TAB, 5 µl N5 × × , 5 µl N7 × × , 1 µl TAE, and a 20 µl reaction volume. Following this, the libraries underwent length sorting (300–700 bp) with DNA clean beads, resulting in a total of 20 mL sample recovery. Subsequently, paired-end sequencing with 150 bp read length was performed using the Illumina NovaSeq 6000 platform.
Sequencing data saturation test
In order to assess sequencing saturation, samples with the minimum amount of sequencing data volume for each species were selected. The sequencing saturation test involved the examination of samples Dd3_4 (D. destructor), Hg1_3 (H. glycines) and Mi5_2 (M. incognita). Using the seqkit29 software (v0.16.1), varying numbers of reads ((10, 50, 100, 200, 300, 500, 1000, 1500, 3000, 5000, (×1000)) were randomly selected for this analysis. The calculation results are shown in Fig. 3. The results indicate that using 3 million reads can achieve saturation and meet satisfy the analysis requirements.

Sequencing data saturation analysis. The X-axis represents the number of randomly selected sampled reads, while the Y-axis shows the number of detected genes.
Gene expression analysis
The reference genome for M. incognita WHF4-1 could be found in our previous work30, and also could be downloaded at ncbi https://www.ncbi.nlm.nih.gov/biosample/33562119. These datasets are applicable to other publicly reference genome versions as well on the wormbase website. Upon obtaining these genome files, the sequenced reads underwent filtering and trimming using fastp31 (v0.20.1). Subsequently, the filtered reads were annotated to their respective species with the specified options (hisat2 -p -x -1 -2 -S –summary-file) using HISAT232 (v2.2.1). Following this preprocessing step, read counts were generated using HTseq33. (v0.6.1) and gene expression levels were calculated as transcripts per kilobase million (TPM) using the RSEM34 (v1.2.28) command. To enable RSEM to perform these calculations, transcriptome indexes required were prepared using the command rsem-prepare-reference rsem-calculate-expression, with the following options (rsem-calculate-expression –paired-end -no-bam-output –alignments -p). To identify the expressed genes, those with TPM > 1 from at least one time point were selected.
General statistics and plots
All statistical analyses and plots were conducted in R, using the following packages: ggplot2 (v3.4.0), reshape235 (v.1.4.4), plyr36 (v1.8.8), and pheatmap (v1.0.12), unless stated otherwise.
Data Records
The raw data supporting the study findings for 162 single embryo libraries have been deposited in NCBI with the SRA accession: SRP44723837. Additional processed gene count data have been deposited on the GEO: GSE26803838. For further inquiry into specific developmental stage data, refer to the accession numbers provided in the Tables 1–3.
Technical Validation
The transcriptome datasets outlined in this study were optimized using established single nematode sequencing technology. In the preliminary experiment, we encountered challenges in achieving a clean cutting operation on small individual nematode eggs, attributed to their smooth egg shells. Consequently, we conducted repeated sampling of 10–20 individual samples in each period and subsequently filtered down to 4 to 5 replicates for further analysis. All input cDNA concentrations were rigorously determined using Qubit® 3.0 Fluorometer, followed by library construction with a cDNA starting amount of 5 ng using Tn5 transposase. Subsequent to quality control, involving the removeal of adapters, it was found that 50% to 90% of the reads in each library could be successfully mapped to the reference genome (Tables 1–3). To assess the repeatability of the data, we computed the TPM value of each gene in every sample and performed clustering through correlation coefficient, the analysis results showed a high correlation between duplicate samples and adjacent development stages, especially evident in the cell stages of the three species (Fig. 4). Notably, we observed significant fluctuations in different samples during the gastrula development period, indicating that the embryo development underwent pronounced fluctuations, indicative of a transitional process between distinct developmental stages, as depicted in Fig. 4. In order to assess the validity of the data, the number of genes expressed at each developmental stage in different species was quantified, offering a preliminary insight into gene dynamic expression as a valuable data resource (Fig. 5). The processed gene count data file has been uploaded to the GEO database, and the resulting information has been tabulated (Tables 1–3). After quality control and alignment verification, the datasets successfully achieved the capture of gene expression dynamics, providing a feasible basis for later multi-version genome alignment.

Test of correlation of data between each sample replicate about three species. The analysis included filtered gene sets only if their TPM ≥ 1. The quality of the data was assessed using replicates from the same stage for biological replicate comparisons, revealing a high correlation in expression values between biological replicates of each species. (a–c) plant-parasitic nematodes generally showed a high correlation between the early and middle stages of embryonic development, samples from the zygote, 2-cell, 4-cell, 8-cell and 24–44 cell stages clustered closely, indicating that early developmental transcriptional profiles are similar.

Statistical analysis of gene expression in three plant parasitic nematodes at different stages of embryo development. The X-axis represents the embryonic development stages, while the Y-axis shows the number of expressed genes. Genes that had an average TPM ≥ 1 across all biological replicates for at least one stage in each developmental phase were considered expressed genes, enabling an examination of overall gene expression in each stage.
Code availability
No custom software code was developed for this study. All bioinformatics tools and pipelines are Follow the manuals and protocols provided by the respective software developers. The software and version have been thoroughly described in the Methods section.
References
Jones, J. T. et al. Top 10 plant-parasitic nematodes in molecular plant pathology. Mol Plant Pathol 14, 946–961 (2013).
Nicol, J. M. et al. in Genomics and Molecular Genetics of Plant-Nematode Interactions. (Springer natyre press, 2011).
Abdel-Rahman, F. & Maggenti, A. R. Embryonic and Postembryonic Development of Meloidogyne californiensis Abdel-Rahman &Maggenti, 1987. J Nematol 19, 505–508 (1987).
Sulston, J. E. & Horvitz, H. R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol 56, 110–156 (1977).
Siddique, S. & Grundler, F. M. Parasitic nematodes manipulate plant development to establish feeding sites. Curr Opin Microbiol 46, 102–108 (2018).
Marcello, M. R. & Singson, A. Fertilization and the oocyte-to-embryo transition in C. elegans. BMB Rep 43, 389–399 (2010).
Dolinski, C., Baldwin, J. G. & Thomas, W. K. Comparative survey of early embryogenesis of Secernentea (Nematoda), with phylogenetic implications. Canadian Journal of Zoology 79, 82–94 (2001).
Haag, E. S., Fitch, D. H. A. & Delattre, M. From “the Worm” to “the Worms” and Back Again: The Evolutionary Developmental Biology of Nematodes. Genetics 210, 397–433 (2018).
Skiba, F. & Schierenberg, E. Cell lineages, developmental timing, and spatial pattern formation in embryos of free-living soil nematodes. Dev Biol 151, 597–610 (1992).
Kiontke, K. & Fitch, D. H. Phenotypic plasticity: different teeth for different feasts. Curr Biol 20, R710–712 (2010).
Malakhov, U. J. N. S. Nematodes: Structure, development, classification and phylogeny. (The university of chicago press, 1994).
Voronov, D. A. & Panchin, Y. V. J. D. Cell lineage in marine nematode Enoplus brevis. Development 125, 143–150 (1998).
Schierenberg, E. Unusual cleavage and gastrulation in a freshwater nematode: developmental and phylogenetic implications. Dev Genes Evol 215, 103–108 (2005).
Thapa, S., Patel, J. A., Reuter-Carlson, U. & Schroeder, N. E. Embryogenesis in the parasitic nematode Heterodera glycines is independent of host-derived hatching stimulation. BMC Dev Biol 17, 2 (2017).
Sulston, J. E., Schierenberg, E., White, J. G. & Thomson, J. N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol 100, 64–119 (1983).
Calderon-Urrea, A. et al. Early development of the root-knot nematode Meloidogyne incognita. BMC Dev Biol 16, 10 (2016).
Kalinka, A. T. et al. Gene expression divergence recapitulates the developmental hourglass model. Nature 468, 811–814 (2010).
Irie, N. & Kuratani, S. Comparative transcriptome analysis reveals vertebrate phylotypic period during organogenesis. Nat Commun 2, 248 (2011).
Gerstein, M. B. et al. Comparative analysis of the transcriptome across distant species. Nature 512, 445–448 (2014).
Grun, D. et al. Conservation of mRNA and protein expression during development of C. elegans. Cell Rep 6, 565–577 (2014).
Levin, M., Hashimshony, T., Wagner, F. & Yanai, I. Developmental milestones punctuate gene expression in the Caenorhabditis embryo. Dev Cell 22, 1101–1108 (2012).
Lu, M. R., Lai, C. K., Liao, B. Y. & Tsai, I. J. Comparative Transcriptomics across Nematode Life Cycles Reveal Gene Expression Conservation and Correlated Evolution in Adjacent Developmental Stages. Genome Biol Evol 12, 1019–1030 (2020).
Yanai, I., Peshkin, L., Jorgensen, P. & Kirschner, M. W. Mapping gene expression in two Xenopus species: evolutionary constraints and developmental flexibility. Dev Cell 20, 483–496 (2011).
Zheng, J. et al. The Ditylenchus destructor genome provides new insights into the evolution of plant parasitic nematodes. Proc Biol Sci 283 (2016).
Lauritis, J. A., Rebois, R. V. & Graney, L. S. Development of Heterodera glycines Ichinohe on Soybean, Glycine max (L.) Merr., under Gnotobiotic Conditions. J Nematol 15, 272–281 (1983).
Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10, 1096–1098 (2013).
Macchietto, M. et al. Comparative Transcriptomics of Steinernema and Caenorhabditis Single Embryos Reveals Orthologous Gene Expression Convergence during Late Embryogenesis. Genome Biol Evol 9, 2681–2696 (2017).
Zalts, H. & Yanai, I. Developmental constraints shape the evolution of the nematode mid-developmental transition. Nat Ecol Evol 1, 113 (2017).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One 11, e0163962 (2016).
Dai, D. et al. Unzipped chromosome-level genomes reveal allopolyploid nematode origin pattern as unreduced gamete hybridization. Nat Commun 14, 7156 (2023).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Wickham, H. Reshaping Data with the reshape Package. Journal of Statistical Software 21, 1–20 (2007).
Wickham, H. The Split-Apply-Combine Strategy for Data Analysis. Journal of Statistical Software 40, 1–29 (2011).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP447238 (2023).
NCBI GEO https://identifiers.org/geo/GSE268038 (2024).
Acknowledgements
We thank all members of the key laboratory for helpful discussions and suggestion of method and experiments.This work was supported by National Natural Science Foundation of China (U20A2040 and 31970076), and the National Natural Science Foundation of China (31970075).
Author information
Authors and Affiliations
Contributions
D.P., M.S. and J.Z. conceived and managed the project. X.W. performed the analysis work and wrote the paper, X.W. and Z.G. collected the samples and library construction, D.D. and C.X. were involved in the data analysis. D.D. was responsible for data sorting and uploading. Z.Z. was involved in the cultivation and collection of plant-parasitic nematodes. All authors have read and approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Guo, Z., Dai, D. et al. High-resolution transcriptome datasets during embryogenesis of plant-parasitic nematodes. Sci Data 11, 690 (2024). https://doi.org/10.1038/s41597-024-03542-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03542-3
- Springer Nature Limited