
Abstract
Cnidium monnieri, a medicinal herb of the Cnidium genus and the Apiaceae family, is among the most important traditional Chinese medicines and is widely distributed in China. However, to date, no C. monnieri-related genomic information has been described. In this study, we assembled the C. monnieri genome of approximately 1210.23 Mb with a contig N50 of 83.14 Mb. Using PacBio HiFi and Hi-C sequencing data, we successfully anchored 93.86% of the assembled sequences to 10 pseudochromosomes (2n = 20). We predicted a total of 37,460 protein-coding genes, with 97.02% of them being functionally annotated in Non-Redundant, Gene Ontology, Kyoto Encyclopedia of Genes and Genomes, and other databases. In addition, we identified 2,778 tRNAs, 4,180 rRNAs, 258 miRNAs, and 1,700 snRNAs in the genome. This is the first reported C. monnieri genome. Hopefully, the availability of this chromosome-level reference genome provides a significant basis for upcoming natural product-related biosynthetic pathway assessment in C. monnieri.
Similar content being viewed by others

Background & Summary
Cnidium monnieri, of the Apiaceae family, is among the most important traditional Chinese medicines. Commonly referred to as “She Chuang Zi”, it has been traditionally used for long in China, Korea, Vietnam, and Japan against various diseases. The first record of C. monnieri could be found in Shennong’s Classic of Materia Medica. The fruit of C. monnieri contains various active ingredients, including volatile oils, coumarins, chromones, glycosides, or terpenoids1, and it retains diverse (e.g., anti-osteoporotic, anti-adipogenic, and anti-fungal) properties2,3,4,5. However, inconsistent C. monnieri quality represents a persistent problem, limiting its widespread application and raising medication safety concerns. Phytochemical analysis revealed significant variations across different geographical regions in the type and content of coumarins6,7, the primary chemical constituents of C. monnieri. These variations could potentially result from a combination of external environmental factors and internal gene regulation. Due to genomic information scarcity, our understanding of the coumarin synthesis mechanism in C. monnieri remains incomplete, hindering our ability to effectively address the underlying causes contributing to significant variations across different geographical regions in the type and content of coumarins.
In this study, we used high-fidelity (HiFi) reads and high-throughput chromosome conformation capture (Hi-C) sequencing technologies to assemble the C. monnieri chromosome-level genome. We revealed a final genome size of 1,210.23 Mb with a scaffold N50 length of 83.14 Mb and successfully anchored 93.86% of the assembled genome sequences to 10 chromosomes (Fig. 1). We identified a total of 36,344 protein-coding genes, all of which were functionally annotated. Technological advancements contributed to the successful completion of several genome sequencing projects, such as those targeting Agastache rugosa8, Hibiscus syriacus9, and Rhododendron vialii10. However, to date, no C. monnieri-related genome information has been made available. To the best of our knowledge, this study describes first the C. monnieri genome. We are convinced that this study will provide significant resources for investigating the biosynthetic mechanisms of this species.

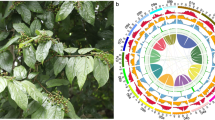
Genome characteristics of C. monnieri. Circos plot from the outer to the inner layers represents the following: (i) Pseudo-chromosomes (Chr1-Chr10); (ii) Gene density; (iii) The density of repeat sequences; (iv) GC content; (v) Each linking line in the center of the circus plot indicates a pair of homologous genes.
Methods
Sample collection, library construction, and sequencing
We extracted high-quality genomic DNA from the aerial parts of C. monnieri using a modified cetyltrimethylammonium bromide (CTAB) method11 and samples collected from Fengtai County (32°33′N, 116°21′E), Anhui Province, China. We used a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), a Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA), and 0.8% agarose gel electrophoresis to determine the concentration and quality of the extracted DNA samples.
We used the aerial parts of C. monnieri to construct a library for Hi-C analyses as described previously12. Briefly, we cross-linked the fresh tissue samples using 3% formaldehyde under vacuum infiltration at 4 °C for 30 min and quenched the cross-linking reaction with a final concentration of 0.375 Mb glycine for 5 min. Next, we lysed the cross-linked samples. We inactivated the endogenous nucleases with 0.3% SDS, then digested the chromatin DNA with 100 U of MboI (NEB), marked it with biotin-14-dCTP (Invitrogen), and ligated it using 50 U of T4 DNA ligase (NEB). After reversing the cross-links, we extracted the ligated DNA using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s instructions. We sheared the purified DNA into 300–500-bp fragments and further blunt-end repaired and A-tailed them, followed by adaptor supplementation, purification through biotin-streptavidin-mediated pull-down, and PCR amplification. Finally, we quantified and sequenced the Hi-C libraries using the MGI-seq platform (BGI, China), generating 850,286,793 raw reads and 255.09 Gb of raw bases. We trimmed the raw data using Trimmomatic with default parameters to truncate sequencing junctions and low-quality fragments. Basic statistics on data quality after trimming using FastQC with default parameters show that 829,780,252 clean reads are generated, along with 247.76 Gb of clean bases, and a clean rate of 97.59%.
RNA sequencing and analysis
We collected the stem, leaf, flower, and seed tissues of C. monnieri plants for RNA extraction. We extracted total RNA from the samples using TRIzol Reagent (Invitrogen, CA, USA) and verified RNA purity and integrity using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). We assessed RNA contamination using 1.5% agarose gel electrophoresis. We used the BGISEQ sequencing platform to obtain RNA sequencing information for C. monnieri. We trimmed raw reads using SOAPnuke (v2.1.0)13, aligned clean reads to the reference genome using HISAT2 (v2.2.1)14 with default parameters and retained only uniquely mapped reads. We estimated the expression values using RSEM (v1.3.3)15 as fragments per kilobase of the exon model per million reads mapped (FPKM). We considered genes with FPKM > 0 expressed and used them for further analysis. We used DESeq2 (v1.22.2)16 to identify differentially expressed genes (DEGs) using FDR < 0.05 and (log2FC > 1 || log2FC < −1).
Genome assembly
We obtained raw reads using DNBSEQ. We subjected the T7 platform to quality control using FastQC and Trimmomatic to filter out adapter sequences and low-quality reads. The initial assembly of the contig genome using Hifiasm (v0.19.5)17,18 resulted in a total length of 1658.64 Mb with a Contig N50 of 78.45 Mb. After using Hi-C scaffolding, the final genome assembly yielded a total length of 1210.23 Mb with a Contig N50 of 83.14 Mb (Table 1).
We obtained the draft genome by assembling HiFi reads. We used Juicer (v1.6) to align the Hi-C reads to the draft assembly, subjected to quality control. We used 3D-DNA (v180922) to anchor primary contigs to the chromosomes. The final C. monnieri genome assembly was 1210.23 Mb with a scaffold N50 of 102.78 Mb. The Hi-C analyses scaffolded 10 pseudomolecules (Fig. 2), anchoring 93.86% of the C. monnieri genome assembly. The average C. monnieri genome assembly GC content was 34.93% (Fig. 1).

Whole genome Hi-C interaction heatmap (with a resolution of 250 kb) of C. monnieri. The strength of the interaction was represented by the color from yellow (low) to red (high).
We evaluated the genome completeness using BUSCO (v5.2.2)19. Our analysis identified 98.6% (single-copy and duplicated genes: 95.1% and 3.5%, respectively), 0.2%, and 1.2% of the 1,367 predicted genes in this genome as complete, fragmented, and missing sequences, respectively (Table 2). These results suggested a highly complete assembled genome.
Functional annotation of protein-coding genes
We inferred gene functions based on the best match of the alignments to the National Center for Biotechnology Information (NCBI) Non-Redundant, TrEMBL20, InterPro21, and Swiss-Prot20 protein databases using BLASTP (NCBI BLAST v2.11.0+)22,23 and the Kyoto Encyclopedia of Genes and Genomes database24 with an E-value threshold of 1E-5. We annotated the protein domains using PfamScan (pfamscan_version)25 and InterProScan (v5.50–84.0)26 based on the InterPro protein databases. We identified the motifs and domains within the gene models using PFAM databases27. We obtained Gene Ontology28 IDs for each gene using Blast2GO29. We functionally annotated approximately 97.02% of the predicted C. monnieri protein-coding genes with known genes, conserved domains, and Gene Ontology terms (Table 3).
Repetitive sequence annotation
To identify the repeated contents in the genome, we used two methods: homology-based and de novo prediction. In the homology-based analysis, we identified known TEs within the C. monnieri genome using RepeatMasker (v4.1.2)30 and the RepBase TE library31,32. We conducted RepeatProteinMask searches using the TE protein database as a query library. For de novo prediction, we constructed a de novo repeat library of the C. monnieri genome using RepeatModeler33 (http://www.repeatmasker.org/RepeatModeler/) and LTR-FINDER33, which could automatically execute two core de novo repeat-finding programs, RECON (v1.08)34 and RepeatScout (v1.0.5)35, to comprehensively conduct, refine, and classify the consensus models of putative interspersed repeats for the C. monnieri genome. Furthermore, we performed a de novo search for long terminal repeat (LTR) retrotransposons in the C. monnieri genome sequences using LTR_FINDER (v1.0.7)33. Moreover, we also identified tandem repeats using the Tandem Repeat Finder (TRF) package36 and non-interspersed repeat sequences, including low-complexity repeats, satellites, and simple repeats, using RepeatMasker. Finally, we merged the library files of the two methods, used a repeatmaker to identify the repeat content, and statistically analyzed the repeated sequence content predicted by the different software methods (Table 4).
Data Records
We deposited the hereby-described relevant data in the National Genomics Data Center (NGDC)37,38, Beijing Institute of Genomics, Chinese Academy of Sciences/China National Center for Bioinformation, under the BioProject accession number PRJCA022794, publicly accessible at https://ngdc.cncb.ac.cn/gwh. The BioSample accession ID is SAMC3313212. We deposited the genomic raw data in the Genome Sequence Archive (GSA) in NGDC under the accession number CRA014484, the C. monnieri genome project in the NCBI database under the BioProject accession ID of PRJNA1065623, the genome assembly at GenBank under the WGS accession ID of JBDIYB00000000039, the genomic raw sequencing data in the SRA at NCBI SRR28903605-SRR2890360640,41, and the transcriptomic raw sequencing data in the SRA at NCBI SRR27600471-SRR2760048242,43,44,45,46,47,48,49,50,51,52,53.
Technical Validation
Genome assembly quality evaluation
To examine assembly integrity and sequencing uniformity, we aligned the HiFi reads in a final assembly using minimap2 (v2.21, parameters: HiFi: -ax map-hifi; ONT: -ax map-ont)54 and BWA55. We mapped a total of 99.73% of the raw reads. The average mapping and average sequencing depth was 79.5 (Table 5). Sequencing data could also be analyzed for GC bias and sample contamination using BWA and minimap2 software (Fig. 3). Moreover, we subjected the assembled genome to BUSCO18 using OrthoDB to evaluate genome completeness. Taken together, our BUSCO analysis revealed that 98.6%, 0.2%, and 1.2% of the 425 single-copy orthologs (in the Viridiplantae_odb10 database) were complete (single-copy and duplicated genes: 95.1% and 3.5%, respectively), fragmented, and missing, respectively (Table 2).

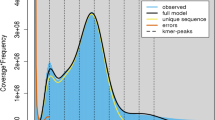
GC content and sequencing depth distribution density plot. The horizontal axis of the figure represents GC content, and the vertical axis represents sequencing depth. The contig coverage depth distribution is displayed on the right, and the GC content distribution is shown above. The large central plot is a scatter plot based on the GC content and coverage depth information of the contigs, where the color scale reflects the density of the plotted points.
To further evaluate the quality of the genome assembly, we used HiFi reads to assess the read-remapping ratio and coverage. We aligned the HiFi reads to the genome using the Juicer (v1.6) software with default parameters and the 3D-DNA (V180922) software for scaffolding. Our analysis revealed that the assembly displayed a high mapping rate and complete genome coverage of 99.73% and 100%, respectively, underscoring the high quality of the assembled genome (Table 5).
To determine prediction accuracy and reliability, we determined the distribution of gene length, CDS length, exon length, and intron length in C. monnieri and other closely related species (Apium graveolens56, Daucus carota57, Peucedanum praeruptorum58, and Notopterygium incisum59). The consistent distribution tendency among all species further supported the ideal annotated gene dataset for C. monnieri (Fig. 4).

Annotated genes comparison of the distribution of gene length, CDS length, exon length, and intron length in C. monnieri with other closely related species. The x-axis represents the length and the y-axis represents the density of genes.
Code availability
We followed the developers’ instructions for the bioinformatics tools used in this study. The software and code used are publicly accessible, with the version and parameters used specified in the Methods section. No custom code was used during the compilation of the dataset.
References
Sun, Y., Yang, A. W. H. & Lenon, G. B. Phytochemistry, ethnopharmacology, pharmacokinetics and toxicology of Cnidium monnieri (L.) Cusson. Int J Mol Sci 21 (2020).
An, J. et al. Natural products for treatment of osteoporosis: the effects and mechanisms on promoting osteoblast-mediated bone formation. Life Sci 147, 46–58 (2016).
Shin, E. et al. Inhibitory effects of coumarins from the stem barks of Fraxinus rhynchophylla on adipocyte differentiation in 3T3-L1 cells. Biol Pharm Bull 33, 1610–1614 (2010).
Wang, C. M. et al. Efficacy of osthol, a potent coumarin compound, in controlling powdery mildew caused by Sphaerotheca fuliginea. J Asian Nat Prod Res 11, 783–791 (2009).
Li, Y. M. et al. Cnidium monnieri: a review of traditional uses, phytochemical and ethnopharmacological properties. Am J Chin Med 43, 835–877 (2015).
Zhang, Y., Bai, P., Zhuang, Y. & Liu, T. Two O-methyltransferases mediate multiple methylation steps in the biosynthesis of coumarins in Cnidium monnieri. J Nat Prod 85 (2022).
Wang, P. et al. Biosynthesis of the plant coumarin osthole by engineered Saccharomyces cerevisiae. ACS Synth Biol 12, 2455–2462 (2023).
Park, H. S. et al. A chromosome-level genome assembly of Korean mint (Agastache rugosa). Sci Data 10, 792 (2023).
Koo, H. et al. Two long read-based genome assembly and annotation of polyploidy woody plants, Hibiscus syriacus L. using PacBio and Nanopore platforms. Sci Data 10, 713 (2023).
Chang, Y., Zhang, R., Ma, Y. & Sun, W. A haplotype-resolved genome assembly of Rhododendron vialii based on PacBio HiFi reads and Hi-C data. Sci Data 10, 451 (2023).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep 15, 8–15 (1997).
Padmarasu, S., Himmelbach, A., Mascher, M. & Stein, N. In situ Hi-C for plants: an improved method to detect long-range chromatin interactions. Methods Mol Biol 1933, 441–472 (2019).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6 (2018).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Driguez, P. et al. LeafGo: Leaf to Genome, a quick workflow to produce high-quality de novo plant genomes using long-read sequencing technology. Genome Biol 22, 256 (2021).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol 1962, 227–245 (2019).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 31, 365–370 (2003).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res 43, D213–221 (2015).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40, D109–114 (2012).
Mistry, J., Bateman, A. & Finn, R. D. Predicting active site residue annotations in the Pfam database. BMC Bioinformatics 8, 298 (2007).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Finn, R. D. et al. The Pfam protein families database. Nucleic Acids Res 36, D281–288 (2008).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000).
Conesa, A. & Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008, 619832 (2008).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics Chapter 4, 4.10.11–14.10.14 (2009).
Jurka, J. Repbase update: a database and an electronic journal of repetitive elements. Trends Genet 16, 418–420 (2000).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110, 462–467 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35, W265–268 (2007).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–358 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res 50, D27–d38 (2022).
Chen, M. et al. Genome Warehouse: a public repository housing genome-scale data. Genom Proteom Bioinf 19, 584–589 (2021).
Wang, Y.-J. Anisodus tanguticus isolate KB-2021, whole genome shotgun sequencing project. Genbank https://identifiers.org/ncbi/insdc:JBDIYB000000000 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28903605 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR28903606 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600471 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600472 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600473 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600474 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600475 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600476 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600477 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600478 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600479 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600480 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600481 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR27600482 (2024).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, M. Y. et al. The genome sequence of celery (Apium graveolens L.), an important leaf vegetable crop rich in apigenin in the Apiaceae family. Hortic Res 7, 9 (2020).
Iorizzo, M. et al. A high-quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat Genet 48, 657–666 (2016).
Song, C. et al. Genome sequencing-based transcriptomic analysis reveals novel genes in Peucedanum praeruptorum. BMC Genom Data 24, 53 (2023).
Li, Q. et al. The chromosome-scale assembly of the Notopterygium incisum genome provides insight into the structural diversity of coumarins. Acta Pharm Sin B 182, 107736 (2024).
Acknowledgements
This work was supported by grants from the ability establishment of sustainable use for valuable Chinese medicine resources (2060302), the open foundation of Shaanxi University of Chinese Medicine state key laboratory of R&D of Characteristic Qin Medicine Resources (SUCM-QM202202), the fund of Traditional Chinese Medicine Institute of Anhui Dabie Mountain (TCMADM-2023-18), and the open research fund of Yunnan characteristic plant extraction laboratory (YKKF2023002). It was also supported by the Fundamental Research Funds for the Central Universities (2632024TD04) and the Sci-Tech Innovation Talent System Construction Program of Shaanxi University of Chinese Medicine (2023).
Author information
Authors and Affiliations
Contributions
Yucheng Zhao and Dingqiao Xu conceived and designed the research. Huanying Tang, Shanyong Yi, Yimeng Feng and Xinyue Zhao collected and prepared the samples. Zixuan Wang and Jiaxin He wrote the manuscript. Zixuan Wang and Jiaxin He assembled the genome. Qi Qi and Kaixuan Wang modified the manuscript. All authors contributed to the article and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, Z., He, J., Qi, Q. et al. Chromosome-level genome assembly of Cnidium monnieri, a highly demanded traditional Chinese medicine. Sci Data 11, 667 (2024). https://doi.org/10.1038/s41597-024-03523-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03523-6
- Springer Nature Limited