
Abstract
The plum fruit moth Grapholita funebrana (Tortricidae, Lepidoptera) is an important pest of many wild and cultivated stone fruits and other plants in the family Rosaceae. Here, we assembled its nuclear and mitochondrial genomes using Illumina, Nanopore, and Hi-C sequencing technologies. The nuclear genome size is 570.9 Mb, with a repeat rate of 51.28%, and a BUCSO completeness of 97.7%. The karyotype for males is 2n = 56. We identified 17,979 protein-coding genes, 5,643 tRNAs, and 94 rRNAs. We also determined the mitochondrial genome of this species and annotated 13 protein-coding genes, 22 tRNAs, and 2 rRNA. These genomes provide resources to understand the genetics, ecology, and genome evolution of the tortricid moths.
Similar content being viewed by others


Background & Summary
The plum fruit moth Grapholita funebrana is an important fruit borer from the family Tortricidae of Lepidoptera1,2. Larvae of G. funebrana cause damage by boring the fruits of many wild and cultivated stone fruits and other plants in the family Rosaceae, such as apricot, cherry, peach, and plum3. This species is native to Europe and currently found in fruit-growing regions of Europe, northern Africa, and Asia4. In the orchards, G. funebrana often co-occur with other fruit borers, such as the oriental fruit moth Grapholita molesta (Busck), the codling moth Cydia pomonella, and peach fruit moth Carposina sasakii Matsumura5. While many studies have focused on the biology and management of fruit borers, research on G. funebrana is lagging behind6,7,8,9,10. In addition, moths from the family Tortricidae are ideal for unveiling the evolution of chromosome fusion11,12. While species from the order Lepidoptera often have a conserved chromosome number of n = 31, in the Tortricidae family, many species have a reduced number of chromosomes due to the fusion of chromosome pairs13,14. Recent research has found that a common ancestor of the suborders Tortricinae and Olethreutinae diverged from the ancestral lepidopteran chromosome pattern due to a fusion of sex chromosomes with autosomes15. The karyotype of tortricid moths was traditionally studied by cytogenetic methods and fluorescence in situ hybridization15. Determining the genome sequences will improve understanding of the molecular evolution of chromosomes of tortricid moths16. Currently, chromosome-level genomes have been published for the C. pomonella16, and G. molesta17, as well as many publicly available assemblies for Tortricidae in the GenBank (https://www.ncbi.nlm.nih.gov/datasets/genome/?taxon=7139).
In this study, we assembled a chromosome-level genome for the G. funebrana as well its mitochondrial genome using Oxford Nanopore Technologies (ONT) long-read sequencing, Illumina short-read sequencing, high-throughput chromatin conformation capture (Hi-C) sequencing, and RNA-sequencing (RNA-seq). We yielded a nuclear genome assembly of 570.9 Mb, with an N50 of 21 Mb. These high-quality genomes will provide invaluable resources for the study of G. funebrana and in-depth investigation of chromosome evolution on macroevolutionary and microevolutionary levels.
Methods
Material and sequencing
Apricot (Prunus armeniaca) fruits with G. funebrana larvae were collected from Yanqing, Beijing, China, and reared in the laboratory for about 30 days to obtain specimens of different developmental stages. To decrease the effect of heterozygosity, a single larva was used for long-read, short-read, and Hi-C library construction. Single larva, pupa, and adult (unknown sex) were collected for the construction of RNA-seq libraries, respectively. All samples were immediately flash-frozen in liquid nitrogen and stored at −80 °C for subsequent experiments.
Genomic DNA was extracted using the Magnetic bead method (Invitrogen, Thermo Fisher Scientific, USA), while RNA was extracted using RNAprep Pure Plus Kit (Tiangen, China), respectively. The quantity of DNA was measured using Qubit 3.0. To generate short-read data for the genome survey, an Illumina library with an insert size of 350 bp was constructed and sequenced on the Illumina NovaSeq 6000 platform. To perform de novo genome assembly, a 15~20 kb ONT library was prepared and sequenced on the ONT platform to generate long-read data. To generate the Hi-C data, tissue from a larva was fixed with paraformaldehyde and digested with restriction enzymes DnpII, generating fragments with sticky ends. These sticky ends were repaired using DNA polymerase and ligated together to form chimeric circles using DNA ligase. The ligated DNAs were then decrosslinked, purified, and sheared into 350 bp insertion size. The Hi-C sequencing library was sequenced on the Illumina NovaSeq 6000 platform to generate 150-bp paired-end reads. Paired-end libraries were constructed using the VAHTSTM mRNA-seq V2 Library Prep Kit (Vazyme, Nanjing, China) and then sequenced on the Illumina NovaSeq 6000 platform with PE reads of 150 bp for genome annotation. A total of 33.7 Gb Illumina short read, 69.7 Gb ONT long-read, 58.3 Gb Hi-C reads, and 21.9 Gb RNA-seq reads data were generated. The raw data of Illumina reads were filtered by Fastp v0.21.018 with default parameters.
Genome survey
Genome survey was performed using a k-mer based method. The k-mer coverage was counted from Illumina short reads using Jellyfish version 2.2.1019 with parameters: ‘count -m 21 -C -s 5 G’. Genome size, heterozygosity, and duplication rate were estimated using GenomeScope version 2.020. The results showed a genome size about 515 Mb, a heterozygosity rate of 1.91%, and a duplication rate of 1.21%.
Genome assembly
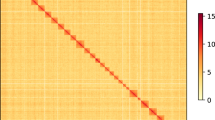
The Nanopore long reads were assembled to the primary set of nuclear genome contigs using NextDenovo v2.5.121 with parameters: ‘read_cutoff = 1k, genome_size = 400 m, pa_correction = 20, nextgraph_options = -a 1’. The contigs contain 215 sequences, with a size of 594 Mb, and N50 of 6.6 Mb. Due to the high error rate of assembly based on ONT reads, the primary contigs were polished using NextPolish 1.4.122 with one round based on long reads and one round based on short reads. To achieve chromosome-level assembly, the polished contigs were anchored into pseudomolecules based on Hi-C reads information. Specifically, the Hi-C reads were mapped to contigs using Chromap 0.2.423 with options: “–preset hic–remove-pcr-duplicates–trim-adapters–SAM”. The SAM output was sorted by read name and output to BAM format using Samtools v1.1724 with options: “sort -n -O BAM”. Yahs v1.2a.125 and Juicerbox 1.22.0126 were then used for unsupervised and supervised scaffolding, respectively. After scaffolding, most contigs (95.3% contigs and 99.86% base-pairs) were anchored into 28 pseudo-chromosomes (Fig. 1a), consistent with the karyotype of most species in the subfamily Olethreutinae. To fill the gaps between contigs, we performed two rounds of polishing based on long- and short-reads using Nextpolish. The final assembly has a genome size of 570.9 Mb, with a N50 of 21 Mb. The assembled genome is 56.9 Mb larger than the estimated genome size. MitoZ v3.6 pipeline27 was performed to assembly using Megahit v1.2928 (“–kmers_megahit 39 59 79 99 119 141–requiring_taxa Lepidoptera”) and annotate mitochondrial genome. The mitochondrial genome of G. funebrana was 15,488 bp in length and contain 13 protein coding genes, 22 tRNA genes and 2 rRNA genes (Fig. 1b).

The interaction heat map of nuclear genome (a), and distribution of genes and read coverage on mitochondrial genome (b).
Genome annotations
For repeat sequence annotation, a species-specific repeat library was generated using RepeatModeler v2.0.429 with options: “-LTRStruct”. The species-specific repeat library, a RepBase database, and a repeat element library for Arthropoda from the Dfam database were then combined and passed to RepeatMasker v4.1.430 for repeat annotation. RepeatMasker was performed with options:” -no_is -norna -xsmall -q”.
For gene structure annotation, we performed a pipeline integrating RNA-seq-based, ab initio, and homolog-based methods. The RNA reads of single larva, pupa and adult libraries were mapped to our final assembly with Hisat v2.2.027 and assembled to transcripts with Stringtie v2.1.231. The transcriptome assemblies and protein sequences of Plutella xylostella (Accession: GCA_932276165.132) were provided as evidence to MAKER v3.01.04 pipeline26 to integrate. SNAP v2013-02-1628 and Augustus v3.2.329 were used to conduct ab initio annotation. Transfer RNA (tRNA) was predicted using tRNAscanSE 2.0.1233 with default parameters, and ribosome RNA (rRNA) was predicted using Barrnap 0.9 (https://github.com/tseemann/barrnap). The above gene models were merged to produce consensus models by EvidenceModeler v2.1.033. Functional annotation of protein-coding genes was evaluated using EggNOG-mapper v234.
Chromosome feature
The gene number, repeat sequence density, and Guanine-Cytosine(GC) content were calculated in 500 Kb non-overlapping sliding windows using Bedtools v2.30.035. The name of the chromosomes was assigned as lepidopteran ancestral linkage groups14, based on homology to Sesia bembeciformis36. The homology was detected using LAST37 alignment. A Circos plot of chromosome feature was generated by TBtools v2.02138 (Fig. 2a).

Chromosome features of Grapholita funebrana genome. (a) Circos plot of GC content, gene count, and repeat content. Chromosomes were labeled using Merian elements according to the homology with the Lepidopteran ancestral linkage groups14. (b) Synteny blocks between the G. funebrana and G. molesta reveal the same number of chromosomes and highly conserved gene order in the two moths. The chromosomes of two genomes were numbered according to their length. The grey lines show the synteny blocks between two genomes.
Data Records
Illumina, Nanopore, Hi-C, and transcriptome data for G. funebrana genome sequencing have been deposited in the NCBI Sequence Read Archive with accession number SRP48223139. The final assembled nuclear genome of G. funebrana has been deposited in the NCBI Genbank with accession number GCA_038095595.140. The mitochondrial genome has been deposited in the NCBI Genbank with accession number PP77602341. The genome assembly and annotation files are available in Figshare42.
Technical Validation
The Hi-C heatmap revealed a well-structured interaction pattern. Short-read sequencing data were mapped to the final assembly with BWA v0.7.1743, revealing a mapping rate of 97.7%. The completeness of G. funebrana genome assembly was evaluated using the BUSCO44 base on the lepidoptera_odb10 database (n = 5286). The completeness of the initial assembly (contig level) was 90.9%, while it increased to 97.7% (97.2% single-copied genes, 0.5% duplicated genes, 0.6% fragmented, and 1.7% missing genes) after polishing with NextPolish22 (Table 1). We identified 14,547 protein-coding genes, 11,673 of which were functionally annotated. The completeness of the annotated gene set was 95.8% (94.8% single-copied genes and 1.0% duplicated genes, 1.1% fragmented, and 3.1% missing genes). A synteny analysis between G. funebrana and G. molesta17 was performed using MCSCAN in JCVI package45. Strong syntenic blocks were found between the two closely related species (Fig. 2b). All evidence strongly supported the completeness and accuracy of G. funebrana genome assembly.
Code availability
No custom scripts or code were used in this study.
References
Li, L.-L. et al. Functional disparity of four pheromone-binding proteins from the plum fruit moth Grapholita funebrana Treitscheke in detection of sex pheromone components. Int. J. Biol. Macromol. 225, 1267–1279 (2023).
Lo Verde, G., Guarino, S., Barone, S. & Rizzo, R. Can mating disruption be a possible route to control plum fruit moth in mediterranean environments? Insects 11, 589 (2020).
Dickler, E. Tortricid pests of pome and stone fruits, eurasian species. in Tortricids Pests, Their Biology, Natural Enemies and Control (eds. van der Geest, L. P. S. & Evenhuis, H. H.) 435–452 (Elsevier, Amsterdam, Netherlands, 1991).
F, K. A taxonomic review of the genus Grapholita and allied genera (Lepidoptera: Tortricidae) in the Palaearctic region. Ent. Scand. Suppl. 55, 110 (1999).
Chen, M. H. & Dorn, S. Reliable and efficient discrimination of four internal fruit-feeding Cydia and Grapholita species (Lepidoptera: Tortricidae) by polymerase chain reaction-restriction fragment length polymorphism. J. Econ. Entomol. 102, 2209–2216 (2009).
Ioriatti, C. et al. Toxicity of emamectin benzoate to Cydia pomonella (L.) and Cydia molesta (Busck) (Lepidoptera: Tortricidae): laboratory and field tests. Pest Manag. Sci. 65, 306–312 (2009).
Liu, J. et al. Reverse chemical ecology guides the screening for Grapholita molesta pheromone synergists. Pest Manag. Sci. 78, 643–652 (2022).
Stelinski, L. L., Il’ichev, A. L. & Gut, L. J. Efficacy and release rate of reservoir pheromone dispensers for simultaneous mating disruption of codling moth and oriental fruit moth (Lepidoptera: Tortricidae). J. Econ. Entomol. 102, 315–323 (2009).
Witzgall, P., Stelinski, L., Gut, L. & Thomson, D. Codling moth management and chemical ecology. Annu. Rev. Entomol. 53, 503–522 (2008).
Wu, Y. et al. Laboratory evaluation of the compatibility of Beauveria bassiana with the egg parasitoid Trichogramma dendrolimi (Hymenoptera: Trichogrammatidae) for joint application against the oriental fruit moth Grapholita molesta (Lepidoptera: Tortricidae). Pest Manag. Sci. 78, 3608–3619 (2022).
Nguyen, P. et al. Neo-sex chromosomes and adaptive potential in tortricid pests. Proc. Natl. Acad. Sci. 110, 6931–6936 (2013).
Sahara, K., Yoshido, A. & Traut, W. Sex chromosome evolution in moths and butterflies. Chromosome Res. 20, 83–94 (2012).
Nguyen, P. & Carabajal Paladino, L. On the neo-sex chromosomes of Lepidoptera. in Evolutionary Biology: Convergent Evolution, Evolution of Complex Traits, Concepts and Methods (ed. Pontarotti, P.) 171–185. https://doi.org/10.1007/978-3-319-41324-2_11 (Springer International Publishing, Cham, 2016).
Wright, C. J., Stevens, L., Mackintosh, A., Lawniczak, M. & Blaxter, M. Comparative genomics reveals the dynamics of chromosome evolution in Lepidoptera. Nat. Ecol. Evol. 1–14, https://doi.org/10.1038/s41559-024-02329-4 (2024).
Šíchová, J., Nguyen, P., Dalíková, M. & Marec, F. Chromosomal evolution in tortricid moths: conserved karyotypes with diverged features. PLoS ONE 8, e64520 (2013).
Wan, F. et al. A chromosome-level genome assembly of Cydia pomonella provides insights into chemical ecology and insecticide resistance. Nat. Commun. 10, 4237 (2019).
Cao, L.-J. et al. Population genomic signatures of the oriental fruit moth related to the Pleistocene climates. Commun. Biol. 5, 142 (2022).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 25, 107 (2024).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Zhang, H. et al. Fast alignment and preprocessing of chromatin profiles with Chromap. Nat. Commun. 12, 6566 (2021).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Zhou, C., McCarthy, S. A. & Durbin, R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics 39, btac808 (2023).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Meng, G., Li, Y., Yang, C. & Liu, S. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47, e63 (2019).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. 25, 4.10.1–4.10.14 (2009).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Genbank https://identifiers.org/ncbi/insdc.gca:GCA_932276165.1 (2022).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096 (2021).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Quinlan, A. R. BEDTools: The swiss-army tool for genome feature analysis. Curr. Protoc. Bioinforma. 47, 11.12.1–11.12.34 (2014).
Genbank https://identifiers.org/ncbi/insdc.gca:GCA_943735995.1 (2022).
Katoh, K. & Frith, M. C. Adding unaligned sequences into an existing alignment using MAFFT and LAST. Bioinformatics 28, 3144–3146 (2012).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202 (2020).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP482231 (2024).
Genbank https://identifiers.org/ncbi/insdc.gca:GCA_038095595.1 (2024).
Genbank http://identifiers.org/ncbi/insdc:PP776023 (2024).
Wei, S.-J. & Yang, F. Genome annotation of Grapholita funebrana. Figshare https://doi.org/10.6084/m9.figshare.24955839.v1 (2024).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Tang, H. et al. Synteny and collinearity in plant genomes. Science 320, 486–488 (2008).
Acknowledgements
This work was supported by National Natural Science Foundation of China (32272543), and Beijing Key Laboratory of Environmentally Friendly Management on Pests of North China Fruits (BZ0432).
Author information
Authors and Affiliations
Contributions
S.J.W. designed the study. J.C.C. contributed to the materials. L.J.C. and F.Y. analysed the data. F.Y. and L.J.C. wrote the manuscript. S.J.W. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, LJ., Yang, F., Chen, JC. et al. Nuclear and mitochondrial genomes of the plum fruit moth Grapholita funebrana. Sci Data 11, 692 (2024). https://doi.org/10.1038/s41597-024-03522-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03522-7
- Springer Nature Limited