
Abstract
The cottony cushion scale, Icerya purchasi, a polyphagous pest, poses a significant threat to the global citrus industry. The hermaphroditic self-fertilization observed in I. purchasi is an exceptionally rare reproductive mode among insects. In this study, we successfully assembled a chromosome-level genome sequence for I. purchasi using PacBio long-reads and the Hi-C technique, resulting in a total size of 1,103.38 Mb and a contig N50 of 12.81 Mb. The genome comprises 14,046 predicted protein-coding genes, with 462,722,633 bp occurrence of repetitive sequences. BUSCO analysis revealed a completeness score of 93.20%. The genome sequence of I. purchasi serves as a crucial resource for comprehending the reproductive modes in insects, with particular emphasis on hermaphroditic self-fertilization.
Similar content being viewed by others


Background & Summary
The cottony cushion scale, Icerya purchasi Maskell (Monophlebidae: Icerya), is a destructive pest well-known for infecting citrus and lemon plants, with a wide distribution across approximately 150 countries in America, Asia, Europe, and Oceania1. Understanding the observed diversity in the reproduction modes across different taxa has become a key focus in evolutionary biology. Scale insects (Hemiptera: Sternorrhyncha: Coccoidea), comprising 8535 species, exhibit significant variation in their reproduction modes, including paternal genome elimination, haplodiploidy, parthenogenesis, diplodiploidy, and hermaphroditism2,3. I. purchasi displays a unique reproductive mode among insects, which is rare in arthropods4,5. Hermaphrodites with a female-like appearance produce sperm and undergo self-fertilization6. Male individuals are scarce in the wild and occasionally mate with hermaphrodites, highlighting the colonization advantages of a selfing organism and the periodic reintroduction of genetic variation6. To date, there are seven chromosome-level genomes belonging to scale insects available from GenBank, two of which have been published as research papers7,8. A high-quality reference genome of I. purchasi would be invaluable for comprehending the mechanism of hermaphroditic self-fertilization.
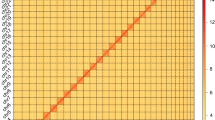
In this study, we generated 61.21 Gb of Illumina short-read sequencing, 36.4 Gb of PacBio long-read sequencing, and 120.71 Gb of High-throughput chromosome conformation capture (Hi-C) sequencing. The final genome assembly size was 1,103.38 Mb, with a contig N50 of 12.81 Mb and scaffold N50 of 601.66 Mb (Fig. 1). The total length of the genome assembly was comparable to the estimated genome size (1,097.12 Mb) based on k = 21 kmer analysis (Fig. 2a). Following Hi-C assembly and manual adjustment of the heat map, a total sequence length of 1,102.86 Mb was mapped onto two chromosomes, representing 99.95% of the genome. Within the sequences mapped to chromosomes, the length of sequences with determined orientation and direction was 1,098.95 Mb, constituting 99.64% of the total length of the mapped chromosome sequences.

The snail plot shows metrics of the I. purchasi genome including the length of the longest contig, N50, N90, base composition.

(a) Estimated genome size based on Illumina short-reads with k = 21 kmer analysis. (b) The heatmap represents two pseudochromosomes of the I. purchasi genome.
Based on the analysis of tandem repeats and interspersed repeats, we identified 462,722,633 bp of transposable element (TE) sequences, comprising 41.94% of the total, along with 35,341,500 bp of tandem repeat sequences, accounting for 3.20% (Fig. 3). Gene prediction involved homology-based prediction, ab initio prediction, and transcript-based prediction, resulting in a total of 14,046 predicted genes (Fig. 4). Further assessment through BUSCO analysis based on the insecta (odb_10) database indicated the presence of 93.20% in our predicted genes, indicating a high integrity of gene prediction. Annotation analysis of the predicted gene sequences against databases such as NR, EggNOG, GO, KEGG, SWISS-PROT, and Pfam yielded comprehensive annotation results, with a total of 89.71% of genes annotated to the databases (Table 1).
Methods
Sample collection and genome sequencing
I. purchasi was collected in 2020 from a lemon tree in Ningde, Fujian Province, China (26°34′47″N, 118°44′15″E). The laboratory colony was cultured on lemon trees for four generations in the insectarium under controlled conditions of 27 ± 1 °C and 60% relative humidity, with a photoperiod of 14:10 (L:D). Adult females and nymphs were collected for sequencing.

The circos plot describes the genomic characteristics of I. purchasi. Ecological photo is displayed in the center of the plot.

The Venn diagram showing the number of genes integrated by EVM that are supported by three prediction methods.
To perform genome sequencing, the samples were washed with 75% ethanol and sterile water, then stored in cryogenic vials with the addition of 95% ethanol. Subsequently, the total DNA from the entire body was extracted using the CATB method9 to construct three different sequencing libraries, followed by sequencing according to standard procedures. Firstly, for genome survey, we used the NEBNext® Ultra™ II DNA Library Prep Kit to build an Illumina short-read library, which was sequenced on the Illumina NovaSeq 6000 platform. Secondly, we constructed PacBio library using the SMRTbell Template Prep Kit and performed HiFi sequencing on the PacBio Sequel II platform. Finally, to generate a chromosome-level genome, we used the Mate-pair Kit to build Hi-C library, which was sequenced on the Illumina NovaSeq 6000 platform.
For transcriptome sequencing, we employed the Trizol method10 to extract RNA from fresh samples. The NEBNext® Ultra™ RNA Library Prep Kit was used to construct RNA library, which was sequenced on the Illumina NovaSeq 6000 platform for transcriptome sequencing. Additionally, the Ligation Sequencing Kit (SQK-LSK110; ONT) was used to construct library, and full-length transcriptome sequencing was completed on the Oxford Nanopore PromethION platform.
Genome assembly
K-mer analysis was conducted using Jellyfish 2.1.411 based on short-read data. Genomescope 2.012 (-k 21 -p 2 -m 100,000) was used to estimate genome size and heterozygosity. The PacBio Sequel II platform generated a total of 36.4 Gb of long reads. These reads were assembled using Hifiasm v0.1413. For chromosomal scaffolding, Hi-C data were aligned to the contig-level genome sequence using Bwa v0.7.1714. Paired reads with mate mapped to a different contig were used to do the Hi-C associated scaffolding. Then, self-ligation, non-ligation and other invalid reads were filtered out. The uniquely mapped data were retained for assembly using LACHESIS15. Any two segments which showed inconsistent connection with information from the raw scaffold were checked manually. These corrected scaffolds were divided into subgroups and sorted and oriented into pseudomolecules with LACHESIS (CLUSTER_MIN_RE_SITES = 35; CLUSTER_MAX_LINK_DENSITY = 2; ORDER_MIN_N_RES_IN_TRUNK = 98; ORDER_MIN_N_RES_IN_SHREDS = 97). After this step, placement and orientation errors featuring obvious discrete chromatin interaction patterns were manually adjusted, and the Hi-C interaction heatmap was constructed using the ggplot216 software in the R package. Finally, high quality assembly at chromosome level was obtained, and a snail plot of genomic features was generated by Blobotootlkit17.
TE annotation
Transposon element (TE) was identified by a combination of homology-based and de novo approaches. Initially, de novo prediction was conducted using RepeatModeler2 v2.0.118, which invoked RECON v1.0.819 and RepeatScout v1.0.620. The predicted results were categorized using RepeatClassifier18 with the assistance of the Dfam database v3.521. Subsequently, LTR _retriever v2.9.022 was utilized for de novo prediction specifically targeting Long Terminal Repeats (LTRs). Full-length long terminal repeat retrotransposons (fl-LTR-RTs) were identified using both LTRharvest v1.5.1023 and LTR_FINDER v1.0724 (ltr_finder -w 2 -C -D), resulting in the generation of high-quality intact fl-LTR-RTs and a non-redundant LTR library. The de novo prediction results were integrated with the known databases to construct a non-redundant species-specific TE library. Employing RepeatMasker v4.1.225 (repeatmasker -nolow -no_is -norna -engine wublast -parallel 8 -qq), homology searches were performed on the library to identify and classify TE sequences in the final chromosome genomes. Concatenated repeat sequences were annotated by MIcroSAtellite identification tool (MISA v2.1)26 and Tandem Repeat Finder v4.0927 (trf 2 7 7 80 10 50 500 -d -h).
Gene prediction and annotation
We integrated three approaches (ab initio prediction, homology search and transcript-based assembly) to annotate protein-coding genes in the genome. Ab initio prediction was conducted using Augustus v3.1.028 and SNAP (2006-07-28)29. GeMoMa v1.730 was utilized for Homology-based prediction, comparing gene features with those of cotton mealybug (P. solenopsis), the fruit fly (Drosophila melanogaster), pea aphids (Acyrthosiphon pisum), and tobacco whitefly (Bemisia tabaci). For transcript-based prediction, RNA sequencing data were mapped to the reference genome using Hisat v2.0. 431 (hisat2–dta -p 10), and assembled using Stringtie v1.2.332 (stringtie -p 2). GeneMarkS-T v5.133 was employed for gene prediction based on assembled transcripts. The PASA v2.4.134 was used to predict genes based on the unigenes (and full-length transcripts from the ONT sequencing) assembled by Trinity v2.1135 (Trinity–genome_guided_bam). Finally, predictions from these three methods were integrated using EVM v1.1.136 and modified with PASA. Gene prediction completeness was assessed using BUSCO v5.2.237 (busco -m prot). Gene functions were deduced through comprehensive sequence alignments against several databases including the NCBI Non-Redundant (NR), EggNOG, KOG, TrEMBL, InterPro, and Swiss-Prot, employing diamond blastp (diamond v0.9.2938) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database with an E-value threshold of 1E-3. Protein domains were annotated via InterProScan v5.3439 utilizing InterPro protein databases, while PFAM databases was used the identification of motifs and domains within gene models. Gene Ontology (GO) IDs for each gene were obtained from TrEMBL, InterPro, and EggNOG.
Data Records
The raw data of genome and transcriptome sequencing were deposited in the Genome Sequence Archive40 (GSA, https://ngdc.cncb.ac.cn/gsa) at the National Genomics Data Center (NGDC) under the accession number CRA01411941. The genome sequence was stored in the Genome Warehouse42 (GWH, https://ngdc.cncb.ac.cn/gwh) with the accession number GWHERBG00000000. All data are linked to BioProject PRJCA022271. The genome sequence was also deposited in the National Center for Biotechnology Information (NCBI) GenBank, with accession number GCA_039619475.143, under BioProject PRJNA1056491. The genome annotations are available from the Figshare repository44.
Technical Validation
High mapping quality were confirmed in our results. After assembling PacBio reads, Bwa12 was used to align Illumina short-reads with the assembled genome, resulted in mapping rate of 99.36%. The coverage of the genome by Illumina short-reads is 99.86% with an average depth of 51. Minimap245 was used to align HiFi reads with the assembled genome, resulted in mapping rate of 98.90%, and achieving a genome coverage of 99.98% with an average depth of 31. A genome preprint on I. purchasi demonstrated similar genome size and composition, with a genome size of 1,098.4 Mb and a GC content of 33.4%46.
Code availability
All software used in this study is in the public domain and its parameters are clearly described in the methodology. If the detailed parameters of the software are not mentioned, the default parameters are used as recommended by the developer. No custom code or scripts were used for the curation and validation of the dataset.
References
García Morales, M. et al. ScaleNet: a literature-based model of scale insect biology and systematics. DATABASE-OXFORD 2016, bav118 (2016).
Nur, U. Evolution of unusual chromosome systems in scale insects (Coccoidea: Homoptera). Insect Cytogenetics, 97–117 (1980).
Ross, L., Pen, I. & Shuker, D. M. Genomic conflict in scale insects: the causes and consequences of bizarre genetic systems. Biol. Rev. 85, 807–828 (2010).
Normark, B. B. The evolution of alternative genetic systems in insects. Annu. Rev. Entomol. 48, 397–423 (2003).
Royer, M. Intersexuality in the Animal Kingdom 135–145 (Springer, 1975).
Mongue, A. J. et al. Sex, males, and hermaphrodites in the scale insect Icerya purchasi. Evolution 75, 2972–2983 (2021).
Li, M. et al. A chromosome-level genome assembly provides new insights into paternal genome elimination in the cotton mealybug Phenacoccus solenopsis. Mol. Ecol. Resour. 20, 1733–1747 (2020).
Yang, P. et al. Genome sequence of the Chinese white wax scale insect Ericerus pela: The first draft genome for the Coccidae family of scale insects. GigaScience 8, giz113 (2019).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19, 11–15 (1987).
Rio, D. C., Ares, M., Hannon, G. J. & Nilsen, T. W. Purification of RNA using TRIzol (TRI reagent). Cold Spring Harbor Protocols 2010, pdb. prot5439 (2010).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. (Springer, 2016).
Challis, R., Richards, E., Rajan, J., Cochrane, G. & Blaxter, M. BlobToolKit–interactive quality assessment of genome assemblies. G3- Genes, Genomes, Genet. 10, 1361–1374 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Wheeler, T. J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 41, D70–D82 (2012).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 1–14 (2008).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4.10. 11–14.10. 14 (2004).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 1–9 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43, e78 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22 (2008).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Chen, T. et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics, Proteomics &. Bioinformatics 19, 578–583 (2021).
NGDC Genome Sequence Archive (GSA) https://ngdc.cncb.ac.cn/gsa/browse/CRA014119 (2024).
Chen, M. et al. Genome warehouse: a public repository housing genome-scale data. Genomics, Proteomics & Bioinformatics 19, 584–589 (2021).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_039619475.1 (2024).
Zhang, L. Chromosome genome annotation information of Icerya purchasi. figshare https://doi.org/10.6084/m9.figshare.24958746.v1 (2024).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Mongue, A. J., Ross, L., Watson, G. W. & Darwin Tree of Life Consortium. The genome sequence of the cottony cushion scale, Icerya purchasi (Maskell, 1879). Wellcome Open Res. 9, 21, https://doi.org/10.12688/wellcomeopenres.20653.1 (2024).
Acknowledgements
This research was supported by National Natural Science Foundation of China (Grant No. 32270499), the Special Investigation Program for National Science and Technology Basic Resources (2022FY100500) and the Fujian Agriculture and Forestry University Science Fund for Distinguished Young Scholars (Kxjq21004).
Author information
Authors and Affiliations
Contributions
X.L.H. and J.D. designed and funded the study, X.B.W. collected and prepared the samples, L.Z. and H.Z. performed the bioinformatic analyses. J.D. and L.Z. drafted the manuscript. X.L.H. and J.D. revised the manuscript. All authors approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, J., Zhang, L., Zhang, H. et al. Chromosome-level genome assembly of the cottony cushion scale Icerya purchasi. Sci Data 11, 639 (2024). https://doi.org/10.1038/s41597-024-03502-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03502-x
- Springer Nature Limited