
Abstract
The Mediterranean mussel, Mytilus galloprovincialis, is a significant marine bivalve species that has ecological and economic importance. This species is robustly resilient and highly invasive. Despite the scientific and commercial interest in studying its biology and aquaculture, there remains a need for a high-quality, chromosome-scale reference genome. In this study, we have assembled a high-quality chromosome-scale reference genome for M. galloprovincialis. The total length of our reference genome is 1.41 Gb, with a scaffold N50 sequence length of 96.9 Mb. BUSCO analysis revealed a 97.5% completeness based on complete BUSCOs. Compared to the four other available M. galloprovincialis assemblies, the assembly described here is dramatically improved in both contiguity and completeness. This new reference genome will greatly contribute to a deeper understanding of the resilience and invasiveness of M. galloprovincialis.
Similar content being viewed by others


Background & Summary
The Mediterranean mussel, Mytilus galloprovincialis Lamarck 1819, is a gregarious species that attaches to rocks or other hard surfaces using byssal threads. The mussel plays a crucial ecological role as an ecosystem engineer by creating habitat and promoting environmental heterogeneity, thereby enhancing local biodiversity1. The mussel is also known to accumulate contaminants in its tissues from the surrounding environment2,3,4,5. As a result, it has been widely used as a reliable bioindicator in various monitoring programs, such as the Mussel Watch Programme6. In addition to its ecological role as an ecosystem engineer and bioindicator, M. galloprovincialis also holds considerable commercial value. M. galloprovincialis is a widely cultivated bivalve species globally. In 2015, the worldwide production of M. galloprovincialis for human consumption exceeded 1.1 million tonnes7.
The mussel M. galloprovincialis is a highly invasive species that originated in the Mediterranean Sea and the eastern Atlantic, extending north to the British Isles. The species has been introduced to different areas via ballast water over the past century, and is now found in temperate coastal regions of both the northern and southern hemispheres (see Fig. 1a for detail). The Global Invasive Species Programme has identified M. galloprovincialis as one of the top 100 worst invasive species globally due to its significant impact on biological diversity8. The invasive success of M. galloprovincialis in central and southern California is believed to be partly attributed to its physiological adaptations that allow it to outperform M. trossulus in high temperatures9. Fields and colleagues9 discovered that a slight alteration in the structure of cytosolic malate dehydrogenases in M. galloprovincialis allows them to function effectively at higher temperatures. Recent advancements in genomic screening have enabled the identification of a greater number of genetic variations associated with differences in environmental conditions, such as temperature.

The global distribution of the Mediterranean mussel, Mytilus galloprovincialis, is shown in the map (a). The green regions indicate the native range, while the red regions indicate the invasive range. The distribution data was compiled from various sources51,52,53,54,55,56,57,58. In the photo (b), gregarious M. galloprovincialis can be seen on a rocky shore in Dalian, China. Photo credit: Guo-dong Han.
Chromosome-scale reference genomes are essential for applying genomics to biology, aquaculture, and biodiversity conservation. They offer improved contiguity and completeness compared to fragmented genome assemblies, enabling the testing of crucial ecological and evolutionary hypotheses. Although there is significant scientific and commercial interest in mussels for both biology and aquaculture purposes, the availability of a high-quality, chromosome-scale reference genome for M. galloprovincialis is currently limited10,11,12. The genome of M. galloprovincialis, similar to other bivalves, is relatively large and complex, and frequently exhibits high heterozygosity13. Particularly, the genome of M. galloprovincialis exhibits high levels of hemizygosity (only one of the two chromosomal pairs encodes a region or block of DNA) compared to other molluscs11,14. Previous attempts at assembly of this species were significantly hindered by these factors.
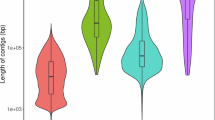
In this study, we used PacBio HiFi technology to sequence and assemble the genome of M. galloprovincialis. We also utilized high-throughput chromosome conformation capture (Hi-C) technology to achieve chromosome-scale scaffolding. As a result, we constructed a high-quality chromosome-scale genome assembly for M. galloprovincialis. The primary assembly is highly continuous, complete, and accurate. Key metrics include a scaffold N50 of 96.9 Mb (Fig. 2b), k-mer completeness of 68.8, and a k-mer-based quality value (QV) of 51.1 (Table 1). Gene annotation analysis using the metazoa_odb10 lineage dataset showed a completeness of 97.5%, indicating high annotation quality. Certain taxonomically restricted genes were not identified during the BUSCO assessment of genome completeness. For instance, myticin was not part of the gene set in metazoa_odb10, but it was found in the genome. Compared to the other available M. galloprovincialis assemblies in GenBank, our assembly significantly improves contiguity and functional completeness (as measured by the number of complete BUSCO notations). Previous karyometric analysis has shown that M. galloprovincialis has 14 chromosomes15,16. The application of Hi-C in this study resulted in 14 long scaffolds in the primary assembly, approaching chromosome-level assembly. The Hi-C contact map suggests that the primary assembly is highly contiguous (Fig. 3).

Characteristics of the M. galloprovincialis genome. (a) The k-mer spectra output is generated by GenomeScope 2.0 using short reads obtained from whole-genome sequencing (WGS). (b) N(x) plots illustrate the contiguity profile of the primary assembly. (c) The cumulative length of the primary assembly is plotted for all scaffolds.

Hi-C contact maps for the primary genome assembly. Hi-C contact map represents the spatial proximity of genomic regions in a linear format. Each cell in the contact map contains sequencing data that confirms the connection between two specific regions. The scaffolds are arranged in order of length and are visually separated by white lines.
The assembly exhibited widespread hemizygous regions, with 8.79% deletions of the total genome (Fig. 4). A total of 18,429 genes were identified that are located within hemizygous regions. The hemizygosity we observed in this study is higher than that of other molluscs (ranging from 0.17% to 6.69%14). However, it is significantly lower than the hemizygosity estimated in a previous study for the same species (36.78% with a genome size of 1.28Gb11). The observed differences may be due to the fact that the sample used in our study was collected from the invasive region, whereas the sample used in the previous study was collected from its native region11. Furthermore, the genome size of M. galloprovincialis could be notably impacted by hemizygous regions. Increased hemizygosity may lead to a reduction in genome size of this species. Therefore, it seems that structural variation, especially large insertion/deletion polymorphisms, play a crucial role in promoting successful invasiveness. Future studies should focus on confirming the significance of structural variation in invasive adaptation by resequencing individuals from a wider range of native and invasive locations.

Chromosomal map of hemizygous loci. Deletions at least 10 kb in length that were not associated with tandem repeats were shown in black.
Our chromosome-level reference genome for M. galloprovincialis provides a more complete understanding of its resilience and invasiveness compared to incomplete and fragmented reference genomes. Previous studies using a fragmented reference genome (GCA_001676915.1) identified a significant number of single nucleotide polymorphisms (SNPs) in M. galloprovincialis, indicating the presence of standing genetic variation that enables rapid adaptation to ocean acidification17. Another study found a large number of SNPs and demonstrated that divergent climatic factors have driven adaptive genetic variation in M. galloprovincialis over the past century, contributing to its successful invasion of various thermal habitats18. Our complete reference genome will further enhance the identification and annotation of standing genetic variation and adaptive genetic variation in this species. Additionally, the whole genome data will be valuable for the aquaculture industry in developing genetic markers for economically important traits. Genotypes in quantitative trait loci (QTLs) are associated with specific phenotypes, such as shell size and body weight, and can be used as markers for selection in breeding programs13. In conclusion, our chromosome-level reference genome, combined with future population genomics studies, will greatly contribute to a deeper understanding of the resilience and invasiveness of M. galloprovincialis and facilitate its application in aquaculture.
Methods
Biological materials
One female mussel was collected from Yangmadao, Shandong Province, China (37.46342°N, 121.59541°E) on June 1, 2022. The mussel had a shell length of 76 mm. The specimen was subsequently taken to Yantai University, where the adductor muscle was dissected and promptly frozen using liquid nitrogen. The adductor muscle tissue was used to prepare Illumina paired-end library, HiFi SMRTbell library and Hi-C library.
Nucleic acid extraction, library preparation and sequencing
The genomic DNA was extracted using the QIAGEN Genomic-tip 100/G kit (QIAGEN, Germany) following the manufacturer’s instructions. DNA quality was assessed using pulsed field gel electrophoresis, and DNA concentration and purity were measured using the Qubit DNA Assay Kit (Invitrogen, USA) and Nanodrop 2000 (Thermo, USA), respectively.
A library with an insert size of 500 bp was prepared using the TruSeq Nano DNA Library kit (Illumina, CA, USA) by randomly fragmenting and ligating adaptors to the DNA sequences. Paired-end sequencing with 150 bp was performed using the NovaSeq 6000 sequencing system (Illumina, CA, USA).
For the HiFi SMRTbell library, 8 μg of DNA was used to prepare library using the SMRTbell Express Template Preparation Kit 2.0 (Pacific Biosciences, USA) following the manufacturer’s recommendations. The library was sequenced using the HiFi SMRTbell sequencing method with an average fragment size of 15 to 20 kb. The DNA library was loaded onto the flow cell with sequencing buffer and loading beads, and sequencing was performed using the DNA Sequencing Reagent Kit (Pacific Biosciences, USA) according to the manual.
To construct Hi-C libraries, the adductor muscle was grinded with liquid nitrogen and cross-linked with 4% formaldehyde solution and then quenched with glycine. The cross-linked sample was lysed and the nuclei were isolated. The nuclei were then solubilized and digested with the restriction enzyme MboI. The DNA ends were marked with biotin-14-dCTP and the cross-linked fragments were ligated. The nuclear complexes were reverse cross-linked and the DNA was purified. Non-ligated fragment ends were treated with T4 DNA polymerase to remove biotin. The sheared fragments were repaired and the Hi-C samples were enriched using streptavidin C1 magnetic beads. The Hi-C libraries were prepared by adding A-tails to the fragment ends and ligating Illumina paired-end (PE) sequencing adapters. The libraries were amplified by PCR and sequenced on an NovaSeq 6000 sequencing system.
Genome assembly
We used Meryl v1.319 to generate k-mer counts (k = 21) from Illumina WGS reads. The resulting k-mer database was then utilized in GenomeScope2.020 to estimate various genome features, such as sequencing error, genome size, heterozygosity, and repeat content. We estimated the genome size to be 1.34 Gb and the nucleotide heterozygosity rate to be 3.27% (Fig. 2a).
For the assembly of the M. galloprovincialis genome, we employed the de novo assembler HiFiasm v0.19.5-r59221. The final output is a diploid assembly comprising two pseudohaplotypes: a primary assembly and an alternate assembly. We used purge_dups v1.2.522 to identify duplicated sequences and overlapping contigs in the assemblies. The assemblies were then scaffolded using Hi-C data with SALSA23. Gaps remaining after scaffolding were closed using PacBio HiFi reads and YAGCloser (https://github.com/merlyescalona/yagcloser). Contamination was checked using BlobToolKit v2.6.524.
The primary assembly was manually curated using Hi-C contact maps. The Hi-C data was aligned to the reference using bwa mem v0.7.17-r118825. Ligation junctions were identified and Hi-C pairs were generated using pairtools v1.0.226. A 50 kb Hi-C matrix was created using cooler v0.9.227 and balanced using hicExplorer v3.628. And PretextMap v0.1.9 (https://github.com/sanger-tol/PretextMap) were used for visualizing the contact maps (Fig. 3).
Structural variation detection
We employed the allelic structural variation detection pipeline29 to determine the hemizygous regions of the primary assembly. Tandem repeats in genome assembly were identify using trf v4.0930. PacBio HiFi reads were aligned to the primary assembly using minimap2 v2.17-r94131. Structural variations in the primary assembly were identified using pbsv v2.9.0 (https://github.com/PacificBiosciences/pbsv). Deletions at least 10 kb in length that were not associated with tandem repeats were visualized using chromoMap v4.1.132. The primary assembly exhibited widespread hemizygous regions, with 8.79% deletions of the total genome length (Fig. 4). When insertions were taken into account, the rate of hemizygosity of the primary assembly increased to 16.32%.
Genome annotation
Prior to gene structure annotation, repetitive sequences in the primary assembly were identified and masked. A de novo canonical database of repetitive elements was constructed using RepeatModeler v2.0.333 with the “-LTRStruct” option. Repeat families specific to Bivalvia were extracted from Repbase (RepeatMaskerEdition-20181026) and Dfam (v3.8) database to create a homology canonical database. These two databases were combined and used with RepeatMasker v4.1.234 to identify and classify repeats in the primary assembly. Approximately 60.42% (854 Mb) of the assembled sequences of M. galloprovincialis were identified as repetitive sequences (Table 2).
Protein-coding genes were identified using a combination of ab initio prediction and transcriptome-assisted methods. The BRAKER2 v2.1.635 gene prediction pipeline was used to predict genes from repeat-masked genome sequences. Transcriptome-assisted annotation was performed using downloaded Illumina RNA-Seq data from seven tissues (PRJNA230138). The RNA-Seq data were aligned to the primary assembly using HISAT2 v2.2.136. The resulting BAM files were merged and used for genome-guided assembly of transcripts using Trinity v2.1.137. The genome-guided RNA-Seq assemblies were then inputted to PASA v2.5.238 for transcriptome database generation. Finally, ab initio gene predictions and transcript alignments were combined into weighted consensus gene structures using EVidenceModeler v1.1.139. Finally, we identifying 58,480 genes in the primary assembly (Table 3). We identified protein-coding regions located within hemizygous regions using bedtools intersect v2.30.040. Genes located within hemizygous regions were determined by identifying at least one exon falling within these regions. A total of 38,301 exons were found to be located within hemizygous regions, and these exons are associated with 18,429 genes.
To perform functional annotation, we compared the predicted protein sequences to the Nr and UniProt database using blastp v2.13.041 with an e-value threshold of 1e-5. Additionally, we used InterProScan v5.5742 and kofam_scan v1.3.0 (https://github.com/takaram/kofam_scan) for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation, respectively. Among the predicted proteins, 54,374, 28,043, 23,011, and 9,547 proteins were matched to the Nr, Uniprot, Go, and KEGG databases, respectively (Table 3).
Data Records
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive43 in National Genomics Data Center (NGDC)44, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA01559745) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. The sequence accessions for the primary and alternate assemblies have been deposited in the NCBI, under accession number JAWDJN00000000046 and JAZKRD00000000047, respectively. The genome annotation file, functional annotations for predicted genes, bed files of hemizygous loci and genes located within hemizygous regions are available in the figshare repository, respectively48,49.
Technical Validation
We used the QV pipeline of Merqury19 to estimate the assembly QV based on k-mer analysis. The script “best_k.sh” in Merqury was employed to determine the optimal k-mer length. Meryl was then utilized to calculate the number of k-mers in the Illumina WGS reads using default settings. The QV evaluation was performed in Merqury using the output from Meryl and the assembly. The findings demonstrated a k-mer completeness of 68.8 and a k-mer-based QV of 51.1, suggesting high levels of completeness and accuracy. The spectra-asm plot illustrates a well-assembled diploid genome (Fig. 5a), with 1-copy k-mers unique to the primary assembly (red) and alternate assembly (blue), and 2-copy k-mers shared by both assemblies (green).

Quality assessment of assembled genome of M. galloprovincialis. (a) Merqury assembly spectrum plots for evaluating k-mer completeness. (b) Comparison of BUSCO scores (metazoa_odb10) for the assemblies of M. galloprovincialis with four other assemblies accessible in the NCBI database.
To evaluate the accuracy of the assemblies and the predicted gene set, we employed BUSCO v5.2.050 with metazoa_odb10 to determine standard metrics of assembly completeness. Gene annotation analysis revealed a completeness rate of 97.5%, signifying high-quality annotation. The ultimate predicted gene set comprised 58,480 genes with a BUSCO value of 97.2%. Our assembly demonstrated superior contiguity and functional completeness compared to other M. galloprovincialis assemblies available in GenBank (Fig. 5b).
Code availability
In this study, software tools were utilized as described in the Method section. All bash command lines and scripts are available at the GitHub repository: https://github.com/HanLab2018/Mytilus-galloprovincialis-genome-assembly.
References
Ramos-Oliveira, C., Sampaio, L., Rubal, M. & Veiga, P. Spatial-temporal variability of Mytilus galloprovincialis Lamarck 1819 populations and their accumulated sediment in northern Portugal. PeerJ 9, e11499, https://doi.org/10.7717/peerj.11499 (2021).
Casas, S. & Bacher, C. Modelling trace metal (Hg and Pb) bioaccumulation in the Mediterranean mussel, Mytilus galloprovincialis, applied to environmental monitoring. J. Sea Res. 56, 168–181, https://doi.org/10.1016/j.seares.2006.03.006 (2006).
Provenza, F. et al. Mussel watch program for microplastics in the Mediterranean Sea: Identification of biomarkers of exposure using Mytilus galloprovincialis. Ecol. Indic. 142, 109212, https://doi.org/10.1016/j.ecolind.2022.109212 (2022).
Soto, M., Ireland, M. P. & Marigómez, I. Changes in mussel biometry on exposure to metals: implications in estimation of metal bioavailability in ‘Mussel-Watch’ programmes. Sci. Total Environ. 247, 175–187, https://doi.org/10.1016/S0048-9697(99)00489-1 (2000).
Sparks, C., Odendaal, J. & Snyman, R. An analysis of historical Mussel Watch Programme data from the west coast of the Cape Peninsula, Cape Town. Mar. Pollut. Bull. 87, 374–380, https://doi.org/10.1016/j.marpolbul.2014.07.047 (2014).
Goldberg, E. D. The mussel watch — A first step in global marine monitoring. Mar. Pollut. Bull. 6, 111, https://doi.org/10.1016/0025-326X(75)90271-4 (1975).
Wijsman, J. W. M., Troost, K., Fang, J. & Roncarati, A. Global Production of Marine Bivalves. Trends and Challenges. in Goods and Services of Marine Bivalves (eds. Smaal, A. C., Ferreira, J. G., Grant, J., Petersen, J. K. & Strand, Ø.) 7–26. https://doi.org/10.1007/978-3-319-96776-9_2 (Springer International Publishing, Cham, 2019).
100 of the World’s Worst Invasive Alien Species: A Selection from the global invasive species database. in Encyclopedia of Biological Invasions (eds. Simberloff, D. & Rejmanek, M.) 715–716. https://doi.org/10.1525/9780520948433-159 (University of California Press, 2019).
Fields, P. A., Rudomin, E. L. & Somero, G. N. Temperature sensitivities of cytosolic malate dehydrogenases from native and invasive species of marine mussels (genus Mytilus): sequence-function linkages and correlations with biogeographic distribution. J. Exp. Biol. 209, 656–667, https://doi.org/10.1242/jeb.02036 (2006).
Murgarella, M. et al. A First Insight into the Genome of the Filter-Feeder Mussel Mytilus galloprovincialis. PLoS ONE 11, e0151561, https://doi.org/10.1371/journal.pone.0151561 (2016).
Gerdol, M. et al. Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel. Genome Biol. 21, 275, https://doi.org/10.1186/s13059-020-02180-3 (2020).
Simon, A. Three new genome assemblies of blue mussel lineages: North and South European Mytilus edulis and Mediterranean Mytilus galloprovincialis. 2022.09.02.506387 Preprint at https://doi.org/10.1101/2022.09.02.506387 (2022).
Takeuchi, T. Molluscan Genomics: Implications for Biology and Aquaculture. Curr. Mol. Biol. Rep. 3, 297–305, https://doi.org/10.1007/s40610-017-0077-3 (2017).
Calcino, A. D., Kenny, N. J. & Gerdol, M. Single individual structural variant detection uncovers widespread hemizygosity in molluscs. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 376, 20200153, https://doi.org/10.1098/rstb.2020.0153 (2021).
Insua, A., Labat, J. P. & Thiriot-Quiévreux, C. Comparative analysis of karyotypes and nucleolar organizer regions in different populations of Mytilus trossulus, Mytilus edulis and Mytilus galloprovincialis. J. Mollus. Stud. 60, 359–360, https://doi.org/10.1093/mollus/60.4.359 (1994).
Pérez-García, C., Morán, P. & Pasantes, J. J. Karyotypic diversification in Mytilus mussels (Bivalvia: Mytilidae) inferred from chromosomal mapping of rRNA and histone gene clusters. BMC Genetics 15, 84 (2014).
Bitter, M. C., Kapsenberg, L., Gattuso, J. P. & Pfister, C. A. Standing genetic variation fuels rapid adaptation to ocean acidification. Nat. Commun. 10, 5821, https://doi.org/10.1038/s41467-019-13767-1 (2019).
Han, G.-D. & Dong, Y.-W. Rapid climate-driven evolution of the invasive species Mytilus galloprovincialis over the past century. Anthr. Coasts 3, 14–29, https://doi.org/10.1139/anc-2019-0012 (2020).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Ghurye, J., Pop, M., Koren, S., Bickhart, D. & Chin, C.-S. Scaffolding of long read assemblies using long range contact information. BMC Genom. 18, 527, https://doi.org/10.1186/s12864-017-3879-z (2017).
Challis, R., Richards, E., Rajan, J., Cochrane, G. & Blaxter, M. BlobToolKit – Interactive Quality Assessment of Genome Assemblies. G3-GENES GENOM. GENET. 10, 1361–1374, https://doi.org/10.1534/g3.119.400908 (2020).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://doi.org/10.48550/arXiv.1303.3997 (2013).
Open2C et al. Pairtools: from sequencing data to chromosome contacts. bioRxiv 2023.02.13.528389 https://doi.org/10.1101/2023.02.13.528389.
Abdennur, N. & Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316, https://doi.org/10.1093/bioinformatics/btz540 (2020).
Ramírez, F. et al. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 9, 189, https://doi.org/10.1038/s41467-017-02525-w (2018).
Sollitto, M. et al. Detecting structural variants and associated gene presence–absence variation phenomena in the genomes of marine organisms. in Marine Genomics: Methods and Protocols (eds. Verde, C. & Giordano, D.) 53–76. https://doi.org/10.1007/978-1-0716-2313-8_4 (Springer US, New York, NY, 2022).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Anand, L. & Rodriguez Lopez, C. M. ChromoMap: an R package for interactive visualization of multi-omics data and annotation of chromosomes. BMC Bioinform. 23, 33, https://doi.org/10.1186/s12859-021-04556-z (2022).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4, 4.10.1–4.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR genom. bioinform. 3, lqaa108, https://doi.org/10.1093/nargab/lqaa108 (2021).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360, https://doi.org/10.1038/nmeth.3317 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652, https://doi.org/10.1038/nbt.1883 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666, https://doi.org/10.1093/nar/gkg770 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, https://doi.org/10.1093/bioinformatics/btq033 (2010).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinf. 19, 578–583, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
CNCB-NGDC MembersPartners Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic. Acids. Res. 50, D27–D38, https://doi.org/10.1093/nar/gkab951 (2022).
Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA015597 (2024).
Han, G. Mytilus galloprovincialis isolate MGYT20220701, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAWDJN000000000 (2024).
Han, G. Mytilus galloprovincialis isolate MGYT20220701, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JAZKRD000000000 (2024).
Han, G. Genome annotation for Mytilus galloprovincialis genome. figshare https://doi.org/10.6084/m9.figshare.25464577.v1 (2024).
Han, G. Hemizygous loci of Mytilus galloprovincialis genome. figshare https://doi.org/10.6084/m9.figshare.25465618.v2 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
McDonald, J. H. & Koehn, R. K. The mussels Mytilus galloprovincialis and M. trossulus on the Pacific coast of North America. Mar. Biol. 99, 111–118, https://doi.org/10.1007/bf00644984 (1988).
Inoue, K. et al. A possible hybrid zone in the Mytilus edulis complex in Japan revealed by PCR markers. Mar. Biol. 128, 91–95, https://doi.org/10.1007/s002270050072 (1997).
Wang, R. Z. Fauna Sinica. Mollusca, Bivalvia: Mytioida. (Science Press, Beijing, China, 1997).
Grant, W. S. & Cherry, M. I. Mytilus galloprovincialis Lmk. in Southern Africa. J. Exp. Mar. Biol. Ecol. 90, 179–191, https://doi.org/10.1016/0022-0981(85)90119-4 (1985).
Gérard, K., Bierne, N., Borsa, P., Chenuil, A. & Féral, J.-P. Pleistocene separation of mitochondrial lineages of Mytilus spp. mussels from Northern and Southern Hemispheres and strong genetic differentiation among southern populations. Mol. Phylogenet. Evol. 49, 84–91, https://doi.org/10.1016/j.ympev.2008.07.006 (2008).
Hilbish, T. J. et al. Origin of the antitropical distribution pattern in marine mussels (Mytilus spp.): routes and timing of transequatorial migration. Mar. Biol. 136, 69–77, https://doi.org/10.1007/s002270050010 (2000).
Toro, J. E., Ojeda, J. A., Vergara, A. M., Castro, G. C. & Alcapán, A. C. Molecular characterization of the Chilean blue mussel (Mytilus chilensis Hupe 1854) demonstrates evidence for the occurrence of Mytilus galloprovincialis in southern Chile. J. Shellfish Res. 24, 1117–1121, https://doi.org/10.2983/0730-8000(2005)24[1117:MCOTCB]2.0.CO;2 (2005).
Lins, D. M. et al. Ecology and genetics of Mytilus galloprovincialis: A threat to bivalve aquaculture in southern Brazil. Aquaculture 540, 736753, https://doi.org/10.1016/j.aquaculture.2021.736753 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China, China (42006107).
Author information
Authors and Affiliations
Contributions
Conception and study design: G.D.H., Laboratory experiments: G.D.H., Sample collection: G.D.H., Data analysis and interpretation: G.D.H., D.D.M., L.N.D., Z.J.Z., Drafting the manuscript: G.D.H., D.D.M., L.N.D., Z.J.Z.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, Gd., Ma, Dd., Du, Ln. et al. Chromosomal-scale genome assembly of the Mediterranean mussel Mytilus galloprovincialis. Sci Data 11, 644 (2024). https://doi.org/10.1038/s41597-024-03497-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03497-5
- Springer Nature Limited