
Abstract
Solanum pimpinellifolium, the closest wild relative of the domesticated tomato, has high potential for use in breeding programs aimed at developing multi-pathogen resistance and quality improvement. We generated a chromosome-level genome assembly of S. pimpinellifolium LA1589, with a size of 833 Mb and a contig N50 of 31 Mb. We anchored 98.80% of the contigs into 12 pseudo-chromosomes, and identified 74.47% of the sequences as repetitive sequences. The genome evaluation revealed BUSCO and LAI score of 98.3% and 14.49, respectively, indicating high quality of this assembly. A total of 41,449 protein-coding genes were predicted in the genome, of which 89.17% were functionally annotated. This high-quality genome assembly serves as a valuable resource for accelerating the biological discovery and molecular breeding of this important horticultural crop.
Similar content being viewed by others

Background & Summary
Tomato (Solanum lycopersicum) is one of the most valuable vegetable crops worldwide. It also serves as a classic model system for studying plant-pathogen interactions and fruit development1,2. Fruit size increased gradually during tomato domestication; however, continued selection reduced the genetic diversity, causing the loss of multiple disease resistance in cultivated species3,4. Thus, wild tomato species have been frequently used as important germplasm donors in modern tomato breeding programs5,6. S. pimpinellifolium, the wild progenitor of the cultivated tomato7, possesses genes that confer resistance to biotic and abiotic stresses8,9; for example, Sm from S. pimpinellifolium PI79532 confers high resistance against gray leaf spot in tomato10; the I gene, also derived from PI79532, confers resistance against Fusarium oxysporum f. sp. lycopersici races 111; Rx4 from S. pimpinellifolium PI128216 confers hypersensitive resistance to bacterial spot race T312; and Ph-3 derived from S. pimpinellifolium L3708, confers resistance to Phytophthora infestans13. These findings indicate the huge potential of S. pimpinellifolium for use in breeding programs to develop disease-resistant varieties.
Whole-genome sequencing improves molecular breeding because high-quality plant genomes facilitate the identification of genetic diversity among different germplasms14,15,16,17. Currently, chromosome-level genome assemblies are available for the cultivated tomatoes, such as S. lycopersicum cv. M8218 and Heinz 170619,20, and wild tomatoes, such as S. pennellii LA071621 and S. galapagense LA043622. All these genome assemblies provide favorable support for the discovery of causal genetic variations underlying the major tomato traits based on comparative genomic analysis. S. pimpinellifolium LA1589 is a wild-type tomato accession with small, red, round fruits (Fig. 1a) that is widely used for trait mapping23,24,25,26. Particularly, the well-established introgression line population from cross of S. lycopersicum cv. E6203 and LA1589 represents one of the widest crosses and serves as an important source for scientists and breeders27. Although the draft genome assembly of this accession was published 10 years ago28, a chromosome-level genome sequence has not yet been published, and thus the vast majority of sequence variations are poorly characterized and their impact on important traits are largely hidden.

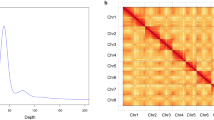
Overview of the S. pimpinellifolium LA1589 genome assembly and features. (a) Morphology of the root, stem, leaf, flower, and fruit of LA1589. (b) Genomescope profile for 21-mers based on Illumina short-reads. (c) Hi-C contact map the chromosome-level assembly of LA1589. (d) Genome features of LA1589. For the circos map, the tracks from outside to inside are: (i) GC content (%); (ii) density of protein-coding genes; (iii) TE density; (iv) LTR density.
In this study, we assembled the chromosome-level genome of S. pimpinellifolium using a combination of short-read sequencing, PacBio sequencing, Hi-C scaffolding, and Bionano optical mapping technologies. The resulting assembly has a total length of 833 Mb, with a contig N50 of 31 Mb, a complete BUSCO value of 98.3%, and a high LAI score of 14.49. The high-quality S. pimpinellifolium genome assembled in this study provides a valuable genetic resource for future efforts to study tomato domestication and promote genome-scale breeding.
Methods
Library construction and genome sequencing
The seeds of S. pimpinellifolium LA1589 were acquired from TGRC (https://tgrc.ucdavis.edu/) and planted in the greenhouse at the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences (Beijing, China). Total genomic DNA was extracted from fresh young leaves using the CTAB method29. A Pacific Biosciences (PacBio) SMRT library was constructed from high molecular weight DNA following the standard SMRTbell library preparation protocol. A total of five SMRT cells were run on the PacBio Sequel system. For short-read sequencing, the paired-end libraries with a 350-bp insert length were constructed and sequenced using the BGISEQ-500 platform. A high-throughput chromosome conformation capture (Hi-C) library was prepared following the proximo Hi-C plant protocol (Phase Genomics) and sequenced using an Illumina NovaSeq. 6000 platform with the paired-end mode. For BioNano optical mapping, genomic DNA was isolated using a BioNano Plant Tissue DNA Isolation Kit. Labelled genomic DNA was then loaded onto the BioNano Saphyr System.
Genome survey
The k-mer frequency method was employed to estimate the genome size. The short-read sequencing produced 104.7 Gb of clean data after filtering out low-quality reads. Jellyfish v2.2.1030 (count -C -m 21; histo -h 40000) was used to compute a histogram of 21 k-mer frequencies. The heterozygosity level was calculated using GenomeScope v1.031. As a result, the estimated genome scale of S. pimpinellifolium was 835.55 Mb, with a heterozygosity rate of 0.08% (Fig. 1b).
Genome assembly and quality assessment
The PacBio sequencing produced 282.3 Gb long reads. Canu v1.832 (genomeSize = 800 m minOverlapLength = 600 minReadLength = 1000) was used to assemble PacBio subreads to PacBio contigs. BioNano optical maps were assembled into consensus physical maps using BioNano Solve v3.1 (https://bionanogenomics.com/). HERA v1.033 was used to extend and connect the contigs, and to fill in gaps in the BioNano hybrid scaffolds. The 128.5 Gb Hi-C reads were mapped to the scaffolds with Bowtie234. Then, HiC-Pro35 was employed to align the pair-end reads and Juicebox36 was used to build the interaction map (Fig. 1c). The scaffolds were further clustered and assigned to different chromosomes. To increase the accuracy of the assembly, Illumina short reads were mapped to genome using BWA v0.7.1537. Next, the genome was corrected using Pilon v1.2438, and three rounds of genome correction were performed. The 833.19-Mb final assembly had a contig N50 length of 31.2 Mb, and approximately 98.87% of the assembled sequence was anchored onto 12 pseudo-chromosomes (Fig. 1d), and showed a greater improvement compared to the previous version of LA1589 genome assembly released in 2012. Moreover, it was also very outstanding when compared with the reference assemblies of S. pennellii LA0716 and S. lycopersicum cv. Heinz 1706 (Table 1).
The completeness of the genome was evaluated using BUSCO (Benchmarking Universal Single-Copy Orthologs) v5.4.539 program with the Solanales odb10 dataset, revealing 98.3% of Solanaceae BUSCOs were captured in this assembly (Table 2). Furthermore, the contiguity of the genome was evaluated by calculating LTR Assembly Index (LAI)40 using LTR_retriever v2.9.941 with default parameters. The LAI value of the genome assembly was 14.49. Collectively, these results indicate a high quality of the S. pimpinellifolium genome assembly.
Repeat annotation
The transposable element (TE) libraries were obtained by running the EDTA pipeline42. In addition, short interspersed nuclear element (SINE) candidates were predicted by the SINE-Finder program v1.043 and integrated into the TE library. RepeatMasker v4.0.744 was used for homologous repeat identification by running against the consensus TE library. Approximately 74.47% of the genome was composed of repetitive sequences (Table 3). LTRs represented the largest proportion (47.45%) of repetitive elements in the genome, of which Gypsy (28.12%) was the most abundant. The insertion time of long terminal repeat (LTR) retrotransposons was estimated as described previously45. In brief, the 5′ and 3′ end terminal repeat sequences of each LTR were extracted and aligned using MUSCLE v3.8.155146. Next, the insertion time of LTR was calculated by T = K/2r, where K is the divergence rate and r is the neutral mutation rate. The results showed that the main burst of Gypsy elements occurred about 0.75 million years ago (MYA), whereas the main burst of Copia elements occurred about 0.6 MYA (Fig. 2), indicating that the amplification of Gypsy elements occurred prior to that of Copia elements and that Gypsy expansion had a major effect on the S. pimpinellifolium genome expansion.

Overall insertion time distribution of LTR elements in the S. pimpinellifolium genome.
Gene prediction and annotation
Protein coding genes (PCGs) in the S. pimpinellifolium genome were annotated using the MAKER pipeline v3.01.0447. Nucleotide and protein sequences from Heinz 1706 v4.0 (https://solgenomics.net/) were used as queries for homology-based predictions. Ab initio gene prediction methods used within MAKER included SNAP v2006-07-2848 and AUGUSTUS v2.5.549. Homology-based and ab initio-based gene prediction resulted in the identification of 41,449 PCGs, which was 6,722 more genes than in the previous version of the genome. Functional annotation of the PCGs was performed using Hayai-Annotation Plants v1.0.250 and KOBAS51. The predicted protein sequences were searched against the InterPro52, Swiss-Prot53, and NR (https://www.ncbi.nlm.nih.gov/protein) databases. In total, 36,960 (89.17%) genes were assigned specific functions (Table 4). Orthologous genes were identified using MCScanX54 and OrthMCL v2.0.955. A total of 29,542 LA1589/Heinz 1706 orthologs were identified.
The first category of non-coding genes, tRNAs, were annotated by tRNAscan-SE v2.0.356. rRNAs were annotated by RNAmmer v1.257. miRNAs and snRNAs were predicted by the cmscan module in INFERNAL v1.1.258 (--cut_ga --rfam --fmt 2) with searches against the Rfam database v14.959. In total, four types of noncoding RNA, including 1073 tRNAs, 698 rRNAs, 582 snRNAs, and 405 miRNAs were identified from the genome.
Data Records
The raw sequencing data generated in this study have been deposited in NCBI Sequence Read Archive with accession number SRP47117760 and in NGDC Genome Sequence Archive with the accession number CRA01244661. The final genome assembly has been deposited in GenBank under accession GCA_034621305.162. The genome annotations are available from the Figshare63.
Technical Validation
The quality of the S. pimpinellifolium assembly was evaluated using three approaches. First, the completeness of the genome assembly was assessed using BUSCO v5.4.5 and 98.30% of the BUSCO genes were complete. Then, the assembly continuity was determined by analyzing the LTR Assembly Index (LAI). The LAI score (14.49) met the quality standard for reference genomes. Additionally, for the assessment of the correctness of the genome assembly, we re-aligned clean Illumina DNA sequencing data against the assembly using BWA v0.7.15, and 99.77% reads could be successfully mapped. All these statistics indicated that this S. pimpinellifolium genome is of high accuracy and completeness.
Code availability
All pipeline and software used in this study were performed to data analysis according to the manuals and protocols. The parameters and the version of the software are described in the Methods section. If no detailed parameters are mentioned for a software, the default parameters were used.
References
Giovannoni, J. J. Genetic regulation of fruit development and ripening. Plant Cell 16, S170–S180 (2004).
Arie, T., Takahashi, H., Kodama, M. & Teraoka, T. Tomato as a model plant for plant-pathogen interactions. Plant Biotechnol 24, 135–147 (2007).
Lin, T. et al. Genomic analyses provide insights into the history of tomato breeding. Nat Genet 46, 1220–1226 (2014).
Schauer, N., Zamir, D. & Fernie, A. R. Metabolic profiling of leaves and fruit of wild species tomato: a survey of the Solanum lycopersicum complex. J Exp Bot 56, 297–307 (2005).
Takei, H. et al. De novo genome assembly of two tomato ancestors, Solanum pimpinellifolium and Solanum lycopersicum var. cerasiforme, by long-read sequencing. DNA Res 28, dsaa028 (2021).
Hake, S. & Richardson, A. Using wild relatives to improve maize. Science 365, 640–641 (2019).
Strickler, S. R. et al. Comparative genomics and phylogenetic discordance of cultivated tomato and close wild relatives. PeerJ 3, e793 (2015).
Kapazoglou, A. et al. Crop wild relatives: a valuable source of tolerance to various abiotic stresses. Plants 12, 328 (2023).
Anderson, T. A. et al. Detection of trait donors and QTL boundaries for early blight resistance using local ancestry inference in a library of genomic sequences for tomato. Plant J 117, 404–415 (2024).
Yang, H. et al. The Sm gene conferring resistance to gray leaf spot disease encodes an NBS-LRR (nucleotide-binding site-leucine-rich repeat) plant resistance protein in tomato. Theor Appl Genet 135, 1467–1476 (2022).
Ori, N. et al. The I2C family from the wilt disease resistance locus I2 belongs to the nucleotide binding, leucine-rich repeat superfamily of plant resistance genes. Plant Cell 9, 521–532 (1997).
Robbins, M. D., Darrigues, A., Sim, S. C., Masud, M. A. & Francis, D. M. Characterization of hypersensitive resistance to bacterial spot race T3 (Xanthomonas perforans) from tomato accession PI 128216. Phytopathology 99, 1037–1044 (2009).
Zhang, C. et al. The Ph-3 gene from Solanum pimpinellifolium encodes CC-NBS-LRR protein conferring resistance to Phytophthora infestans. Theor Appl Genet 127, 1353–1364 (2014).
Gladman, N., Goodwin, S., Chougule, K., Richard, M. W. & Ware, D. Era of gapless plant genomes: innovations in sequencing and mapping technologies revolutionize genomics and breeding. Curr Opin Biotechnol 79, 102886 (2023).
Tian, T. et al. Genome assembly and genetic dissection of a prominent drought-resistant maize germplasm. Nat Genet 55, 496–506 (2023).
Jiang, L. et al. Chromosome-scale genome assembly-assisted identification of Mi-9 gene in Solanum arcanum accession LA2157, conferring heat-stable resistance to Meloidogyne incognita. Plant Biotechnol J 21, 1496–1509 (2023).
Wang, X. et al. Genome of Solanum pimpinellifolium provides insights into structural variants during tomato breeding. Nat Commun 11, 5817 (2020).
Alonge, M. et al. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol 20, 224 (2019).
Hosmani, P. S. et al. An improved de novo assembly and annotation of the tomato reference genome using single-molecule sequencing, Hi-C proximity ligation and optical maps. bioRxiv, 767764 (2019).
Su, X. et al. A high-continuity and annotated tomato reference genome. BMC Genomics 22, 898 (2021).
Bolger, A. et al. The genome of the stress-tolerant wild tomato species Solanum pennellii. Nat Genet 46, 1034–1038 (2014).
Li, N. et al. Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species. Nat Genet 55, 852–860 (2023).
Frary, A. et al. Development of a set of PCR-based anchor markers encompassing the tomato genome and evaluation of their usefulness for genetics and breeding experiments. Theor Appl Genet 111, 291–312 (2005).
Doganlar, S., Frary, A., Ku, H. M. & Tanksley, S. D. Mapping quantitative trait loci in inbred backcross lines of Lycopersicon pimpinellifolium (LA1589). Genome 45, 1189–1202 (2002).
Colak, N. G., Eken, N. T., Ulger, M., Frary, A. & Doganlar, S. Exploring wild alleles from Solanum pimpinellifolium with the potential to improve tomato flavor compounds. Plant Sci 298, 110567 (2020).
Van Der Knaap, E., Lippman, Z. B. & Tanksley, S. D. Extremely elongated tomato fruit controlled by four quantitative trait loci with epistatic interactions. Theor Appl Genet 104, 241–247 (2002).
Liu, J. et al. A natural variation in SlSCaBP8 promoter contributes to the loss of saline-alkaline tolerance during tomato improvement. Hortic Res 11, uhae055 (2024).
The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Inglis, P. W., Pappas, M., Resende, L. V. & Grattapaglia, D. Fast and inexpensive protocols for consistent extraction of high quality DNA and RNA from challenging plant and fungal samples for high-throughput SNP genotyping and sequencing applications. PLoS One 13, e0206085 (2018).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736 (2017).
Du, H. & Liang, C. Assembly of chromosome-scale contigs by efficiently resolving repetitive sequences with long reads. Nat Commun 10, 5360 (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259 (2015).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3, 99–101 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol Biol 1962, 227–245 (2019).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res 46, e126 (2018).
Ou, S. & Jiang, N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol 176, 1410–1422 (2018).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol 20, 275 (2019).
Wenke, T. et al. Targeted identification of short interspersed nuclear element families shows their widespread existence and extreme heterogeneity in plant genomes. Plant Cell 23, 3117–3128 (2011).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics 25, 4.10.11–4.10.14 (2009).
Wang, Z. et al. A chromosome-level reference genome of Ensete glaucum gives insight into diversity and chromosomal and repetitive sequence evolution in the Musaceae. Gigascience 11, 1–21 (2022).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004).
Campbell, M. S. et al. MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol 164, 513–524 (2014).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 1–19 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–W439 (2006).
Ghelfi, A., Shirasawa, K., Hirakawa, H. & Isobe, S. Hayai-Annotation Plants: an ultra-fast and comprehensive functional gene annotation system in plants. Bioinformatics 35, 4427–4429 (2019).
Bu, D. et al. KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res 49, W317–W325 (2021).
Mitchell, A. L. et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 47, D351–D360 (2019).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res 28, 45–48 (2000).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49 (2012).
Li, L., Stoeckert, C. J. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–2189 (2003).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res 49, 9077–9096 (2021).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35, 3100–3108 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res 49, D192–D200 (2021).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP471177 (2023).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA012446 (2023).
NCBI GenBank, https://identifiers.org/ncbi/insdc.gca:GCA_034621305.1 (2023).
Han, H. Y. Chromosome-level genome assembly of Solanum pimpinellifolium. Figshare https://doi.org/10.6084/m9.figshare.24605586 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32000364), the Natural Science Foundation of Shandong Province (ZR2020JQ12), the National Key Research and Development Program of China (2022YFD1201700), and the Qingdao Municipal Science and Technology Huimin Demonstration Project (23-2-8-xdny-15-nsh).
Author information
Authors and Affiliations
Contributions
C.L. and X.M. conceived this study. H.H., X.L. and T.L. collected the samples and performed the experiments. H.H., Q.C., J.Z., H.Z., L.D. and X.M. implemented the computational pipeline and performed the bioinformatics analyses. C.L. and X.M. wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, H., Li, X., Li, T. et al. Chromosome-level genome assembly of Solanum pimpinellifolium. Sci Data 11, 577 (2024). https://doi.org/10.1038/s41597-024-03442-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03442-6
- Springer Nature Limited