
Abstract
Single-cell methods offer a high-resolution approach for characterizing cell populations. Many studies rely on single-cell transcriptomics to draw conclusions regarding cell state and behavior, with the underlying assumption that transcriptomic readouts largely parallel their protein counterparts and subsequent activity. However, the relationship between transcriptomic and proteomic measurements is imprecise, and thus datasets that probe the extent of their concordance will be useful to refine such conclusions. Additionally, novel single-cell analysis tools often lack appropriate gold standard datasets for the purposes of assessment. Integrative (combining the two data modalities) and predictive (using one modality to improve results from the other) approaches in particular, would benefit from transcriptomic and proteomic data from the same sample of cells. For these reasons, we performed single-cell RNA sequencing, mass cytometry, and flow cytometry on a split-sample of human peripheral blood mononuclear cells. We directly compare the proportions of specific cell types resolved by each technique, and further describe the extent to which protein and mRNA measurements correlate within distinct cell types.
Similar content being viewed by others


Background & Summary
Single-cell techniques have revolutionized the resolution at which biology is studied in the lab. Such approaches overcome issues of averaging and subsequent masking of cell-cell heterogeneity experienced by bulk techniques, and thus have become staples in omics research. In particular, single-cell RNA sequencing (scRNA-seq)1,2,3, a high-throughput method to profile transcriptomes, has assisted in cell-type identification4, trajectory inference (TI) modeling5,6,7,8, and gene regulatory network reconstruction9,10,11, to name a few applications. Mass cytometry12,13,14, a high-throughput cytometry method with the ability to simultaneously measure over 40 parameters, has similar applications in cell-type identification15, TI modeling16, as well as differential signaling and cytokine expression studies14.
Transcriptomic data from scRNA-seq is commonly used as a proxy for studying the proteome, as its genomic-scale readout offers a breadth of detail that proteomic approaches are unable to quantify within a single cell. Though broad expression patterns generally associate well with cellular state, the correlation between individual protein expression and corresponding mRNA may be tenuous and even differ amongst proteins or between different cell types17,18. These differences can arise from biological sources, including post-transcriptional regulation, or even technical biases, including dropout in scRNA-seq. Thus, improving our understanding of the relationship between measured mRNA and protein content may aid in refining the conclusions drawn from scRNA-seq.
Additionally, integration of scRNA-seq and cytometry is highly enticing as these data modalities are seemingly complementary, with scRNA-seq measuring a large number of features for a relatively low number of observations and vice versa for mass cytometry. Moreover, predictive methods that use one modality to refine the results of the other are equally desirable. Indeed, several such computational approaches have been reported, including COMET19, which utilizes scRNA-seq data to infer protein marker panels capable of distinguishing specific cell populations. As these approaches are developed, datasets of both readouts from the same population of cells will be useful as a gold standard for validation.
Here, we performed scRNA-seq, mass cytometry, and flow cytometry on a single, split sample of human peripheral blood mononuclear cells (PBMCs). We further compare the ability of scRNA-seq and mass cytometry in resolving distinct cell types and cell states, describe the extent to which protein and mRNA correlate, and quantify and compare cell-type composition based on data from each technique. Because of the split-sample nature of the work, this is a valuable dataset for the purposes of assessing integrative analyses and resolving measured mRNA-protein relationships. In the development of analysis tools capable of isolating and identifying rare populations, PBMCs are an excellent data source given the existence of well-documented subpopulations and small fraction of certain cell types such as dendritic cells. Such data may be further down-sampled or sub-sampled so as to mimic a sample with even rarer populations. Down-sampling may also enable the exploration of properties of each data modality with respect to accuracy and precision in estimating population structure in the presence of technical factors. Finally, while not presented here, we expect that this dataset can be aggregated with other existing PBMC data in meta-analyses that uncover variation in cell type and states in human PBMCs.
Methods
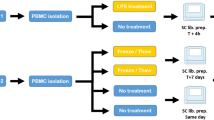
Human PBMCs were obtained from a donor, who had provided written informed consent (IRB 15328), at University of Virginia School of Medicine, Heart Center.
Split-sample preparation for scRNA-seq, CyToF, and flow cytometry
PBMCs were thawed in RPMI 1640 with 5% FBS, and incubated at 37 °C for 1 hr for recovery to ground state. 3 × 105 cells were set aside for scRNA-sequencing. The remaining cells (~7.5 × 106) were divided evenly for mass cytometry and flow cytometry. Cells allocated for scRNA-seq were strained and washed with PBS containing 0.4% BSA. Cell concentration was adjusted to ~500 cells/μL before proceeding with the 10x sequencing protocol.
Next, cells allocated for mass cytometry were fixed. Briefly, cells were incubated with cisplatin (10 µM in PBS) then quenched with cell staining medium (CSM; 0.5% BSA, 0.02% NaN3 in PBS). The cells were strained with a 100 micron nylon strainer before being fixed at room temperature for 10 minutes in 1.6% paraformaldehyde and subsequently stored at −80 °C in CSM. The sample was thawed and stained with metal-conjugated antibodies. Samples are first blocked with 10% donkey serum, stained with surface antibody metal-conjugated antibody cocktail (Table 1), then methanol permeabilized for 10 minutes at 4 °C before being stained for intracellular markers. After staining, samples are incubated with Iridium intercalator for DNA staining overnight at 4 °C before being analyzed on CyTOF mass cytometer (Standard Biotools). Normalization beads containing Lanthanum-139, Praseodymium-141, Terbium-159, Thulium-169, and Lutetium-175 are added to stained samples to perform normalization as previously described20. Stained samples and normalization bead mixtures are then filtered through a 40 micron filter and subsequently analyzed across several runs at a rate of ~250 cells per second on the mass cytometer. After measurement, samples are normalized20 and de-barcoded21 to individual FCS files. FCS files are gated for bead removal, debris clean up, and DNA intercalator.
Finally, cells allocated for flow cytometry were blocked with FcBlock (BD, Catalog No. 564219), before they were further divided evenly into six tubes. Primary antibody incubation of each tube was as follows: Tubes 1 and 2, no primary antibody; Tubes 3–6 anti-CD3 (Thermo Fisher, Catalog No. MHCD0300), anti-CD19 (Thermo Fisher, Catalog No. 14-0199-80), anti-CD56 (Thermo Fisher, Catalog No. 14-0567-80), anti-CD14 (Thermo Fisher, Catalog No. 14-0149-80) respectively. Tubes were placed on ice for 30 minutes and washed twice with FACS buffer. Cells were then incubated with secondary antibody (Thermo Fisher, Catalog No. A-11001) for an additional 30 minutes and washed twice before resuspension in FACS buffer. Flow cytometry was carried out on a BD LSR II flow cytometer and analyzed using FlowJo.
scRNA-seq data processing
Quality control filtering, normalization, clustering, and differential gene expression analysis was performed using Scanpy22 (version 1.8.2). Genes were excluded if they were detected in less than 3 cells; cells were excluded if their mitochondrial gene content exceeded 10% of their total reads or if they had fewer than 200 unique genes in order remove data from any prematurely lysed cells or from ambient RNA. Thresholds were chosen based on manually detecting steep changes in corresponding distributions, aligning with currently accepted practices22,23. Of note, varying these thresholds did not significantly change results of downstream analysis (Supplementary Fig. 1, Supplementary Table 1). Filtering resulted in 2653 cells and 15998 genes. The data was then normalized and log transformed and highly variable genes were identified (3004 genes). The data was then scaled and PCA was performed. Cells were clustered using the Leiden algorithm and visualized on a UMAP embedding (Fig. 1a). Further cell type classification was performed via SingleCellNet24, using sampled data from Zheng et al. as reference data (Fig. 1b). Finally, we annotated cell identity based on these classification results and expression of marker genes (Fig. 1c).

scRNA-seq analysis. (a) Clustering result of the scRNA-seq data. (b) SingleCellNet classification score heatmap. Reference data was taken from Zheng et al. (c) Select marker gene expression.
Clusters ‘0’ and ‘1’ expressed CD3D and CD4, and were subsequently annotated as CD4 T cells. Clusters ‘3’ and ‘4’, which expressed CD3D and CD8, classified strongly as CD8 cytotoxic T cells, and were thus annotated as CD8 T cells. Clusters ‘5.0’ and ‘5.1’ expressed CD19, classified strongly as B cells, and were annotated as B cells. Cluster ‘6’, which expressed NCAM1 and KLRD1, classified strongly as natural killer (NK) cells, and were annotated as such. Clusters ‘2.0’ and ‘2.1’ expressed CD14 and CD68, did not express FCGR3A, and classified strongly as monocytes. Both clusters were annotated as CD16- monocytes. Cluster ‘7’ showed markedly lower expression of CD14, high expression of FCGR3A and MS4A7, and classified as monocytes. This cluster was annotated as CD16+ monocyte. Cluster ‘2.2’ did not express CD14 or FCGR3A, but did express CD68, and was annotated as dendritic cells (DC). Finally, the smallest cluster, cluster ‘8’, expressed ‘PPBP’ and is likely a small group of platelets and was annotated as megakaryocyte-lineage (Mk). To note, these annotations can be further divided into finer sub-populations should users choose to refine the clustering, use a different reference dataset for classification, or widen the field of marker genes to analyze.
CyToF data processing
Gating to remove debris and subsequent arcsin normalization was done on Cytobank. Leiden clustering and UMAP visualization was performed using Scanpy (Fig. 2a). No other normalization or dimension reduction was performed, and cell annotation was based on marker expression (Fig. 2b,c).

mass cytometry analysis. (a) Clustering result of the mass cytometry data. (b) Average transformed expression of select markers in each cluster. (c) Full mass cytometry panel expression.
Briefly, clusters ‘0’, ‘1’, ‘9.0’, and ‘9.1’ were CD3+ CD4+, and were annotated as CD4 T cells. Clusters ‘2’, ‘5’, ‘6’, and ‘8.0’, were CD3+ and CD8a+, and were annotated as CD8 T cells. Clusters ‘4’ and ‘11’ were CD19+ CD20+ CD79b+ HLADR+, and were annotated as B cell. Cluster ‘14’ exhibited lower levels of CD19 and CD20, but was also annotated as B cell. Clusters ‘3.0’, ‘3,1’, ‘3.2’, and ‘3.4’ were CD56+ and were labeled as NK cells. Cluster ‘3.3’ were both CD56+ and CD3+, and were labeled as NKT cells. Cluster ‘7.0’ was CD14+ and CD16-, and was annotated as CD16- monocytes. Cluster ‘7.1’ and ‘12’ were HLADR+ and CD68+, and were annotated as DCs. Finally, we found isolated two populations in the CyToF data that we were unable to resolve or detect in the scRNA-seq population. First, Clusters ‘10’ and ‘13’ were CD3+ and both CD4- and CD8a-. We labeled these as double negative T cells (DN T cell). In contrast, cluster ‘15’ was CD3+ CD4+ CD8a+, and was labeled as double positive T cells (DP T cell). To note, based on our Ab panel, users of this dataset can further divide these broad annotations into finer subpopulations.
Quantifying differences in scRNA-seq and mass cytometry
Based on our cluster annotations, we quantified the percentage of each population in the scRNA-seq, mass cytometry, and flow cytometry data (Fig. 3a,b). Importantly, despite the split-sample nature of the three datasets, there was variation in the proportions of specific cell populations. Notably, while mass cytometry and flow cytometry largely agreed in percentage of T cells, scRNA-seq detected a lower percentage of the same population. As described above, this difference was further exacerbated by the DN and DP T cells that were not detected in the scRNA-seq data. In contrast, the scRNA-seq data exhibited a larger proportion of monocytes than both the mass cytometry and flow cytometry data. To note, we did not resolve a CD16+ monocyte population in the mass cytometry data. Finally, while the scRNA-seq and mass cytometry data exhibited a roughly equal proportion of NK cells, the flow cytometry data had a larger percentage of the same population. To note, differences in cell type percentages measured by scRNA-seq and cytometry have been previously reported in bone marrow mononuclear cells (BMMCs)25.

scRNA-seq and mass cytometry comparison. (a) Percentage of given cell types in scRNA-seq and mass cytometry data. (b) Percentage of given cell types in scRNA-seq, mass cytometry, and flow cytometry data. T cell percentage includes cells annotated as CD4 T, CD8 T, DN T, and DP T. Monocyte percentage includes CD16+ and CD16- monocytes. (c) Correlation of CyToF and scRNA-seq measurements in each cell type.
To broadly estimate the correlation between scRNA-seq and CyToF measurements, we examined the normalized and log-transformed mass cytometry measurements and compared them to the normalized and log-transformed expression of the corresponding genes in the scRNA-seq data across the different cell types (Fig. 3c). Overall, the correlation between the mass cytometry and scRNA-seq measurements was relatively weak (r2 = 0.47–0.66). Taken together, these brief analyses suggest an imprecise concordance between scRNA-seq and mass cytometry measurements. This finding may have broader impacts including suggesting the need for careful consideration in applications such as the identification of rare populations and cell states, which may be obscured using one data modality over another.
Data Records
The CyToF data26 is available at https://flowrepository.org/id/FR-FCM-Z6ZN. The flow cytometry data27 is available at https://flowrepository.org/id/FR-FCM-Z6ZX. The scRNA-seq data is available at GEO under accession GSE22543128.
Technical Validation
Common quality control metrics were calculated in Scanpy (including genes per cell, UMI per cell, and percent mitochondrial gene transcripts), and cells were filtered based on these metrics (see Methods). Subsequent singleCellNet classification results of the transcriptomic data largely agreed with the marker gene expression and corresponding cell type annotation (Fig. 1). Additionally, the percentages of each cell type apparent in the scRNA-seq, mass cytometry, and flow cytometry datasets are in line with previously established percentages3,25,26,,29,30. Of note, reported PBMC composition proportions vary, likely reflecting both biological and technical variability. For example, one source estimates 70–85% T cells, 5–10% B cells, 5–20% NK cells, 10–20% monocytes, 1-2% dendritic cells, and another measured 49–77% T cells, 6–17% B cells, 7–35% NK cells, 6–12% monocytes, 0.6–1.5% dendritic cells. However, given that our measurements fall within these ranges, we believe this dataset is an accurate representation of human PBMCs. Further, the split sample nature of this dataset ensures that RNA-seq and cytometry measurements correspond to the same sample state.
Usage Notes
Both raw counts and processed (normalized, log-transformed, with UMAP coordinates) scRNA-seq data are available as H5AD files. Both are stored as AnnData objects. Cluster annotation can be accessed from the ‘.obs’ slot. For the raw dataset, counts data can be found in the ‘.X’ slot. For the processed dataset, the full normalized, log-transformed expression matrix is stored in the ‘.raw’ slot, while the expression matrix subset for highly variable genes is stored in the ‘.X’ slot.
Similarly, the processed CyToF data is available as an H5AD file. Normalized and asinh-transformed data is stored in the ‘.X’ slot. Cluster annotations can be accessed from the ‘.obs’ slot.
References
Klein, A. M. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015).
Macosko, E. Z. et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Villani, A.-C. et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, (2017).
Trapnell, C. Defining cell types and states with single-cell genomics. Genome Res. 25, 1491–1498 (2015).
Qiu, X. et al. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982 (2017).
Street, K. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19, 477 (2018).
Haghverdi, L., Büttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Qiu, X. et al. Inferring Causal Gene Regulatory Networks from Coupled Single-Cell Expression Dynamics Using Scribe. Cell Syst. 10, 265–274.e11 (2020).
Su, E. Y., Spangler, A., Bian, Q., Kasamoto, J. Y. & Cahan, P. Reconstruction of dynamic regulatory networks reveals signaling-induced topology changes associated with germ layer specification. Stem Cell Rep. 17, 427–442 (2022).
Bandura, D. R. et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 81, 6813–6822 (2009).
Ornatsky, O. et al. Highly multiparametric analysis by mass cytometry. J. Immunol. Methods 361, 1–20 (2010).
Bendall, S. C. et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696 (2011).
Levine, J. H. et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197 (2015).
Bendall, S. C. et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 157, 714–725 (2014).
Vogel, C. & Marcotte, E. M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 (2012).
de Sousa Abreu, R., Penalva, L. O., Marcotte, E. M. & Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 5, 1512–1526 (2009).
Delaney, C. et al. Combinatorial prediction of marker panels from single-cell transcriptomic data. Mol. Syst. Biol. 15, e9005 (2019).
Finck, R. et al. Normalization of mass cytometry data with bead standards. Cytometry A 83, 483–494 (2013).
Fread, K. I., Strickland, W. D., Nolan, G. P. & Zunder, E. R. An updated debarcoding tool for mass cytometry with cell type-specific and cell sample-specific stringency adjustment. Pac Symp Biocomput 22, 588–598 (2017).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Heumos, L. et al. Best practices for single-cell analysis across modalities. Nat. Rev. Genet. 24, 550–572 (2023).
Tan, Y. & Cahan, P. SingleCellNet: A Computational Tool to Classify Single Cell RNA-Seq Data Across Platforms and Across Species. Cell Syst. 9, 207–213.e2 (2019).
Oetjen, K. A. et al. Human bone marrow assessment by single-cell RNA sequencing, mass cytometry, and flow cytometry. JCI Insight 3, (2018).
Zunder, E. FlowRepository https://flowrepository.org/id/FR-FCM-Z6ZN (2023).
Zunder, E. FlowRepository https://flowrepository.org/id/FR-FCM-Z6ZX (2023).
Su, E. Y., Fread, K., Goggin, S., Zunder, E. R. & Cahan, P. GEO. https://identifiers.org/geo/GSE225431 (2023).
Autissier, P., Soulas, C., Burdo, T. H. & Williams, K. C. Evaluation of a 12-color flow cytometry panel to study lymphocyte, monocyte, and dendritic cell subsets in humans. Cytometry A 77, 410–419 (2010).
Kleiveland, C. R. in The impact of food bioactives on health (eds. Verhoeckx, K. et al.) 161–167, https://doi.org/10.1007/978-3-319-16104-4_15 (Springer International Publishing, 2015).
Acknowledgements
We thank Coleen A. McNamara for providing the PBMCs and Chantel McSkimming for advice on the antibody panel. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health to PC under Award No. R35GM124725. EYS was supported by the NSF Graduate Research Fellowship under Grant No. DGE-1746891. ERZ was supported by NIH/NINDS Grant No. 5R01NS111220. KF was supported by NIH/NIGMS Grant No. 5T32GM136615. SG was supported by NIH Grant No. 5T32LM012416.
Author information
Authors and Affiliations
Contributions
E.Y.S. and K.F. performed the experiments with assistance from S.G. E.Y.S. wrote the manuscript with assistance from K.F. P.C. and E.R.Z. conceptualized and supervised the project.
Corresponding authors
Ethics declarations
Competing interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, E.Y., Fread, K., Goggin, S. et al. Direct comparison of mass cytometry and single-cell RNA sequencing of human peripheral blood mononuclear cells. Sci Data 11, 559 (2024). https://doi.org/10.1038/s41597-024-03399-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03399-6
- Springer Nature Limited