
Abstract
Freshwater mussels of the order Unionida are a global conservation concern. Species of this group are strictly freshwater, sessile, slow-growing animals and, extremely sensitive to environmental changes. Human-mediated changes in freshwater habitats are imposing enormous pressure on the survival of freshwater mussels. Although a few flagship species are protected in Europe, other highly imperilled species receive much less attention. Moreover, knowledge about biology, ecology, and evolution and proper conservation assessments of many European species are still sparse. This knowledge gap is further aggravated by the lack of genomic resources available, which are key tools for conservation. Here we present the transcriptome assembly of Unio elongatulus C. Pfeiffer, 1825, one of the least studied European freshwater mussels. Using the individual sequencing outputs from eight physiologically representative mussel tissues, we provide an annotated panel of tissue-specific Relative Gene Expression profiles. These resources are pivotal to studying the species’ biological and ecological features, as well as helping to understand its vulnerability to current and future threats.
Similar content being viewed by others


Background & Summary
Being the animal group with the highest extinction rate (6.3%) and one of the most imperilled taxa (~43% threatened species)1, freshwater mussels of the order Unionida have become a global conservation concern2,3,4,5. This order contains the most diverse group of strictly freshwater bivalves6,7, which share several biological traits that make them extremely vulnerable to ecological disturbance2,4,5. Most freshwater mussels have long life spans with slow growth rates, a sessile adult stage with limited vagility, and a highly specialised life cycle that includes an obligatory parasitic larval stage (glochidia) that must attach to freshwater fish for food and dispersal8,9. Throughout the many stages of their complex life cycle (e.g., sperm release to larval release), mussels are particularly vulnerable to external ecological factors (e.g., water and sediment quality and the number and diversity of host fish). Consequently, the stability of these ecological factors is key to the survival of freshwater mussels2,8,10. Freshwater habitats are among the most intensively modified ecosystems, with massive pressures on the reproduction and recruitment of freshwater mussels that cannot adapt to the pace of these changes2,4,8,11,12,13. Nearly 50% of freshwater mussel species are threatened in Europe (IUCN 2023). Steep population declines and local extinctions have been widely documented, with long-term surveys revealing no halt to these trends2,14. In addition, European conservation actions and funding have largely been absorbed by a few flagship species, with almost 33 million € invested in Life projects focused on the conservation of just three freshwater mussel species15. Conversely, other species that are highly imperilled or experiencing severe population declines receive much less attention2,8,14. Perhaps more worryingly, some European species still lack a comprehensive understanding of their evolutionary, biological, and ecological characteristics or even adequate conservation assessments. This is the case of the species Unio elongatulus C. Pfeiffer, 1825, whose taxonomy has been difficult to resolve until recently2,16,17,18.
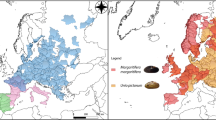
At the end of the 20th century, the species was considered to be threatened and has since been legally protected in EU countries (listed in the Bern Convention [Appendix III] and the Habitats Directive [Annex V]). However, the concept of U. elongatulus at the time of the introduction of these directives now includes three different species that are currently recognised as valid species, i.e. U. elongatulus, Unio ravoisieri Deshayes, 1848, and Unio mancus Lamarck, 1819. Due to this unstable taxonomy, until recently the distribution of the species was poorly defined and often misplaced2,16,17 and its conservation status was unclear and therefore not assessed by the IUCN. With the advent of molecular tools, U. elongatulus has been separated from U. mancus and U. ravoisieri and its distribution is now restricted to rivers and lakes draining into the Adriatic Sea in Italy (from the Ofanto River in the South to the Po River in the North Adriatic) and the Balkans (Croatia and Albania), as well as some Tyrrhenian Sea drainages in Northern Italy (e.g. Arno and Serchio) (Fig. 1)16,17,18. Furthermore, due to the recent redefinition of the species, most aspects of its biology and ecology are equally understudied, hindering a full understanding of its vulnerability and the anticipation and/or planning of conservation measures2,16,17.

When applied to the study of non-model organisms, genomic methods provide highly informative tools to enhance the success of conservation efforts19,20,21,22. Genomic approaches provide robust inferences about genetic diversity, population structure and connectivity and selective and adaptive traits, which in turn allow the identification of significant conservation units, traits and genetic elements and provide a comprehensive picture of population health19,20,21,22,23,24,25. In addition, genomic tools significantly increase the accuracy of predicting the impact of anthropogenic threats and climate change on natural populations21,23,24,25. Nevertheless, the availability of genomic resources is still scarce for many non-model organisms26. This is the case for freshwater mussels with nearly a thousand known species, for which only six whole genome assemblies27,28,29,30,31,32,33,34 and fewer than 20 transcriptomes35,36,37,38,39,40,41,42,43,44,45,46,47,48 are available. The generation of novel genomic resources for the study of freshwater mussels is therefore of fundamental importance. While genome assemblies are undoubtedly the most powerful tools for understanding the biological and evolutionary traits of species, functionally informative methods such as transcriptomics are also important to provide information on gene structure and expression profiles49. This is particularly significant for poorly known species, as these resources represent easily accessible and affordable approaches to accessing highly informative biological, ecological, and evolutionary genetic traits. Freshwater mussels transcriptomics has been instrumental in investigating responses to thermal stress, water depletion, pollution, and bacterial infection (e.g35,37,39,40,41), traits that are intrinsically linked to their global conservation.
Here we present a highly comprehensive multi-tissue transcriptome assembly for one of the least studied freshwater mussels in Europe, U. elongatulus. In addition, we provide a well-annotated panel of tissue-specific relative gene expression profiles, using the individual sequencing outputs of the eight functionally representative tissues, i.e., adductor muscle, foot, gill, gonad, gut, hepatopancreas, mantle and palps. Altogether, these resources will provide a key tool to explore and decipher many biological and ecological aspects of this freshwater mussel species, which in turn will help to understand its vulnerability to current and future threats and shed light on the evolutionary patterns of European freshwater mussels.
Methods
Animal sampling
One individual of U. elongatulus was collected from Lake Maggiore in Italy (Table 1) and transported alive to the laboratory, where the tissues (i.e., adductor muscle, foot, gill, gonad, gut, hepatopancreas, mantle and palps) were immediately dissected, flash frozen and stored at −80 °C, at the CIIMAR tissue and mussels collection under voucher code BIV10776. The shell was also deposited at CIIMAR under the same voucher code.
RNA extraction, library construction, and sequencing
Total RNA of each tissue was extracted with the NZY Total RNA Isolation Kit (NZYTech, Lda. - Genes and Enzymes), according to the manufacturer’s guidelines. A DS-11 Series spectrophotometer/fluorometer was used to estimate RNA concentration (ng/μl) and absorbance measurements (OD260/280 ratio values) (adductor muscle 154.88 ng/μl, foot 824.48 ng/μl, gill 742.08 ng/μl, gonad 1234.08 ng/μl, gut 354.08 ng/μl, hepatopancreas 2993.28 ng/μl, mantle 441.28 ng/μl and palps 342.08 ng/μl). RNA extractions from the eight tissues were sent to Macrogen, Inc., where strand-specific libraries were built (insert size of 250–300 bp) and total RNA was sequenced using 150 bp paired-end reads on the Illumina HiSeq. 4000 platform.
Read processing and de novo transcriptome assembly
Raw sequencing reads for each tissue were inspected with FastQC (version 0.11.8) software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), filtered for Illumina adaptors and quality using Trimmomatic (version 0.38)50 (LEADING:5 TRAILING:5 SLIDINGWINDOW:5:20 MINLEN:36) and corrected for random sequencing errors using a Rcorrector (version 1.0.3)51 (default parameters). The trimmed and corrected reads were concatenated and used for de novo assembly of the entire transcriptome using Trinity (version 2.13.2)52,53 (default parameters). The resulting assembly was screened for putative contaminations by blast search against the Univec (Download; 02/04/2019) and NCBI-nt (Download; 24/08/2021)54 nucleotide databases using Blast-n (version 2.11.0)55. Assembled transcripts matching the phylum Mollusca (NCBI: taxid 6447) or with no match at all were retained. Conversely, transcripts matching other taxa with over 100 bp alignment length, an e-value of 1e-5 or lower, and over 90% of identity score, were flagged as contaminants and excluded. Moreover, all transcripts with any match to the Univec database were also excluded from the assembly.
Redundancy removal
Redundancy from the filtered assembly was removed using the hierarchical contig clustering method applied by Corset (version 1.0.9)56. Briefly, quality treated reads for each tissue were mapped to the transcriptome assembly using Bowtie2 (version 2.3.5) (parameter: --no-mixed --no-discordant --end-to --end --all --score-min L, −0.1, −0.1) and after Corset used to exclude redundancies and transcripts with fewer than 10 reads mapped. The general characteristics, structural integrity, and completeness of the transcriptome (before and after Corset) were assessed using TransRate (version 1.0.3)57 and Benchmarking Universal Single-Copy Orthologs tool (BUSCO version 3.0.2). For BUSCO, completeness estimations were performed using the lineage-specific libraries available for Eukaryota and Metazoa58.
Open reading frame prediction and transcriptome annotation
Open reading frames (ORFs) prediction was performed using Transdecoder (version 5.3.0) (https://transdecoder.github.io/), homology and protein searches being performed in the PFAM59 and UniProtKB/Swiss-Prot60 databases59 using the Blast-p (version 2.12.0)55 and hmmscan of hmmer2 (version 2.4i)61, respectively. Subsequently, a structural annotation file was produced using Gtf/Gff Analysis Toolkit (AGAT) (version 0.8.0)62, supplying the software with the Transdecoder gff output file and the fasta transcriptome assembly. This allowed the generation of protein, transcript, cds and mrdn fasta files with standardised feature names. Functional annotation was performed using InterProScan (version 5.44.80) and Blast-n/p/x searches against several databases. Protein sequences were queried against the NCBI protein databases NCBI-RefSeq Database (Download; 10/03/2022)63, NCBI-nr – non-redundant database (Download; 15/12/2021)54 and InterPro database (Download; 30/03/2019), using the Blast-p/x tools from DIAMOND (version version 2.0.13)64. Transcripts were queried against the NCBI-nt and NCBI-nr databases using the Blast-n tool of NCBI and the Blast-x tool from DIAMOND. All blast (outfmt6 files) and the InterProScan (tsv file) outputs were combined into the gff3 annotation file using AGAT. Gene names were assigned, per sequence, based on the best blast hit (Gene symbol – NCBI Accession Number) and following the hierarchical order: 1- Blast-p Hit in RefSeq database; 2 - Blast-p Hit in NCBI-nr database; 3 - Blast-x Hit in NCBI-nr database; 4 - Blast-n Hit in NCBI-nt database.
Read alignment and tissues specific relative gene expression analysis
For each of the individual sequencing output, Relative Gene Expression (RGE) analyses were conducted using the tool “align_and_estimate_abundance.pl”, implemented through Trinity, specifying “RSEM” as the estimation method and “Bowtie2” as an aligner, using the non-redundant assembly as a reference (Parameters: --est_method RSEM --aln_method bowtie2 --trinity_mode).
Data Records
The sequencing outputs and transcriptome assembly were deposited in NCBI under the BioProject accession number PRJNA1030315: BioSample accession SAMN37904383; Transcriptome Shotgun Assembly (TSA) accession GKPW0000000065; Sequence Read Archive (SRA) accessions SRR26451330, SRR26451329, SRR26451328, SRR26451327, SRR26451325, SRR26451324, SRR26451323, SRR2645132666. The remaining information was uploaded to figshare67. In detail, the files uploaded to figshare include, the filtered trinity redundant assemblies (Uel_trinity_filtered.fasta), the non-redundant transcriptomes (Uel_transcriptome.fa), transcript files (Uel_genes.fa), messenger RNA file (Uel_mrna.fa), open reading frames predictions (Uel_cds.fa), open reading frames proteins predictions (Uel_proteins.fa) as well as the annotation files (Uel_Annotation_sorted.gff3.gz) and the Relative Gene Expression Count tables for all tissues (*_RSEM.isoforms.results).
Technical Validation
Raw datasets and pre-assembly processing quality control
Overall, the raw sequencing output for each tissue was evenly distributed, resulting in almost 22 million reads (M) for adductor muscle, 20 M for foot, 23 M for gill, 23 M for gonad, 19 M for gut, 23 M for hepatopancreas, 22 M for mantle, and 22 M for palps. The overall quality of the raw data was substantially good as demonstrated by the fastqc report (Fig. 2), thus resulting in a low number of low-quality reads being removed during quality trimming (Trimmomatic), less than 2% for each tissue dataset (Table 2). Nevertheless, trimming and error correction (Rcorrector) improved the quality of the reads (Fig. 2).

Bioinformatics pipeline applied for the transcriptome assembly and annotation. Auxiliary representative figures were created with BioRender.com.
Transcriptome assembly metrics
The Trinity transcriptome assembler, used with default parameters, is a very fast and effective approach for unionid transcriptome assemblies35,38,39,40,41,48. This is demonstrated again in our multi-tissue de novo transcriptome (Table 3). The quality of the Trinity output, even before any redundancy removal, is evidenced by the Transrate statistical report, with an N50 length of 1,035 in a total of 1,950,805 transcripts, as well as by the completeness report generated using BUSCO, which reported 100% and 99.2% total genes found (complete + fragmented) for the lineage-specific libraries available for Eukaryota and Metazoa, respectively (Table 3). These values are within the range reported for other Unionida transcriptome assembly projects35,36,37,38,39,41,43,44,45,47, including other congeneric European species48.
Post-assembly processing and annotation verification
Before proceeding with transcript redundancy removal, the assembly was screened and cleaned of putative contaminations, using an approach developed by the team and previously successfully tested on freshwater mussels’ transcriptomes48,68,69. This approach identifies sequences based on homology and only excludes transcripts with well-supported matches between different databases (i.e., NCBI-nt and Univec), thus avoiding the exclusion of unambiguous matches. Using this approach, a total of 5,758 were identified as likely contaminations and removed from the assembly, which was then used for redundancy removal using Bowtie2/Corset. Bowtie2 mapping percentages per tissue were as follows: 89.30% for adductor muscle, 89.22% for foot, 87.46% for gill, 87.69% for gonad, 84.20% for gut, 89.21% for hepatopancreas, 85.87% for mantle, and 87.48% for palps. Corset uses a hierarchical clustering of contig sharing read alignments, resulting in unbiased redundancy exclusion of total transcripts, rather than removing the largest isoform based on protein-coding transcripts only56 and has been successfully applied by the team to freshwater mussel transcriptome assemblies48. Corset was highly efficient in redundancy removal, removing almost 80% of the total number of transcripts and reducing the reported BUSCO’s duplicated genes from around 60% to 2%, with a residual impact on the number of total genes found (both above 90%) (Table 3). These results, reinforce the importance of removing redundancy after Trinity, which despite producing highly complete assemblies, also produces many duplicated and/or low-read-supported transcripts. In the end, Corset significantly reduced the initial assembly, while maintaining its completeness and removing putative errors introduced during the assembly. Open reading frames (ORF) prediction was performed on the decontaminated non-redundant assembly using TransDecoder, resulting in a total of 63,067 ORF (Table 4). Functional annotation was performed on all the predicted amino acid sequences using a well-established pipeline within the team27,33,48,70,71, and the results are presented in Table 5, which shows the detailed hit list with several different databases used for annotation. In total, 30,605 and 34,958 transcripts were functionally annotated by InterProScan and Blast search, respectively (Table 5). These values are very similar to the number of functionally annotated genes reported for the genome assemblies of two congeneric European species, i.e., Unio delphinus (32,089)70 and Unio pictorum (34,137)33, fitting well within the values reported for other Unionida genome assemblies27,28,29,30,34,71. Conversely, the numbers reported here are slightly higher than those reported in the recently published transcriptomes of four Unio species, i.e., Unio pictorum (InterProScan:14,723; Blast:24,194), Unio mancus (InterProScan:14,971; Blast:24,775), Unio crassus (InterProScan:20,432; Blast:51,937) and Unio delphinus (InterProScan:20,637; Blast:32,688)48. However, the transcriptome assemblies were only based on a single tissue (i.e., gill), which probably explains the reduced number of annotated transcripts. Overall, these results confirm the global quality and completeness of the transcriptome assembly presented here and highlight the importance of using a multi-tissue approach for transcriptome assemblies in freshwater mussels to ensure the obstinances of a representative panel of transcripts. Finally, we used the sequencing outputs for each sample tissue to generate and provide the tissue-specific relative gene expression profiles. The full RGE tables are available in FigShare, where the quantification of gene expression is displayed using both Fragments Per Kilobase of transcript per Million (FKPM) and Transcripts Per Million (TPM) values (Gene IDs according to the gff annotation file). The number of genes with TPM and FKPM values > 1 identified by the two methods and for each tissue is presented in Table 5.
The present results provide an important genomic resource that will serve as an essential tool for future studies investigating the biology of one of the least studied freshwater mussel species in Europe, as well as contributing to the broader understanding of the biology and evolution of freshwater mussels as a whole.
Code availability
All software with respective versions and parameters used to generate the resources presented here (i.e., transcriptome assembly, pre- and post-assembly processing stages, and transcriptome annotation) are listed in the Methods section. Software programs without associated parameters were used with the default settings.
References
Díaz, S. et al. IPBES, 2019: Summary for policymakers of the global assessment report on biodiversity and ecosystem services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services. Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services (2019).
Lopes-Lima, M. et al. Conservation status of freshwater mussels in Europe: state of the art and future challenges. Biological Reviews 92, 572–607 (2017).
Lopes-Lima, M. et al. Conservation of freshwater bivalves at the global scale: diversity, threats and research needs. Hydrobiologia 810, 1–14 (2018).
Aldridge, D. C. et al. Freshwater mussel conservation: A global horizon scan of emerging threats and opportunities. Glob Chang Biol 29, 575–589 (2023).
Sousa, R. et al. A roadmap for the conservation of freshwater mussels in Europe. Conservation Biology 37, e13994 (2023).
Graf, D. L. & Cummings, K. S. A ‘big data’ approach to global freshwater mussel diversity (Bivalvia: Unionoida), with an updated checklist of genera and species. Journal of Molluscan Studies 87 (2021).
Graf, D. L. & Cummings, K. S. Freshwater mussel (Mollusca: Bivalvia: Unionoida) richness and endemism in the ecoregions of Africa and Madagascar based on comprehensive museum sampling. Hydrobiologia 678, 17–36 (2011).
Riccardi, N. et al. Microcondylaea bonellii, a Testimonial for Neglected Endangered Species. in Imperiled: The Encyclopedia of Conservation 436–446, https://doi.org/10.1016/B978-0-12-821139-7.00180-X (Elsevier, 2022).
Modesto, V. et al. Fish and mussels: Importance of fish for freshwater mussel conservation. Fish and Fisheries 19, 244–259 (2018).
Lydeard, C. et al. The Global Decline of Nonmarine Mollusks. Bioscience 54, 321 (2006).
Reid, A. J. et al. Emerging threats and persistent conservation challenges for freshwater biodiversity. Biological Reviews, https://doi.org/10.1111/brv.12480 (2018).
Strayer, D. L. Freshwater Mussel Ecology: A Multifactor Approach to Distribution and Abundance. vol. 1 (Univ of California Press, 2008).
Vörösmarty, C. J. et al. Global threats to human water security and river biodiversity. Nature 2010 467:7315 467, 555–561 (2010).
Lopes-Lima, M. et al. The silent extinction of freshwater mussels in Portugal. Biol Conserv 285, 110244 (2023).
Mammola, S. et al. Towards a taxonomically unbiased European Union biodiversity strategy for 2030. Proceedings of the Royal Society B 287 (2020).
MARRONE, F. et al. Diversity and taxonomy of the genus Unio Philipsson in Italy, with the designation of a neotype for Unio elongatulus C. Pfeiffer (Mollusca, Bivalvia, Unionidae). Zootaxa 4545 (2019).
Froufe, E. et al. Lifting the curtain on the freshwater mussel diversity of the Italian Peninsula and Croatian Adriatic coast. Biodivers Conserv 26, 3255–3274 (2017).
Araujo, R., Buckley, D., Nagel, K. O., García-Jiménez, R. & Machordom, A. Species boundaries, geographic distribution and evolutionary history of the Western palaearctic freshwater mussels unio (Bivalvia: Unionidae). Zool J Linn Soc 182, 275–299 (2018).
Allendorf, F. W., Hohenlohe, P. A. & Luikart, G. Genomics and the future of conservation genetics. Nature Reviews Genetics 2010 11:10 11, 697–709 (2010).
Formenti, G. et al. The era of reference genomes in conservation genomics. Trends Ecol Evol 37, 197–202 (2022).
Hohenlohe, P. A., Funk, W. C. & Rajora, O. P. Population genomics for wildlife conservation and management. Mol Ecol 30, 62–82 (2021).
Meek, M. H. & Larson, W. A. The future is now: Amplicon sequencing and sequence capture usher in the conservation genomics era. Mol Ecol Resour 19, 795–803 (2019).
Oppen, M. J. Hvan & Coleman, M. A. Advancing the protection of marine life through genomics. PLoS Biol 20, e3001801 (2022).
Paez, S. et al. Reference genomes for conservation. Science (1979) 377, 364–366 (2022).
Bertorelle, G. et al. Genetic load: genomic estimates and applications in non-model animals. Nature Reviews Genetics 2022 23:8 23, 492–503 (2022).
Hotaling, S., Kelley, J. L. & Frandsen, P. B. Toward a genome sequence for every animal: Where are we now? Proceedings of the National Academy of Sciences 118, e2109019118 (2021).
Gomes-dos-Santos, A. et al. The Crown Pearl: a draft genome assembly of the European freshwater pearl mussel Margaritifera margaritifera (Linnaeus, 1758). DNA Research 28 (2021).
Smith, C. H. A High-Quality Reference Genome for a Parasitic Bivalve with Doubly Uniparental Inheritance (Bivalvia: Unionida). Genome Biol Evol 13 (2021).
Rogers, R. L. et al. Gene family amplification facilitates adaptation in freshwater unionid bivalve Megalonaias nervosa. Mol Ecol 30, 1155–1173 (2021).
Renaut, S. et al. Genome Survey of the Freshwater Mussel Venustaconcha ellipsiformis (Bivalvia: Unionida) Using a Hybrid De Novo Assembly Approach. Genome Biol Evol 10, 1637–1646 (2018).
Gomes-dos-Santos, A. et al. PacBio Hi-Fi genome assembly of the Iberian dolphin freshwater mussel Unio delphinus Spengler, 1793. bioRxiv 2023.01.16.524251, https://doi.org/10.1101/2023.01.16.524251 (2023).
Gomes-dos-Santos, A. et al. The Crown Pearl V2: an improved genome assembly of the European freshwater pearl mussel Margaritifera margaritifera (Linnaeus, 1758). bioRxiv 2023.02.11.528107, https://doi.org/10.1101/2023.02.11.528107 (2023).
Gomes-dos-Santos, A. et al. A PacBio Hi-Fi genome assembly of the Painter’s Mussel Unio pictorum (Linnaeus, 1758). Genome Biol Evol, https://doi.org/10.1093/gbe/evad116 (2023).
Bai, Z. et al. The First High-Quality Genome Assembly of Freshwater Pearl Mussel Sinohyriopsis cumingii: New Insights into Pearl Biomineralization. International Journal of Molecular Sciences. 25(6), 3146, https://doi.org/10.3390/ijms25063146 (2024).
Roznere, I., Sinn, B. T. & Watters, G. T. The Amblema plicata Transcriptome as a Resource to Assess Environmental Impacts on Freshwater Mussels. Freshwater Mollusk Biology and Conservation 21, 57–64 (2018).
Wang, R. et al. Rapid development of molecular resources for a freshwater mussel, Villosa lienosa (Bivalvia:Unionidae), using an RNA-seq-based approach 31, 695–708, https://doi.org/10.1899/11-149.1 (2015).
Luo, Y. et al. Transcriptomic Profiling of Differential Responses to Drought in Two Freshwater Mussel Species, the Giant Floater Pyganodon grandis and the Pondhorn Uniomerus tetralasmus. PLoS One 9, e89481 (2014).
Patnaik, B. B. et al. Sequencing, De Novo Assembly, and Annotation of the Transcriptome of the Endangered Freshwater Pearl Bivalve, Cristaria plicata, Provides Novel Insights into Functional Genes and Marker Discovery. PLoS One 11, e0148622 (2016).
Wang, X., Liu, Z. & Wu, W. Transcriptome analysis of the freshwater pearl mussel (Cristaria plicata) mantle unravels genes involved in the formation of shell and pearl. Molecular Genetics and Genomics 292, 343–352 (2017).
Yang, Q. et al. Histopathology, antioxidant responses, transcriptome and gene expression analysis in triangle sail mussel Hyriopsis cumingii after bacterial infection. Dev Comp Immunol 124, 104175 (2021).
Bertucci, A. et al. Transcriptomic responses of the endangered freshwater mussel Margaritifera margaritifera to trace metal contamination in the Dronne River, France. Environmental Science and Pollution Research 24, 27145–27159 (2017).
Robertson, L. S., Galbraith, H. S., Iwanowicz, D., Blakeslee, C. J. & Cornman, R. S. RNA sequencing analysis of transcriptional change in the freshwater mussel Elliptio complanata after environmentally relevant sodium chloride exposure. Environ Toxicol Chem 36, 2352–2366 (2017).
Capt, C. et al. Deciphering the Link between Doubly Uniparental Inheritance of mtDNA and Sex Determination in Bivalves: Clues from Comparative Transcriptomics. Genome Biol Evol 10, 577–590 (2018).
Huang, D., Shen, J., Li, J. & Bai, Z. Integrated transcriptome analysis of immunological responses in the pearl sac of the triangle sail mussel (Hyriopsis cumingii) after mantle implantation. Fish Shellfish Immunol 90, 385–394 (2019).
Capt, C., Renaut, S., Stewart, D. T., Johnson, N. A. & Breton, S. Putative Mitochondrial Sex Determination in the Bivalvia: Insights From a Hybrid Transcriptome Assembly in Freshwater Mussels. Front Genet 10, 840 (2019).
Chen, X., Bai, Z. & Li, J. The Mantle Exosome and MicroRNAs of Hyriopsis cumingii Involved in Nacre Color Formation. Marine Biotechnology 21, 634–642 (2019).
Cornman, R. S., Robertson, L. S., Galbraith, H. & Blakeslee, C. Transcriptomic Analysis of the Mussel Elliptio complanata Identifies Candidate Stress-Response Genes and an Abundance of Novel or Noncoding Transcripts. PLoS One 9, e112420 (2014).
Gomes-dos-Santos, A. et al. The gill transcriptome of threatened European freshwater mussels. Sci Data 9, 494 (2022).
Stephan, T. et al. Darwinian genomics and diversity in the tree of life. Proc Natl Acad Sci USA 119 (2022).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Song, L. & Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. Gigascience 4, 48 (2015).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 2011 29:7 29, 644–652 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8, 1494–1512 (2013).
Agarwala, R. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 44, D7–D19 (2016).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinformatics 10, 1–9 (2009).
Davidson, N. M. & Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol 15, 1–14 (2014).
Lang-Unnasch, N. Purification and properties of Plasmodium falciparum malate dehydrogenase. Mol Biochem Parasitol 50, 17–25 (1992).
Simão, F. A., Waterhouse, R. M., Ioannidis, P. & Kriventseva, E. v. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res 40, D290–D301 (2012).
Bateman, A. et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res 45, D158–D169 (2017).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39, W29–W37 (2011).
Dainat, J., Hereñú, D. & Pucholt, P. AGAT: Another Gff Analysis Toolkit to handle annotations in any GTF/GFF format. Zenodo https://doi.org/10.5281/zenodo.4205393 (2020).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35, D61–D65 (2007).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015).
Gomes-dos-Santos, A. et al. TSA: Unio elongatulus isolate BIV10776, transcriptome shotgun assembly. GenBank https://identifiers.org/ncbi/insdc:GKPW01000000 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP467529 (2023).
Gomes-dos-Santos, A. The transcriptome assembly of the European freshwater mussel Unio elongatulus C. Pfeiffer, 1825. figshare https://doi.org/10.6084/m9.figshare.24422431 (2023).
Machado, A. M. et al. The male and female gonad transcriptome of the edible sea urchin, Paracentrotus lividus: Identification of sex-related and lipid biosynthesis genes. Aquac Rep 22, 100936 (2022).
Machado, A. M. et al. Liver transcriptome resources of four commercially exploited teleost species. Sci Data 7, 1–9 (2020).
Gomes-dos-Santos, A. et al. PacBio Hi-Fi genome assembly of the Iberian dolphin freshwater mussel Unio delphinus Spengler, 1793. Sci Data 10, 340 (2023).
Gomes-dos-Santos, A. et al. The Crown Pearl V2: an improved genome assembly of the European freshwater pearl mussel Margaritifera margaritifera (Linnaeus, 1758). GigaByte 2023, 1–14 (2023).
Lehner, B. & Grill, G. Global river hydrography and network routing: Baseline data and new approaches to study the world’s large river systems. Hydrol Process 27, 2171–2186 (2013).
Acknowledgements
This work was support by FCT - Fundação para a Ciência e Tecnologia, I.P. by project "Bivalves no limite: genómica da adaptação em cenários de alteraçoes climáticas" with the reference PTDC/CTA-AMB/3065/2020 and DOI identifier https://doi.org/10.54499/PTDC/CTA-AMB/3065/2020. EF was also support by the FCT, with the project reference CEECINST/00027/2021/CP2789/CT0003 and DOI identifier https://doi.org/10.54499/CEECINST/00027/2021/CP2789/CT0003. FCT also supported MLL (2020.03608.CEECIND) and provided additional strategic funding (UIDB/04423/2020 and UIDP/04423/2020).
Author information
Authors and Affiliations
Contributions
A.G.S. and E.F. designed and conceived this work. N.R. and M.L.L. collected the sample. A.G.S., M.H. and E.S.S.F. conducted the lab experiments. A.G.S. carry on all the analysis. A.G.S. and E.F. wrote the first version of the manuscript. All authors read, revised, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gomes-dos-Santos, A., Fonseca, E., Riccardi, N. et al. The transcriptome assembly of the European freshwater mussel Unio elongatulus C. Pfeiffer, 1825. Sci Data 11, 377 (2024). https://doi.org/10.1038/s41597-024-03226-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03226-y
- Springer Nature Limited