
Abstract
Echiura is a distinctive family of unsegmented sausage-shaped marine worms whose phylogenetic relationship still needs strong evidence from the phylogenomic analysis. In this family, Urechis unicinctus is known for its high nutritional and medicinal value and adaptation to harsh intertidal conditions. Herein, we combined PacBio long-read, short-read Illumina and Hi-C sequencing, generating a high-quality chromosome-level genome assembly of U. unicinctus. The assembled genome spans ~1,138.6 Mb with a scaffold N50 of 68.3 Mb, of which 1,113.8 Mb (97.82%) were anchored into 17 pseudo-chromosomes. The BUSCO analysis demonstrated the completeness of the genome assembly and gene model prediction are 93.5% and 91.5%, respectively. A total of 482.1 Mb repetitive sequences, 21,524 protein-coding genes, 1,535 miRNAs, 3,431 tRNAs, 124 rRNAs, and 348 snRNAs were annotated. This study significantly improves the quality of U. unicinctus genome assembly, sets the footsteps for molecular breeding and further study in genome evolution, genetic and molecular biology of U. unicinctus.
Similar content being viewed by others


Background & Summary
Echiura, commonly referred to as spoon worms, are bilaterally symmetrical and coelomate marine invertebrates with a sausage-shaped body living in burrows in the sediments. They possess annelid-like morphological features, including the ladder-like nervous system, the ultrastructure of cuticle and chaetae, and the larval nervous system segments1, however, they have secondarily lost segmentation as adults, providing a particularly important model for understanding the mechanism underlying segment formation and secondary loss2. Furthermore, the evolutionary relationship between the Echiura and Annelida is in a long- standing controversy. Given the lack of segmentation as adults, Echiura has generally been regarded as a separate phyla closely related to Annelida. Recently, some researchers advised the inclusion of Echiura into Annelida based on the increasing amounts of morphological, molecular phylogenetic and phylogenomic evidence, including expressed sequence tags, transcriptome, and mitochondrial genome3,4,5,6,7,8. As solid evidence for a better understanding of their deep-level evolutionary relationships, phylogenomic analysis from high-quality chromosome-level genome data of the Echiura is still lacking.
Urechis unicinctus (also called the penis fish or the fat innkeeper worm), belonging to the Echiura, is a deposit-feeding burrowing organism that inhabits the intertidal zones along the Korean and Japanese coast and Bohai Gulf on the northeast coast of China. The intertidal zones are peculiar and dynamic areas which are vulnerable to a host of stressors, like steep gradients in temperature and oxygen concentration, threats from pathogen infections, pollution, and toxic substances9. The U. unicinctuss without adaptive immunity can survive in such harsh environments, providing an exciting resource for investigating environmental adaptative evolution. In addition, this endemic Echiuran species has essential ecological and socioeconomic significance and has become an important cultured aquaculture species due to its desirable flavour, nutrient-rich and high medicinal values in Asian countries, especially in China, Japan, and Korea10. The first draft genome assembly of U. unicinctuss based on Illumina short reads was published in 202111. However, due to the limitations of the sequencing technique and assembly algorithm, the genome assembly with a contig N50 length of 0.458 kb and scaffold N50 length of 0.517 kb remains highly fragmented (Table 1), which lags far behind the demand for further study of genetic and molecular in U. unicinctuss. Hence, a high-quality chromosome-scale genome assembly of U. unicinctuss are essential in elucidating its genome evolution and adaptive evolution, and providing theoretical support for the species’ culture.
Here, we present a high-quality chromosome-scale genome assembly of U. unicinctus obtained by combining Illumina, PacBio, and high-throughput chromosome conformation capture (Hi-C) sequencing technology toolkits. The U. unicinctus genome, with a total size of ~1,138.6 Mb, was assembled into 1,394 scaffolds (N50 = 68.3 Mb). A total of 1,113.8 Mb assembled sequences (97.82%) were further anchored to 17 pseudochromosomes (Fig. 1). The quality of the genome assembly is significantly higher than that of the previously published version (NCBI accession No. PRJNA603659), with contig N50 being ~1,160 times higher and scaffold N50 being ~135,472 times higher (Table 1)11. The completeness, accuracy, and contiguity of the genome assembly were evaluated by Benchmarking Universal Single-Copy Ortholog (BUSCO) analysis, Core Eukaryotic Genes Mapping Approach (CEGMA), re-alignment between clean Illumina reads and the genome assembly, and SNP identification. Of the assembled genome, 482.1 Mb (42.34%) were repetitive sequences with a dominance of DNA elements. Additionally, a total of 21,524 protein-coding genes were annotated, of which 99.5% could be functionally annotated. This chromosome-level genome assembly builds the foundation for the understanding of genome evolution and evolutionary adaption and provides a valuable tool for further studies on the genetic and molecular biology of U. unicinctus.

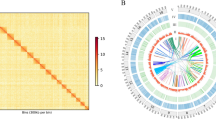
Overview of U. unicinctus genome. (a) Circos plot of genomic features. From outer to inner ring: (A) gene density; (B) GC content; (C) transposable element abundance; (D) Tandem repeat density. Insert: image of adult U. unicinctus. (b) Pseudochromosomes interaction heatmap of U. unicinctus based on Hi-C assembly. Color block indicates the intensity of interaction from yellow (low) to red (high).
Methods
Samples collection and whole-genome sequencing
Adult U. unicinctus samples were obtained from the field of Xiyan, Yantai, Shandong Province, China (121°25’E, 37°56’N), and genomic DNA extracted from the muscle tissue was collected for whole-genome sequencing using a QIAGEN DNeasy Blood & TissueKit (QIAGEN, Shanghai, China). Paired-end Illumina sequence library with insert size 350 bp and 10× Genomics linked-read library were sequenced by Illumina HiSeq X Ten platform with 97.74 Gb of short- read sequencing data (Table 1). For long-read sequencing, a library with an insert of 20 kb was constructed using SMRTbell Template Prep Kits, followed by PacBio single-molecule real- time (SMRT) sequencing using Pacbio Sequel Platform (Pacific Biosciences, Menlo Park, USA), generating approximately 142.1 Gb of long-read raw data.
Transcriptome sequencing
For transcriptome sequencing, four tissues, including intestines, gonads, blood, and muscle, were sampled from the same individual and stored in liquid nitrogen. RNA was extracted from these tissues and used for transcriptome sequencing, respectively. The cDNA paired- end libraries were prepared and sequenced on an Illumina HiSeq X sequencer (Paired-end 350 bp reads). Approximately 26.51 Gb of clean data were yielded from the RNA-seq raw data after quality control using fastp v. 0.21.012 (Table 1).
Genome size estimation and assembly
Jellyfish v. 2.1.3 method13 with k-mer distribution was employed to calculate k-mer frequency (k = 17) based on the high-quality paired-end reads (with an insert size of 350 bp). The distribution of 17-mer depends on the characteristic of the genome and follows a Poisson’s distribution. The genome size was estimated to be 1,396.33 MB with K-mer depth of 58. The genome heterozygosity and repeat ratio are 1.25% and 53.86%, respectively (Table 2).
The WTDBG software v. 2.5, https://github.com/ruanjue/wtdbg) was used to assemble the contig of the U. unicinctus genome with parameters setting as ‘--node-drop 0.20 --node-len 2304 --node-max 150 -s 0.05 -e 3′. Then, Racon v. 1.3.114 with default parameters was used to correct errors of contigs assembly by PacBio data. The resulting contigs were connected to super-scaffolds by 10× Genomics linked-read data using the fragScaff software v. 140324 with parameters setting as ‘-maxCore 200 -m 3000 -q 30 -C 5’15. Lastly, pilon v. 1.22 with parameters setting as ‘-Xmx300G --diploid --threads 20’16 was used to perform the second round of error correction with short paired-end reads generated from Illumina Hiseq X Ten Platforms. The total length of the contig assembly was 1130.4 Mb with the contig N50 size of 528.1 Kb (Table 3). For the scaffolding step, SSPACE v. 3.017 was first used to construct scaffolds using HiSeq data from all the mate-pair libraries (2 kb, 5 kb, 10 kb and 20 kb). FragScaff v. 140324 was further applied to build superscaffolds using the barcoded sequencing reads, generating a genome with a scaffold N50 size of 1080.3 Kb. The total length of this version is 1146.5 Mb.
Hi-C library construction, sequencing and pseudo-chromosome anchoring
The Hi-C library was constructed by a standard protocol described previously with certain modifications18. Briefly, the mussel tissue of U. unicinctus was fixed with 1% formaldehyde solution in MS buffer (10 mM potassium phosphate, pH 7.0; 50 mM NaCl; 0.1 M sucrose), and the nuclei were enriched from flow-through and subsequently digested with HindIII restriction enzyme (NEB). Biotin-labeled DNAs were ligated and purified, followed by fragmenting to a size of 300–500 bp. After a quality control process, the constructed Hi-C library was sequenced on an Illumina HiSeq X Ten sequencer with paired-end 350 bp. In total, 159.47 Gb of high-quality Hi-C data with 132.89 × coverage was acquired (Table 1).
The clean Hi-C paired-end reads were assembled using ALLHIC v. 0.9.819 containing five steps, namely pruning, partition, rescue, optimizing and building, with the following parameter settings: “allhic partition --pairsfile group.clean.pairs.txt --contigfile group.clean.countsGATC.txt -K 26 --minREs 50 --maxlinkdensity 3 --NonInformativeRabio 0”. Ultimately, the size of chromosome-level genome assembly is ~1113.8 Mb, of which 97.82% were anchored into 17 pseudo-chromosomes ranging from 41.8 Mb to 86.6 Mb in length (Fig. 1b and Table 4), containing 7,429 contigs with N50 of 531.5 kb and 1,394 scaffolds with N50 of 68.3 Mb (Table 5).
Annotation of repeats and non-coding RNA (ncRNA)
Homologous comparison and de novo prediction were applied to annotate the repeated sequences in the assembled genome. For homologous comparison, the RepeatMasker v. 4.0.720 and the associated RepeatProteinMask v. 4.0521 were performed to align against Repbase database22. For ab initio prediction, LTR_FINDER v.1.0723, RepeatScout v. 1.0524 and RepeatModeler v. 1.0525 were first used for de novo candidate database constructing of repetitive elements. The repeated sequences were annotated using RepeatMasker v. 4.0.7 Tandem repeat sequences were de novo predicted using TRF v. 4.07b26. In total, 482.1 Mb repetitive sequences were annotated, accounting for 42.34% of the assembled U. unicinctus genome (Table 6). Among the repetitive sequences, DNA transposons (DNA), long interspersed elements (LINE), short interspersed nuclear elements (SINEs), and long terminal repeats (LTRs) accounted for 20.72%, 4.26%, 0.25%, and 10.60% of the whole genome, respectively (Table 7).
For ncRNA annotation, the tRNAs were predicted using tRNAscan-SE v. 1.3.1 software27, and the rRNAs fragments were identified by searching against the Human rRNA database using BLAST with an E-value of 1E-10. Other ncRNAs, including microRNAs (miRNA) and small nuclear RNAs (snRNAs) were predicted by INFERNAL v. 1.1rc428 using Rfam database29. Finally, a total of 5,908 ncRNAs were annotated, including 1,535 miRNAs, 3,431 tRNAs, 124 rRNAs, and 348 snRNAs in U. unicinctus genome(Table 8).
Protein-coding gene prediction and function annotation
The structure of protein-coding genes were predicted by homology-based prediction, de novo prediction and transcriptome-based methods. For homologous annotation, the protein sequences of Helobdella robusta (GCA000326865.1), Capitella teleta (GCA000328365.1), Lottia gigantea (GCA000327385.1), Crassostrea gigas (GCA000297895.2), Mizuhopecten yessoensis (GCA002113885.2), Octopus bimaculoides (GCA001194135.2), Drosophila melanogaster (GCA000001215.4), Anopheles gambiae (GCA000005575.1), Caenorhabditis elegans (GCA004526295.1), Mnemiopsis leidyi (GCA000226015.1), Nematostella vectensis (GCA_932526225.1), Trichoplax adhaerens (GCA000150275.1), Branchiostoma floridae (GCA015852565.1), Homo sapiens (GCA000001405.29) were downloaded from the NCBI’s Genbank database, and aligned against U. unicinctus genome using TBLASTN v. 2.2.2630. The matching proteins were conjoined by Solar software v. 0.9.631, and then aligned to homologous genome sequences for structural prediction by GeneWise v. 2.4.132 (referred to “Homolog” in Table 9). Clean data of RNA-sequencing (RNA-seq) derived from intestines, blood, gonad, and muscle were assembled with Trinity (v2.0)33, and were then aligned against U. unicinctus genome using Program to Assemble Spliced Alignment (PASA)34 (referred to “PASA” in Table 9). Simultaneously, Augustus v. 3.2.335, GeneID v. 1.436, GeneScan37, GlimmerHMM v. 3.0.338, and SNAP39 were employed for ab initio prediction, in which Augustus, SNAP, and GlimmerHMM were trained by homolog set gene models (referred to “De novo” in Table 9). Additionally, RNA-seq reads were directly mapped to U. unicinctus genome using Tophat v. 2.0.1340. The mapped reads were assembled into gene models (RNAseq-Cufflinks-set) by Cufflinks v. 2.1.141 (referred to “Cufflinks” in Table 9). Finally, the gene models were integrated by EvidenceModeler v. 1.1.142. We set the Weights for each type of evidence as follows: PASA-T-set > Homology-set > Cufflinks-set > Augustus > GeneID = SNAP = GlimmerHMM = GeneScan. In order to get the information of untranslated regions (UTRs) and alternatively spliced sites, PASA2 was used to update the final gene models (referred to “Pasa-update” in Table 9). In total, 21,524 protein-coding genes were predicted in the U. unicinctus genome with an average transcript and coding sequence (CDS) length of 5,391.7 bp and 1,291.94 bp, respectively (Table 9).
For functional annotation, the predicted protein sequences were aligned against SwissProt43, NCBI’s non-redundant protein sequence databases (NR), InterPro44, Gene Ontology (GO)45, Kyoto Encyclopedia of Genes and Genomes (KEGG)46 and Pfam protein databases47 by BLASTP (E-value ≤ 1E-05) with the matched rates of 74.1%, 93.2%, 74.2%, 98.5%, 90.6%, and 67.8%, respectively (Table 10). InterproScan tool48 in coordination with InterPro database was applied to predict protein function based on the conserved protein domains and functional sites. In total, 21,408 genes were functionally annotated by at least one database, accounting for 99.5% of all predicted genes, among which 123,56 (68.33%) were supported by all six databases (Fig. 2).

Venn diagram of functional annotation of the U. unicinctus protein-coding genes. The Venn diagram shows the shared and unique annotations among NR, KEGG, SwissProt, and InterPro.
Data Records
All raw genomic sequencing data (Illumina, PacBio, Hi-C, 10× genomics) were deposited in the NCBI Sequence Read Archive (SRA) database with accession numbers SRR2589312949 and SRP45820150. Four transcriptome data from intestine, blood, gonad, and muscle were submitted to the NCBI SRA database with accession numbers SRR25683611, SRR25683610, SRR25683609, and SRR25683608, respectively, under accession the BioProject number PRJNA100651451. The final chromosome assembly was deposited in the GenBank at the NCBI (JAXDRA000000000)52. The sequences of CDS, and protein and results of genome annotation, including repeat sequences, protein-coding regions, and ncRNA annotation, are available in figshare53.
Technical Validation
Quality validation of sequencing data
The quality control of Illumina, 10 × genomic and transcriptome sequencing data was assessed using FastQC quality control (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). The Q20 of Illumina sequencing data was greater than 95.92%, and Q30 was greater than 89.67%. The Q20 of 10× genomic sequencing data was greater than 92.71%, and Q30 was greater than 85.77%. The Q20 of transcriptome sequencing data was larger than 97.67%, and Q30 was larger than 93.41%. The low-quality reads (<Q20) were filtered to ensure the reliability of the data used in subsequent analyses.
Assessment of the genome assembly
The quality of the genome assembly was assessed by four methods as follows: (i) The evaluation of the genome assembly by BUSCO v. 5.1.254 suggested a high level of completeness (93.5%). Of 954 metazoa BUSCO genes, 92.7% were complete and single-copy, 0.8% were complete and duplicated, 4.0% were fragmented, and 2.5% were missing (Table 11). Additionally, completeness of the gene model prediction was also evaluated by BUSCO v. 5.1.2, generating a score of 91.6% (4.7% fragmented and 3.7% missing BUSCOs) (Table 12); (ii) Employing CEGMA (RRID: SCR 015055)55, we detected 240 (96.77%) of 248 core eukaryotic genes were detected in the genome assembly, including 221 (89.11%) complete genes. (iii) The clean short reads generated by the Illumina platform were mapped to the assembled U. unicinctus genome using BWA with parameters setting as ‘-o 1 -i 15’56. The results showed a mapping rate of 97.83% and a coverage rate of 93.90%; (iv) To evaluate the accuracy of the assembly at a single base level, variant calling with SAMTOOLS v. 0.1.19 was performed57. A total of 8,064,289 SNPs, including 7,985,055 heterozygous SNPs and 79,234 homozygous SNPs, were identified with a homozygous rate of 0.0081% (Table 13). All these results suggested the high completeness and accuracy of the U. unicinctus genome assembly.
Code availability
The software and pipelines used in this study were executed following the developers’ instructions, and the versions and parameters of these bioinformatic tools were described in the Methods section. If the parameter is not provided, the default value is used. No custom script or code was used.
References
Hessling, R. Metameric organisation of the nervous system in developmental stages of Urechis caupo (Echiura) and its phylogenetic implications. Zoomorphology 121, 221–234 (2002).
Hou, X. et al. Transcriptome Analysis of larval segment formation and secondary loss in the echiuran worm Urechis unicinctus. Int. J. Mol. Sci. 20, 1806 (2019).
Capa, M. & Hutchings, P. Annelid diversity: historical overview and future perspectives. Diversity 13, 129 (2021).
Struck, T. H. et al. Phylogenomic analyses unravel annelid evolution. Nature 471, 95–98 (2011).
Struck, T. H. et al. Annelid phylogeny and the status of Sipuncula and Echiura. BMC Evol. Biol. 7, 57 (2007).
Weigert, A. et al. Illuminating the base of the annelid tree using transcriptomics. Mol. Biol. 257 Evol. 31, 1391–1401 (2014).
Andrade, S. C. S. et al. Articulating “Archiannelids”: phylogenomics and annelid relationships, with emphasis on meiofaunal taxa. Mol. Biol. Evol. 32, 2860–2875 (2015).
Wu, Z. et al. Phylogenetic analyses of complete mitochondrial genome of Urechis unicinctus (Echiura) support that echiurans are derived annelids. Mol. Phylogen. Evol. 52, 558–562 (2009).
Patil, M. P. et al. Effect of Bacillus Subtilis zeolite used for sediment remediation on sulfide, phosphate, and nitrogen control in a microcosm. Int. J. Env. Res. Public Health 19, 4163 (2022).
Abe, H. et al. Swimming behavior of the spoon worm Urechis unicinctus (Annelida, Echiura). Zoology 117, 216–223 (2014).
Jiao, X., Shi, J., Qin, S., Zhao, D. & Wang, Y. Draft genome sequence data of Urechis unicinctus, a marine echiuroid worm. Data Brief 36, 107032 (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Adey, A. et al. In vitro, long-range sequence information for de novo genome assembly via transposase contiguity. Genome Res. 24, 2041–2049 (2014).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Belton, J.-M. et al. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal- scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4–10 (2004).
Bergman, C. M. & Quesneville, H. Discovering and detecting transposable elements in genome sequences. Brief. bioinformatics 8, 382–392 (2007).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile Dna 6, 1–6 (2015).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, 265–268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, 351–358 (2005).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Li, Y.-h et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 32, 1045–1052 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Yu, X.-J., Zheng, H.-K., Wang, J., Wang, W. & Su, B. Detecting lineage-specific adaptive evolution of brain-expressed genes in human using rhesus macaque as outgroup. Genomics 88, 745–751 (2006).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, 465–467 (2005).
Guigó, R., Knudsen, S., Drake, N. & Smith, T. Prediction of gene structure. J. Mol. Biol. 226, 141–157 (1992).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinform. 5, 1–9 (2004).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, 1–13 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22 (2008).
Apweiler, R. et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32, 115–119 (2004).
Finn, R. D. et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 45, 190–199 (2017).
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D. & Cherry, J. M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, 457–462 (2016).
Jaina, M. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, 412-419 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRR25893129 (2023).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP458201 (2023).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP455724 (2023).
Cheng, Y., Chen, J., Chen, R. & Chen, J. A chromosome-level genome assembly of the Echiura Urechis unicinctus. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_034190875.2 (2023).
Cheng, Y. Chromosome-level genome assembly of the Echiura Urechis unicinctus. figshare https://doi.org/10.6084/m9.figshare.24079509.v3 (2023).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Acknowledgements
This study was funded by the Fujian Provincial Central Guidance Local Science and Technology Development Project (Grant No. 2021L3031), the Project of Department of Fujian Science and Technology in Fujian Province (Grant No. 2020J01868), and the National Natural Science Foundation of China (Grant No. 31902352).
Author information
Authors and Affiliations
Contributions
Jianming Chen designed and conceived this work; Yunying Cheng, Ruanni Chen, and Jinlin Chen analyzed the data and conducted the statistical analysis; Jinlin Chen and Ruanni Chen collected the materials for sequencing; Wanlong Huang constructed the phylogenetic tree. Yunying Cheng wrote the manuscript. Yunying Cheng and Wanlong Huang revised the manuscript. All authors have read, edited, and approved the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, Y., Chen, R., Chen, J. et al. A chromosome-level genome assembly of the Echiura Urechis unicinctus. Sci Data 11, 90 (2024). https://doi.org/10.1038/s41597-023-02885-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02885-7
- Springer Nature Limited