
Abstract
Multidimensional NMR spectra are the basis for studying proteins by NMR spectroscopy and crucial for the development and evaluation of methods for biomolecular NMR data analysis. Nevertheless, in contrast to derived data such as chemical shift assignments in the BMRB and protein structures in the PDB databases, this primary data is in general not publicly archived. To change this unsatisfactory situation, we present a standardized set of solution NMR data comprising 1329 2–4-dimensional NMR spectra and associated reference (chemical shift assignments, structures) and derived (peak lists, restraints for structure calculation, etc.) annotations. With the 100-protein NMR spectra dataset that was originally compiled for the development of the ARTINA deep learning-based spectra analysis method, 100 protein structures can be reproduced from their original experimental data. The 100-protein NMR spectra dataset is expected to help the development of computational methods for NMR spectroscopy, in particular machine learning approaches, and enable consistent and objective comparisons of these methods.
Similar content being viewed by others


Background & Summary
The fundamental data produced by biomolecular NMR spectroscopy are multidimensional NMR spectra. All NMR-based information is derived from these spectra, generally by chemical shift assignment followed by a variety of analyses yielding information on the structure, dynamics, interactions, and mechanisms of proteins and other biomolecules1. Despite the central importance of NMR spectra, these are so far not systematically archived in public databases and are therefore not readily available to other researchers. This contrasts with the situation for certain derived data, namely chemical shift assignments and three-dimensional structures, which are archived since many years and abundant in well-established databases, i.e., the Biological Magnetic Resonance Data Bank (BMRB)2 for chemical shifts and the Protein Data Bank (PDB)3 for protein structures. Although the BMRB does support deposition of time-domain NMR data and peak lists, this feature of the data archive is not extensively used by the community. Some standardized data sets are collected in a few study-specific data repositories4,5 and recommendations for organizing such data have been developed6. However, there is not yet a community-wide effort to collect and validate the NMR time-domain data that support the BMRB and PDB archives of derived chemical shifts and structures.
The fact that NMR spectra, which underlie protein structure determinations by NMR, are in general not available hampers NMR studies, particularly methods development for NMR data analysis. Due to a lack of large-scale primary NMR datasets, NMR data analysis methods are generally developed on the basis of NMR data from just a few proteins that happen to be in the researchers’ hands. This presumably results in sub-optimally parametrized methods, loss of statistical significance in validation, and, importantly, lack of comparability with other methods, since different approaches are not evaluated on the same, standardized data. Machine learning-based methods that require large training and testing datasets exacerbate this problem. Hence, in machine learning large-scale benchmark datasets are standard for methods development and evaluation, e.g., for image classification7.
During our recent development of the machine learning-based ARTINA workflow8 and the NMRtist webserver9 for automated NMR peak picking, chemical shift assignment, and protein structure determination we became acutely aware of the lack of benchmark data sets that include the complete sets of spectra for the assignment and structure determination of a protein. Even though more than 20,000 chemical shift lists and about 14,000 NMR structures have been deposited in the BMRB and PDB databases, respectively, it was a cumbersome task, requiring months of manual work, to collect and standardize the comparatively small number of spectra that had been used previously for the structure determination of just 100 of these proteins (Supplementary Table 1, Supplementary Table 2). Currently, these data are organized differently in every NMR research group and often even by every individual within a group with annotations and naming conventions of the spectra being variable in format and often even internally inconsistent or ambiguous. This makes it difficult to collect, standardize and annotate NMR datasets obtained from individuals and groups. We therefore developed software which assists in this process and allowed us to integrate spectra from different sources.
Here we present a 100-protein NMR spectra dataset that comprises 1329 2D–4D NMR spectra (Supplementary Table 3, Supplementary Table 4), as well as associated reference data (chemical shift assignments, distance restraints, and protein structures collected from the BMRB and PDB databases) and derived data (e.g., expected list of peaks calculated for each NMR spectrum). Our dataset allows to recapitulate the entire process of structure determination with NMR spectroscopy for 100 proteins, reproducing all steps from visual analysis of raw spectra to the calculation of the protein structure. The spectra data is standardized and has been converted to the most popular formats in the field, such as UCSF Sparky10, NMRPipe11 and XEASY12.
Primary NMR data may either be stored as time-domain (free induction decays, FIDs) or frequency-domain (spectra) data. While the two are essentially equivalent, unknown details of the data processing (for instance, apodization functions, baseline correction, etc.) impede a strict mathematical one-to-one correspondence or invertibility. The ARTINA data set provides spectra rather than time-domain data because (i) time-domain data was not available for many of the 100 proteins, (ii) NMR data analysis for assignment, structure determination, and other investigations works almost always in the frequency domain, and (iii) time-domain data needs to be accompanied by a comprehensive set of parameters (parameters of the measurement on the spectrometer, and parameters/scripts for different data processing software packages) in order to reproduce the corresponding spectrum, which poses additional challenges for standardization.
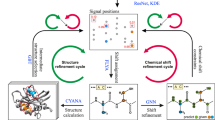
The 100-protein NMR spectra dataset (in the following referred to as ‘the Dataset’) covers a wide range of proteins typically studied with NMR spectroscopy, ranging from small domains (35 residues, 4.1 kDa) to larger systems (175 residues, 20.3 kDa). All these proteins have well-defined tertiary structure, but their sequences may also include unstructured regions (Fig. 1).

NMR spectra analysis workflow associated with the Dataset. Each protein record contains the protein sequence and a set of 2D–4D spectra, which undergo visual spectrum analysis (peak picking), yielding the coordinates of signals in the NMR spectra. Subsequently, identified signals are assigned to atoms in the protein sequence (chemical shift assignment). Assignments can then be used, for instance, to obtain interatomic distance restraints and to determine the three-dimensional protein structure. The Dataset documents all steps of this analysis for 100 proteins with (i) experimental, (ii) experimentally derived, and (iii) in-silico data, as indicated in the diagram.
To the best of our knowledge, the Dataset constitutes the largest standardized source of NMR primary data. In the past, similar datasets were used, but they consisted of fewer spectra and frequently did not cover all steps of protein structure determination with NMR. One of the prominent examples was the community-wide Critical Assessment of Automated Structure Determination by NMR (CASD-NMR) experiment5,13, for which 20 NMR spectra (NOESY type only) of 10 protein targets were provided. The benchmark presented here contains therefore 10 times more protein targets and over 65 times more spectra than the CASD-NMR dataset that was prepared to host this popular event in the NMR community. Other reference points are provided by publications presenting new computational methods in the field. For example, the FLYA algorithm14 was originally evaluated with 3 proteins (46 spectra), WaVPeak15 with 8 proteins (32 spectra), and PICKY16 with 8 proteins (32 spectra).
Further insights into the Dataset come from considering the NMR structure determination process (Fig. 1). It starts from a set of NMR spectra in the frequency domain, consisting of different experiment types recorded for the same protein. In total, 25 distinct experiment types are present among the 1329 NMR spectra in the Dataset. Both in the conventional manual and the automated ARTINA protocol, these spectra undergo visual analysis to identify signals coordinates. As reference information for this step of analysis, we provide for each benchmark spectrum lists of expected peak positions back-calculated from knowledge of the magnetization transfer rules, the protein sequence, the ground truth chemical shift deposited in the BMRB, and (for NOESY) the protein structure deposited in the PDB. In the next step of the analysis, identified signals are mapped to atoms in the protein sequence, yielding list of chemical shifts. Finally, interatomic distance restraints and possibly other conformational restraints are collected, from which the protein structure is calculated. In the Dataset, we provide lists of manually identified chemical shifts, distance restraints, and the protein structure in a standardized form, extracted from the public PDB and BMRB repositories. This non-primary experimental data is complemented with derived annotations, including in-silico predictions, such as AlphaFold17 structure models and UCBShift18-predicted chemical shift lists, that facilitate the development of hybrid approaches for experimental data analysis.
Methods
Spectra data acquisition
To collect NMR spectra, we explored four data sources. First, we implemented specialized crawler software that systematically scanned the FTP server of the BMRB database, extracting files relevant for this project, i.e., either spectra files with frequency-domain data or time-domain data accompanied by processing scripts. These files were converted to data formats available in the Dataset without any alteration of the original data. We did not perform any additional spectra processing steps to improve the deposited data. Time-domain data was used only if no frequency-domain data was available and if the author of the original measurement had uploaded the NMRpipe11 processing script to the BMRB. Notably, a large portion of the data extracted by our BMRB crawler software had been measured within the Northeast Structural Genomics Initiative (NESG)19.
The above data acquisition channel was complemented with a volunteer data upload initiative. To this end, we established a temporary web portal that allowed researchers to upload their published NMR spectra, thereby contributing to the Dataset. Guidelines for data submission were provided for quality and consistency, and each uploaded dataset was manually verified before inclusion in the Dataset. Finally, the diversity of the benchmark dataset was enhanced by including measurements of the authors and spectra recorded in their collaboration network. In total, the data originates from 13 different last authors of PDB depositions (Supplementary Table 2), including 78 referring to the Northeast Structural Genomics Initiative (NESG)19 and 3 from the RIKEN Structural Genomics/Proteomics Initiative (RSGI)20.
A broad range of high-field NMR instruments (600–950 MHz) and experimental setups was used to measure the spectra in the Dataset. This ensures that the dataset is representative for equipment used in the field and increases the chance that computational approaches developed with its support will generalize well to future applications. It is worth mentioning that the Dataset does not include any proprietary data, and all spectra were either publicly available, shared voluntarily by researchers, or generated by the authors and their collaborators. Supplementary Table 2 provides, for each benchmark protein, the names of the authors of the original measurements and the manual structure determination, the literature reference (if available), NMR instruments used to acquire the spectra, etc. Manually determined shift assignments and structures are available from the BMRB and PDB for 97 out of 100 proteins. Papers describing the manual structure determination have been published for 34 proteins21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49, and NOE and possibly other restraints for the manual structure determination have been deposited in the PDB for 94 of the 100 proteins.
Spectra data standardization
A common challenge in NMR data analysis are systematic shifts, which may originate from different relative referencing of spectra. In manual data analysis, it constitutes only a minor obstacle, because two differently referenced spectra are consistent with each other, and their expected signal positions are merely shifted by constant. Popular software packages allow for the correction of these systematic shifts while visualizing spectra, solving the problem entirely from the user perspective, but leaving the source data intact. Therefore, we had to reference all 1329 2D–4D NMR spectra to their corresponding BMRB/PDB depositions. For each spectrum we back-calculated coordinates of expected signals from the corresponding BMRB chemical shift list and PDB structure. Afterwards we determined the optimal reference shifts for each spectrum by maximizing the sum of the absolute intensities of the spectrum at the expected peak positions, \(o\left({\boldsymbol{w}}\right)={\sum }_{n}\left|s\left({{\boldsymbol{p}}}_{n}+{\boldsymbol{w}}\right)\right|\), where the summation runs over all peaks back-calculated from BMRB shifts and the PDB structure, pn denotes the position of the n-th expected peak in the spectrum, the vector w collects the systematic spectrum reference shifts in each dimension, and s(∙) is the intensity of the spectrum at the given position. Since the reference shifts w are typically small and the digital resolution of the spectrum is finite, the optimal reference shifts could be determined by a simple exhaustive search procedure with a finer spacing than the digital resolution of the spectrum. The correctness of the referencing was manually verified for each spectrum, as described in the Technical Validation section below.
Subsequently, we unified the spectra properties, following the standards of the CYANA/FLYA library14. Experiment types and axis labels in each spectrum file were set as specified in Supplementary Table 3 and the overall spectrum intensity was normalized by rescaling each spectrum with a constant to obtain an approximate median value of 100 for the absolute intensities of the scaled spectrum data points50.
Standardization and preparation of other data
For each spectrum included in the Dataset, we calculated signals that are expected to be observed14,51 based on the reported chemical shift assignments deposited in the BMRB and the structures in the PDB. Lists of generated cross-peaks are available in three different variants: expected peaks, expected assigned peaks folded and expected assigned peaks unfolded. The first list stores the assignments of all signals that are expected based on the pulse sequence, the protein sequence and, in case of NOESY spectra, the PDB structure14, regardless of whether their chemical shift assignment is available in the BMRB or not. This list does not contain peak coordinates. The second and third variant of back-calculated peak lists contain folded or unfolded signal coordinates together with atoms assigned to each dimension of every cross-peak. Cross-peaks with missing assignments in the BMRB, and therefore unknown signal coordinates in the spectrum, are not included in these two peak lists. Peaks and their positions are therefore set according to the chemical shift assignments deposited in the BMRB; these are not the experimental peak lists that have been deposited, for only a small fraction of all spectra, in the BMRB by the original depositors of the data. Lists of expected assigned peaks were prepared according to the formal magnetization transfer rules in the CYANA/FLYA library14 (Supplementary Table 3). For spectra with purely through-bond magnetization transfer, the peak lists were generated using only the protein sequence as input, whereas for NOESY spectra the manually determined structure from the PDB was used in addition to obtain NOESY cross peaks for short 1H-1H distances. If an NOE involved groups of degenerate 1H shifts, e.g., for methyl groups, the r–6-summed distance52 was used. These generated peak lists were used to match the chemical shift referencing of the spectra and to verify data consistency.
Each protein record in the Dataset contains reference chemical shift assignments, which were acquired from the BMRB database, as well as a reference protein structure, acquired from the PDB database. To ensure consistency of the reference data, we unified the residue numberings in all data files to match those of the BMRB deposition.
To further facilitate the use of the Dataset for the development and evaluation of computational approaches in protein NMR spectroscopy, we prepared AlphaFold17 and UCBShift18 predictions of structures and chemical shifts. The latter comprises HN, Hα, Hβ, Cα, Cβ, C’, and N shifts predicted using the AlphaFold structure and can serve as prior information for computational approaches under development.
Data Records
Each of the 100 protein records in the Dataset (Supplementary Table 1, Supplementary Table 2) is comprised of the data specified in Tables 1–5. It is available from the long-term ETH Research Collection53 and from https://nmrdb.ethz.ch. Data for each protein is stored in a directory entry that is named with the PDB code or with an abbreviated protein name if the structure of the protein has not been deposited in the PDB. Figure 2 provides an overview of the dataset, including, for each of the 100 proteins, the spectra available, the chemical shift assignment completeness from the BMRB, and the sequence length and secondary structure composition from the PDB.

Overview of the Dataset comprising 100 proteins and 1329 spectra. Proteins are ordered by sequence length and identified by their PDB code or abbreviated name if not deposited in the PDB (left). The left panel shows the spectra available for each protein. Where multiple spectra are available for a given spectrum type, they typically have been acquired by separate measurement of aliphatic and aromatic 13C nuclei or with H2O and D2O solvent. The middle panel shows the completeness of the chemical shift assignments deposited in the BMRB. The right panel shows secondary structure elements and well-defined regions plotted versus the residue number. Well-defined regions, which are used for all RMSD calculations, were determined by CYRANGE66.
For each protein, the subdirectory ‘spectra’ contains the multidimensional NMR spectra, each represented with the data listed in Table 1. All spectra are named by the spectrum type according to FLYA conventions14 (Supplementary Table 3), possibly followed by tags @ALI or @ARO to indicate that the spectrum contains only aliphatic or aromatic 13C signals, respectively.
A complete list of the 1329 spectra is given in Supplementary Table 4. Statistics about spectra and assignments are given in Fig. 3. About 80% of the spectra are three-dimensional, 18% are 2D,and 2% 4D (all HC-HC NOESY experiments). The dataset includes 3D 13C-edited [1H-1H] NOESY and 15N-edited [1H-1H] NOESY spectra for all 100 proteins, and for each protein the complete set of spectra of various types that was used for the original backbone and sidechain chemical shift assignment deposited in the BMRB. About 51% of the spectra were recorded at 600 MHz 1H frequency, 3% at lower frequency (500 MHz), 4% at 700–750 MHz, and 42% at higher frequencies of 800–900 MHz.

Statistics for data records in the Dataset. (a) Distribution of experiment types, spectrum dimensionality (2D–4D), and spectrometer frequency. (b) Distribution of number of data points and chemical shift ranges (ppm) across different dimensions for common 3D experiment types in the Dataset (Supplementary Table 4). For each spectrum type, the bottom row features histograms that represent the number of spectra with the specified number of data points in the given dimension, as indicated by the dimension label in the lower left corner. The upper row provides information about the chemical shift range in the spectrum file and the distribution of expected peaks in each dimension. The red line gives the number of spectra for which a given chemical shift value falls within the experimental spectral width in the given dimension. Similarly, the green line represents the number of spectra for which a given chemical shift value coincides (within tolerance) with at least one expected peak position based on the (unfolded) chemical shift assignments from the BMRB. Where the green line exceeds the red line, it indicates that folding is typically applied along that spectral axis.
Another factor that plays an important role in the development of computational approaches for NMR spectra analysis are the number of data points in each dimension of the spectrum and the type of aliasing (folding) used in the experiment. Such information can be extracted directly from data provided in the Dataset. Figure 3 presents a summary for eight popular experiment types. Folding is typically applied to a 13C dimension in sidechain assignment spectra (axis labels C for HCCH-TOCSY or C1 for CCHTOCSY) and the 15N or 13C dimension in 3D NOESY experiments. Most triple-resonance NMR data used for determining backbone resonance assignments are collected using the constant-time frequency labeling approach54, which avoids the need for folding to improve digital resolution in these indirect dimensions.
NMR measurements are complemented with reference data, stored in the ‘others’ subdirectory, comprising the protein sequence, the manually determined structure from the PDB database, and the manually determined chemical shift assignments from the BMRB (Table 2).
Expected peak lists for each spectrum are in the subdirectory ‘peak_lists’ (Table 3). These are not experimental peak lists (which are not available from BMRB or PDB depositions for most spectra) but lists of peaks expected based on sequence and experiment type (see Methods). As an example, the back-calculated peak list for the [1H,13C]-HSQC spectrum of the protein with PDB code 1VDY is overlayed on the spectrum in Fig. 4.

Contour plot of a [1H,13C]-HSQC spectrum for the protein 1VDY. Positions of peaks back-calculated from the chemical shifts deposited in the BMRB are indicated by blue crosses.
Additional derived data are available in the subdirectory ‘others’ (Table 4). Files ARTINA*.* are the result of fully automated spectra analysis8. These comprise chemical shift assignments for all atoms and the subset of “strong” (reliable) assignments14 as well as the three-dimensional structures obtained by ARTINA using only sequence and spectra as input. Additionally, we included, in each data record, structure predictions by AlphaFold17 and chemical shift predictions by UCBShift18 using the AlphaFold structure as input.
The subdirectory ‘recalc’ contains the manually determined conformational restraints deposited in the PDB, if available, and data from a recalculation of the PDB structure with CYANA52,55 using the available NOE distance restraints, hydrogen bond distance restraints, and torsion angle restraints (Table 5).
Technical Validation
Data stored in the Dataset underwent rigorous qualitative and quantitative validation, as well as consistency checks, which verified that different data modalities (i.e., chemical shift lists obtained from the BMRB, and spectra acquired on-line from the web portal) are consistent with each other.
The first data validation procedure involved the spectra files and expected assigned peaks lists stored in each Dataset record. For each multidimensional spectrum, we calculated all 2D projections of the spectral data along one (3D spectra) or two (4D spectra) spectrum axes. Afterwards, we visualized each projection as a contour plot overlaid with the expected peaks back-calculated from the reference chemical shifts in the BMRB and the structure in the PDB (Fig. 4; Supplementary Fig. 1). In this way we prepared 3593 data visualizations, which were inspected manually and are included in the Dataset together with the source spectra files (Table 1). This verification procedure ensured consistency between raw spectral data, corresponding reference data (shift lists, protein structure), and derived data (expected peak lists). All spectra in the Dataset agree well with the corresponding reference data from BMRB/PDB. Subsequently, we performed a similar round of qualitative evaluation using randomly selected 2D planes (3279 in total) instead of projections from the multidimensional spectra (Supplementary Fig. 2).
In quantitative data validation, we used as input 1329 2D–4D spectra to automatically reproduce 100 protein structures and assignments with the ARTINA algorithm8. The results (Fig. 5) indicate good agreement between the automated spectra analysis and the manual annotations deposited in the BMRB and PDB databases. Consistency of the ARTINA result, obtained exclusively from spectra and sequence, was confirmed by 91.36% identical chemical shift assignments and a median RMSD of 1.44 Å (Supplementary Table 5) with respect to the manually determined chemical shift assignments and structures deposited in the BMRB and PDB, respectively8.

Data validation by automated spectrum analysis with ARTINA. The three panels show, for 100 proteins, the backbone RMSD between the ARTINA structure and the NMR structure deposited in the PDB, as well as the accuracy of the backbone and sidechain assignment by ARTINA relative to the assignments deposited in the BMRB. Proteins presented in bar plots are sorted clockwise by sequence length. Box plots present the distribution and the median of these quantities.
The accuracy of the AlphaFold models was validated by the RMSD to the structures deposited in the PDB (Supplementary Table 5). The median accuracy of the AlphaFold models on the benchmark dataset is 0.96 Å for the backbone atoms. There are only four proteins with RMSD above 2 Å and all RMSDs are below 3 Å. Excellent agreement between AlphaFold models and NMR NOESY data, including for some of the proteins in the Dataset, has been described elsewhere56,57.
Validations of the standardized manually determined conformational restraints deposited in the PDB were performed by recalculating the structures using CYANA. Structure recalculation could be performed for 96 out of 100 proteins for which more than 2 manually determined NOE distance restraints per residue are available from the PDB. Statistics of the reported conformational restraints and the structures deposited in the PDB or recalculated from this data are given in Supplementary Table 6. Consistency of the interpretation of the restraints and the recalculation of the structures (e.g., regarding residue numbering, atom nomenclature, and handling of degenerate or non-stereospecifically assigned atoms) is confirmed by low CYANA target function values for the recalculated structures (0.01–14.27 Å2, median 0.28 Å2) and small backbone RMSD values between the deposited and recalculated structures (0.25–1.98 Å, median 0.79 Å).
The above analysis confirmed consistency of all data modalities, namely NMR spectra, expected peak lists, manual chemical shift assignments, manually solved protein structures, and AlphaFold predictions. This agrees with our previous study8, in which the same spectra dataset was used to develop ARTINA method.
Usage Notes
Since NMR spectra are the fundamental result of NMR measurements, the Dataset facilitates the development of computational methods and software packages for protein NMR, covering a broad range of downstream tasks, ranging from automated visual spectrum analysis to hybrid approaches for assignment and protein structure determination that merge in-silico predictions (e.g., AlphaFold) with experimental data58,59, as well as testing of software for manual NMR data analysis.
We believe that one of the primary applications of the Dataset will involve the development of new computational approaches to classical problems in NMR spectroscopy, such as automated peak picking, chemical shift assignment, structure determination, as well as spectrum quality enhancement. The Dataset opens new avenues in these areas by providing enough data for the training of deep learning architectures, which have so far been used rarely in NMR spectroscopy, mainly because of the lack of adequate training/benchmark data. In addition, the Dataset provides a means to establish links between core machine learning problems and protein NMR, such as the adaptation of well-performing model architectures to NMR spectroscopy.
Likewise, the Dataset may find its application in the development and testing of software packages designed for manual analysis, storage, or validation of NMR data. In essence, the Dataset serves as a robust testing ground, enabling software developers to evaluate and refine their tools, ultimately contributing to the advancement of their software packages and NMR research generally.
The Dataset also opens new opportunities for comparative studies of existing methods in NMR spectroscopy. By providing a large, comprehensive, and standardized dataset, it allows for systematic and unbiased evaluations of current methods in terms of accuracy, efficiency, and robustness. It enables the identification of potential limitations and areas for improvement in current methodologies. This, in turn, can drive the refinement and optimization of existing tools, ultimately contributing to the advancement of computational approaches in the field of protein NMR spectroscopy.
After the recent breakthroughs in deep learning-based protein structure prediction, the research community seeks hybrid approaches that combine in-silico methods with experimental data60. The Dataset facilitates these studies by providing primary experimental data as well as ground truth protein folds and the reference structure quality, obtained by automated data analysis with the ARTINA method.
Finally, the Dataset can be used for educational purposes, serving as a resource for both students and researchers who seek to deepen their understanding of NMR spectroscopy and computational methods for protein structure determination. The comprehensive nature of the Dataset allows for a wide range of applications, from basic spectrum analysis to advanced structure determination exercises. The Dataset can serve as a basis for practical assignments and projects in courses related to bioinformatics, cheminformatics, and structural biology.
In summary, the Dataset addresses a crucial gap in biomolecular NMR research by providing a large-scale, standardized set of spectra, reference data, and derived annotations for 100 proteins. It is significantly larger and more comprehensive than previous datasets that were typically compiled ad hoc for the development of specific methods or for a smaller number of spectral data types4,5,61. The Dataset not only enables reproduction of the entire structure determination process, but also facilitates the development and evaluation of new computational and machine learning-based approaches, holding the promise of becoming a crucial asset for future computational methods development in NMR spectroscopy.
It may be surprising that more than 30 years after the initial development of NMR protein structure determination, and with currently about 14,000 NMR-derived structures in the PDB and a similar number of chemical shift assignment datasets in the BMRB, no comparable collection of primary, time- or frequency-domain NMR data exists. In contrast, it has long been universal practice in X-ray crystallography to deposit the primary data, i.e., the structure factors, along with the protein structure derived from it. One reason for the scarcity of NMR primary data depositions (available for < 0.5% of the NMR structures in the PDB) is that this data is significantly more complex than the structure factors in X-ray crystallography: a protein structure determination requires a series of multidimensional spectra of different types, a variety of binary encoded formats and variants thereof are in use for time- and frequency-domain NMR data, the data size is typically 2–4 orders of magnitude larger than for structure factors, and, significantly, the connection between spectra and structure is much less direct than in X-ray crystallography, where the degree of agreement between structure factors and the structure model derived from them can be quantified readily, e.g., by R-factors. In the absence of a fully automated method such as ARTINA, any verification of an NMR protein structure from spectra required a significant amount of manual work and would therefore have been undertaken only rarely even if the primary data were available. With machine learning-based methods like ARTINA, the situation has changed: NMR analyses of many proteins can now be done efficiently, and, at the same time, the demand for large amounts of standardized training data has become acute. While some solutions to this data management challenge have been proposed6,62,63,64, none have been adopted by the wider biomolecular NMR community. The Dataset constitutes a significant step towards solving this problem. In the medium and long term, however, it would be best to establish the simultaneous deposition of primary NMR data (including time-domain FID data, frequency-domain spectra, and peak lists) together with chemical shift assignments and structures in public databases as a standard in structural biology. The necessary standardization and validation will likely require the public databases to provide seamless automated procedures such as those that have been developed, in part, for the preparation of the 100-protein NMR spectra data set.
Code availability
No custom code is required to access and use the ARTINA spectra database.
References
Wüthrich, K. NMR studies of structure and function of biological macromolecules (Nobel Lecture). J. Biomol. NMR 27, 13–39, https://doi.org/10.1023/a:1024733922459 (2003).
Hoch, J. C. et al. Biological Magnetic Resonance Data Bank. Nucleic Acids Res. 51, D368–D376, https://doi.org/10.1093/nar/gkac1050 (2023).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242, https://doi.org/10.1093/nar/28.1.235 (2000).
Everett, J. K. et al. A community resource of experimental data for NMR / X-ray crystal structure pairs. Protein Sci. 25, 30–45, https://doi.org/10.1002/pro.2774 (2016).
Rosato, A. et al. The second round of Critical Assessment of Automated Structure Determination of Proteins by NMR: CASD-NMR-2013. J. Biomol. NMR 62, 413–424, https://doi.org/10.1007/s10858-015-9953-4 (2015).
Fraga, K. J. et al. SpecDB: A relational database for archiving biomolecular NMR spectral data. J. Magn. Reson. 342, 107268, https://doi.org/10.1016/j.jmr.2022.107268 (2022).
Deng, J., et al ImageNet: A large-scale hierarchical image database. In Proc. IEEE-Computer-Society Conference on Computer Vision and Pattern Recognition Workshops, 248–255, https://doi.org/10.1109/cvpr.2009.5206848 (2009).
Klukowski, P., Riek, R. & Güntert, P. Rapid protein assignments and structures from raw NMR spectra with the deep learning technique ARTINA. Nat. Commun. 13, 6151, https://doi.org/10.1038/s41467-022-33879-5 (2022).
Klukowski, P., Riek, R. & Güntert, P. NMRtist: an online platform for automated biomolecular NMR spectra analysis. Bioinformatics 39, btad066, https://doi.org/10.1093/bioinformatics/btad066 (2023).
Goddard, T. D. & Kneller, D. G. (University of California, San Francisco; 2001).
Delaglio, F. et al. NMRPipe - A multidimensional spectral processing system based on Unix pipes. J. Biomol. NMR 6, 277–293, https://doi.org/10.1007/BF00197809 (1995).
Bartels, C., Xia, T. H., Billeter, M., Güntert, P. & Wüthrich, K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 6, 1–10, https://doi.org/10.1007/BF00417486 (1995).
Rosato, A. et al. CASD-NMR: critical assessment of automated structure determination by NMR. Nat. Methods 6, 625–626, https://doi.org/10.1038/nmeth0909-625 (2009).
Schmidt, E. & Güntert, P. A new algorithm for reliable and general NMR resonance assignment. J. Am. Chem. Soc. 134, 12817–12829, https://doi.org/10.1021/ja305091n (2012).
Liu, Z., Abbas, A., Jing, B. Y. & Gao, X. WaVPeak: picking NMR peaks through wavelet-based smoothing and volume-based filtering. Bioinformatics 28, 914–920, https://doi.org/10.1093/bioinformatics/bts078 (2012).
Alipanahi, B., Gao, X., Karakoc, E., Donaldson, L. & Li, M. PICKY: A novel SVD-based NMR spectra peak picking method. Bioinformatics 25, i268–i275, https://doi.org/10.1093/bioinformatics/btp225 (2009).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589, https://doi.org/10.1038/s41586-021-03819-2 (2021).
Li, J., Bennett, K. C., Liu, Y. C., Martin, M. V. & Head-Gordon, T. Accurate prediction of chemical shifts for aqueous protein structure on “Real World” data. Chem. Sci. 11, 3180–3191, https://doi.org/10.1039/c9sc06561j (2020).
Wunderlich, Z. et al. The protein target list of the Northeast Structural Genomics Consortium. Proteins 56, 181–187, https://doi.org/10.1002/prot.20091 (2004).
Kobayashi, N. et al. An automated system designed for large scale NMR data deposition and annotation: application to over 600 assigned chemical shift data entries to the BioMagResBank from the Riken Structural Genomics/Proteomics Initiative internal database. J. Biomol. NMR 53, 311–320, https://doi.org/10.1007/s10858-012-9641-6 (2012).
Strotz, D. et al. Protein allostery at atomic resolution. Angew. Chem. Int. Ed. 59, 22132–22139, https://doi.org/10.1002/anie.202008734 (2020).
Aramini, J. M. et al. Solution NMR structure of the SOS response protein YnzC from Bacillus subtilis. Proteins 72, 526–530, https://doi.org/10.1002/prot.22064 (2008).
Heikkinen, H. A., Backlund, S. M. & Iwaï, H. NMR structure determinations of small proteins using only one fractionally 20% 13C and uniformly 100% 15N-labeled sample. Molecules 26, 747, https://doi.org/10.3390/molecules26030747 (2021).
Aramini, J. M. et al. Solution NMR structure of the plasmid-encoded fimbriae regulatory protein PefI from Salmonella enterica serovar Typhimurium. Proteins 79, 335–339, https://doi.org/10.1002/prot.22869 (2011).
Skrisovska, L. & Allain, F. H. T. Improved segmental isotope labeling methods for the NMR study of multidomain or large proteins: Application to the RRMs of Npl3p and hnRNP L. J. Mol. Biol. 375, 151–164, https://doi.org/10.1016/j.jmb.2007.09.030 (2008).
Auweter, S. D. et al. Molecular basis of RNA recognition by the human alternative splicing factor Fox-1. EMBO J. 25, 163–173, https://doi.org/10.1038/sj.emboj.7600918 (2006).
Koga, N. et al. Principles for designing ideal protein structures. Nature 491, 222–227, https://doi.org/10.1038/nature11600 (2012).
Ramelot, T. A. et al. Solution structure of 4’-phosphopantetheine-GmACP3 from Geobacter metallireducens: A specialized acyl carrier protein with atypical structural features and a putative role in lipopolysaccharide biosynthesis. Biochemistry 50, 1442–1453, https://doi.org/10.1021/bi101932s (2011).
Barraud, P., Banerjee, S., Mohamed, W. I., Jantsch, M. F. & Allain, F. H. T. A bimodular nuclear localization signal assembled via an extended double-stranded RNA-binding domain acts as an RNA-sensing signal for transportin 1. Proc. Natl. Acad. Sci. USA 111, E1852–E1861, https://doi.org/10.1073/pnas.1323698111 (2014).
Moursy, A., Allain, F. H. T. & Cléry, A. Characterization of the RNA recognition mode of hnRNP G extends its role in SMN2 splicing regulation. Nucleic Acids Res. 42, 6659–6672, https://doi.org/10.1093/nar/gku244 (2014).
Ramelot, T. A. et al. NMR structure and MD simulations of the AAA protease intermembrane space domain indicates peripheral membrane localization within the hexaoligomer. FEBS Lett. 587, 3522–3528, https://doi.org/10.1016/j.febslet.2013.09.009 (2013).
Oeemig, J. S., Ollila, O. H. S. & Iwai, H. NMR structure of the C-terminal domain of TonB protein from Pseudomonas aeruginosa. PeerJ 6, 19, https://doi.org/10.7717/peerj.5412 (2018).
Daubner, G. M., Cléry, A., Jayne, S., Stevenin, J. & Allain, F. H. T. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 31, 162–174, https://doi.org/10.1038/emboj.2011.367 (2012).
Ramelot, T. A. et al. Structure of a specialized acyl carrier protein essential for lipid A biosynthesis with very long-chain fatty acids in open and closed conformations. Biochemistry 51, 7239–7249, https://doi.org/10.1021/bi300546b (2012).
Blatter, M. et al. The signature of the five-stranded vRRM fold defined by functional, structural and computational analysis of the hnRNP L protein. J. Mol. Biol. 427, 3001–3022, https://doi.org/10.1016/j.jmb.2015.05.020 (2015).
Aramini, J. M. et al. Structural basis of O6-alkylguanine recognition by a bacterial alkyltransferase-like DNA repair protein. J. Biol. Chem. 285, 13736–13741, https://doi.org/10.1074/jbc.M109.093591 (2010).
Scott, A. et al. Solution structure of the Src homology 2 domain from the human feline sarcoma oncogene Fes. J. Biomol. NMR 31, 357–361, https://doi.org/10.1007/s10858-005-0946-6 (2005).
Boudet, J. et al. A small helical bundle prepares primer synthesis by binding two nucleotides that enhance sequence-specific recognition of the DNA template. Cell 176, 154–166, https://doi.org/10.1016/j.cell.2018.11.031 (2019).
Yang, Y. H. et al. Solution NMR structure of photosystem II reaction center protein Psb28 from Synechocystis sp. strain PCC 6803. Proteins 79, 340–344, https://doi.org/10.1002/prot.22876 (2011).
Lytle, B. L. et al. Solution structure of a ubiquitin-like domain from tubulin-binding cofactor B. J. Biol. Chem. 279, 46787–46793, https://doi.org/10.1074/jbc.M409422200 (2004).
Ramelot, T. A. et al. Solution NMR structure of BT_0084, a conjugative transposon lipoprotein from Bacteroides thetaiotamicron. Proteins 80, 667–670, https://doi.org/10.1002/prot.23235 (2012).
Feldmann, E. A. et al. Solution NMR and X-ray crystal structures of Pseudomonas syringae Pspto_3016 from protein domain family PF04237 (DUF419) adopt a “double wing” DNA binding motif. J. Struct. Funct. Genom. 13, 155–162, https://doi.org/10.1007/s10969-012-9140-8 (2012).
Vinarov, D. A. et al. Cell-free protein production and labeling protocol for NMR-based structural proteomics. Nat. Methods 1, 149–153, https://doi.org/10.1038/nmeth716 (2004).
Pantoja-Uceda, D. et al. Solution structure of the rhodanese homology domain At4g01050(175-295) from Arabidopsis thaliana. Protein Sci. 14, 224–230, https://doi.org/10.1110/ps.041138705 (2005).
Aramini, J. M. et al. Solution NMR structure of the NlpC/P60 domain of lipoprotein Spr from Escherichia coli: Structural evidence for a novel cysteine peptidase catalytic triad. Biochemistry 47, 9715–9717, https://doi.org/10.1021/bi8010779 (2008).
Tang, Y. F. et al. Protein structure determination by combining sparse NMR data with evolutionary couplings. Nat. Methods 12, 751–U786, https://doi.org/10.1038/nmeth.3455 (2015).
Bhattacharjee, A. et al. Structural basis for complement evasion by Lyme disease pathogen Borrelia burgdorferi. J. Biol. Chem. 288, 18685–18695, https://doi.org/10.1074/jbc.M113.459040 (2013).
Rossi, P. et al. Structural elucidation of the Cys-His-Glu-Asn proteolytic relay in the secreted CHAP domain enzyme from the human pathogen Staphylococcus saprophyticus. Proteins 74, 515–519, https://doi.org/10.1002/prot.22267 (2009).
Lytle, B. L. et al. Solution structure of Arabidopsis thaliana protein At5g39720.1, a member of the AlG2-like protein family. Acta Crystallogr. F-Struct. Biol. Commun. 62, 490–493, https://doi.org/10.1107/s1744309106015946 (2006).
Güntert, P., Dötsch, V., Wider, G. & Wüthrich, K. Processing of multidimensional NMR data with the new software PROSA. J. Biomol. NMR 2, 619–629, https://doi.org/10.1007/BF02192850 (1992).
Bartels, C., Güntert, P., Billeter, M. & Wüthrich, K. GARANT - A general algorithm for resonance assignment of multidimensional nuclear magnetic resonance spectra. J. Comput. Chem. 18, 139–149, 10.1002/(SICI)1096-987X(19970115)18:1<139::AID-JCC13>3.0.CO;2-H (1997).
Güntert, P. & Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 62, 453–471, https://doi.org/10.1007/s10858-015-9924-9 (2015).
Klukowski, P., Riek, R. & Güntert, P. The 100-protein NMR spectra dataset: A resource for biomolecular NMR data analysis. ETH Research Collection, https://doi.org/10.3929/ethz-b-000630211 (2023).
Vuister, G. W. & Bax, A. Resolution enhancement and spectral editing of uniformly 13C enriched proteins by homonuclear broadband 13C-13C decoupling. J. Magn. Reson. 98, 428–435, https://doi.org/10.1016/0022-2364(92)90144-V (1992).
Güntert, P., Mumenthaler, C. & Wüthrich, K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298, https://doi.org/10.1006/jmbi.1997.1284 (1997).
Li, E. H. et al. Blind assessment of monomeric AlphaFold2 protein structure models with experimental NMR data. J. Magn. Reson. 352, 107481, https://doi.org/10.1016/j.jmr.2023.107481 (2023).
Tejero, R., Huang, Y. J., Ramelot, T. A. & Montelione, G. T. AlphaFold Models of Small Proteins Rival the Accuracy of Solution NMR Structures. Front. Mol. Biosci. 9, 18, https://doi.org/10.3389/fmolb.2022.877000 (2022).
Klukowski, P., Riek, R. & Güntert, P. Time-optimized protein NMR assignment with an integrative deep learning approach using AlphaFold and chemical shift prediction. Sci. Adv. 9, eadi9323, https://doi.org/10.1126/sciadv.adi9323 (2023).
Wetton, H., Klukowski, P., Riek, R. & Güntert, P. Chemical shift transfer: An effective strategy for protein NMR assignment with ARTINA. Front. Mol. Biosci. 10, 1244029, https://doi.org/10.3389/fmolb.2023.1244029 (2023).
Stahl, K., Graziadei, A., Dau, T., Brock, O. & Rappsilber, J. Protein structure prediction with in-cell photo-crosslinking mass spectrometry and deep learning. Nat. Biotechnol, https://doi.org/10.1038/s41587-023-01704-z (2023).
López-Méndez, B. & Güntert, P. Automated protein structure determination from NMR spectra. J. Am. Chem. Soc. 128, 13112–13122, https://doi.org/10.1021/ja061136l (2006).
Baran, M. C., Moseley, H. N. B., Sahota, G. & Montelione, G. T. SPINS: Standardized protein NMR storage. A data dictionary and object-oriented relational database for archiving protein NMR spectra. J. Biomol. NMR 24, 113–121, https://doi.org/10.1023/a:1020940806745 (2002).
Vranken, W. F. et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696, https://doi.org/10.1002/prot.20449 (2005).
Fenwick, M., Hoch, J. C., Ulrich, E. & Gryk, M. R. CONNJUR R: An annotation strategy for fostering reproducibility in bio-NMR: protein spectral assignment. J. Biomol. NMR 63, 141–150, https://doi.org/10.1007/s10858-015-9964-1 (2015).
Gutmanas, A. et al. NMR Exchange Format: a unified and open standard for representation of NMR restraint data. Nat. Struct. Mol. Biol. 22, 433–434, https://doi.org/10.1038/nsmb.3041 (2015).
Kirchner, D. K. & Güntert, P. Objective identification of residue ranges for the superposition of protein structures. BMC Bioinformatics 12, 170, https://doi.org/10.1186/1471-2105-12-170 (2011).
Acknowledgements
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 891690 (P.K.), and Grants-in-Aid for Scientific Research of the Japan Society for the Promotion of Science (P.G., 20K06508 and 23K05660). G.T.M. and T.A.R. acknowledge support by National Institutes of Health grant R35-GM141818.
Funding
Open access funding provided by Swiss Federal Institute of Technology Zurich.
Author information
Authors and Affiliations
Contributions
P.K. and P.G. prepared the database, performed technical validation, and wrote the manuscript draft. F.F.D., F.H.T.A., H.I., H.K., G.T.M., T.A.R., R.R., and P.G. contributed experimental data. R.R. and P.G. supervised the research. All authors reviewed and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
G.T.M. is a founder of Nexomics Biosciences, Inc. This does not represent a conflict of interest for this study. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klukowski, P., Damberger, F.F., Allain, F.HT. et al. The 100-protein NMR spectra dataset: A resource for biomolecular NMR data analysis. Sci Data 11, 30 (2024). https://doi.org/10.1038/s41597-023-02879-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02879-5
- Springer Nature Limited