
Abstract
Fusarium verticillioides is a filamentous fungus that causes plant diseases and harms human health through cancer-inducing mycotoxin and life-threatening Fusariosis. Given its threat to agriculture and public health, genome assembly of this fungus is critical to our understanding of its pathobiology and developing antifungal drugs. Here, we report a gap-free genome assembly of F. verticillioides using PacBio HiFi data and high-throughput chromosome capture (Hi-C) sequencing data. The assembled 42.0 Mb sequence contains eleven gapless chromosomes capturing all centromeres and 19 of all 22 telomeres. This assembly represents a significant improvement over previous version on contiguity (contig N50: 4.3 Mb), completeness (BUSCO score: 99.0%) and correctness (QV: 88.8). A total of 15,230 protein-coding genes were predicted, 6.2% of which are newly annotated genes. In addition, we identified three-dimension chromatin structures such as TADs-like structures and chromatin loops based on Hi-C data of ultra-high coverage. This gap-free genome of F. verticillioides is an excellent resource for further panoramic understanding mechanisms of fungal genome evolution, mycotoxin production and pathogenesis on plant and human host.
Similar content being viewed by others


Background & Summary
Fusarium verticillioides, a filamentous fungus belonging to Fusarium fujikuroi species complex, causes Fusarium ear rot of maize, a major crop worldwide. Besides yield loss, various mycotoxins are produced during fungal infection of maize, reducing the quality of corn products. The best characterized F. verticillioides mycotoxins are fumonisins, a group of polyketide mycotoxins associated with esophageal cancer and neural tube birth defects in human populations consuming the contaminated maize products1. Although F. veticillioides is considered nonpathogenic to healthy human being, it can become a serious threat to individuals with compromised immune system such as those infected by undergoing organ transplants2. Human infection by F. verticillioides commonly known as Fusariosis has been a surging life threat to the immunocompromised patients due to limited options of antifungal drugs for treatment and emergence of multi-drug resistance3. Therefore, elucidation of molecular mechanisms underlying fungal pathogenesis and antifungal resistance in F. verticillioides is crucial to both agricultural safety and public health.
Despite the importance of this fungus, its complete genome sequence has not been assembled and thoroughly analyzed, impeding dissection of molecular and evolutionary mechanisms underlying its pathogenesis, secondary metabolism and drug resistance. The first genome assembly of F. verticillioides strain 7600 was released in 20104 with a contig N50 of 392.3 kb. Recently, several updated versions of F. verticillioides genome assemblies are available in NCBI (National Center for Biotechnology Information) genome database. Although these genome assemblies have since facilitated the genetic studies of fungal biological processes, they are highly fragmented with several hundreds to thousands of contigs. The fact that F. verticillioides has 11 chromosomes suggests the presence of gaps in these assembly versions. Furthermore, no telomere and centromere sequences have been reported in any F. verticillioides genome assembly available, leaving these essential and complex genomic regions unexplored. A complete genome sequence for F. verticillioides would enable accurate characterization of the fungal genome function, regulation and evolution, shedding light on mechanisms of growth, development, pathogenicity and mycotoxin production.
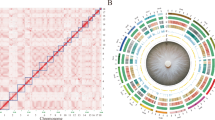
Here, we aim to produce a gap-free reference genome of F. verticillioides, and update the genome annotations based on the improved genome assembly. We sequenced the genome of F. verticillioides strain 7600 to produce high-fidelity (HiFi) long reads of PacBio (Pacific Biosciences, CA) single-molecule real-time (SMRT) sequencing, and Hi-C (high throughput chromatin conformation capture) data using Illumina pair-end sequencing. In total, we generated 4.1 Gb (~96.7X coverage) PacBio HiFi raw reads with a N50 of 10.0 kb. and 53.8 Gb Hi-C data (Illumina paired-end reads, ~1,272X coverage) (Table 1). For genome assembly, HiFi data were assembled using multiple tools including Hifiasm5, HiCanu6, NextDenovo (https://github.com/Nextomics/NextDenovo) and Flye7 to obtain draft genome assemblies which were individually polished using Nextpolish (v.1.4.0)8 followed by assembly merge using quickmerge (https://github.com/mahulchak/quickmerge) (Table 2). Then, Hi-C data were used to anchor the contigs onto chromosomes using Juicer9 and 3d-DNA pipeline10. The final genome assembly (42.0 Mb) contains 11 gap-free chromosomes (Figure 1a,b) with a contig N50 of 4.3 Mb, a significant improvement (+989.5%) compared to the previous version GCA_000149555.1 (contig N50 = 392.4 kb) (Table 3).

Overview of the gap-free reference genome and annotation of Fusarium verticillioides strain 7600. (a) Circos plot showing the gene features at 10 kb windows across the 11 chromosomes in F. verticillioides strain 7600. From outer to inner ring: ① chromosome ideogram, ② GC content, ③ gene density, ④ exon density, ⑤ TE (transposable element) density, ⑥ Simple repeat density, ⑦ t-RNA density, ⑧ Colony morphology photographed after 6-day incubation at 25 °C. (b) High-throughput chromatin conformation capture (Hi-C) interaction map of F. verticillioides strain 7600 visualizes the number of chromosome interactions within and between 11 chromosomes. (c) Violin plots of genomic features, including gene length, CDS length, exon length, mRNA length, three prime UTR (untranslated region), five prime UTR, tRNA length, TE class I, TE class II.
For the gap-free assembly, we performed genome annotation to predict protein-coding genes and repeat elements. To see how much a nearly complete genome sequence improves genome annotations, the same annotation pipeline and RNA-seq data were applied to annotation of both our assembly and the previous version GCA_000149555.1. For protein-coding genes, the two genome assemblies were comparable where our assembly encodes 15,230 genes, a slight increase (+6.2%) compared to the previous assembly (Table 3; Fig. 1c). Comparing the two annotations revealed 15,056 genes present in both genome assemblies while 75 and 174 genes were uniquely annotated using previous and our genome assembly, respectively. The new genome assembly contains 2.8% (1,164,494 bp) repeat content, higher than the previous version (1.7%, 708,545 bp). Specifically, our assembly contains 120,266 bp LTR (long terminal repeat) element (+102.9%) and 102,640 bp DNA transposon (+2,608.2%) (Table 3).
Compared to previous genome assemblies, this gap-free genome assembly of F. verticillioides contained all centromeres on 11 chromosomes (Fig. 2a), thanks to the highly accurate HiFi sequence data and improved assembly algorithms. To validate the centromere regions, we mapped the HiFi reads and RNA-seq reads to the gap-free assembly. We found a decent coverage of HiFi reads throughout the assembly including the centromeres (Figure 2b,c) and telomeres (Figure 2d,e) which contained no protein-coding genes and little RNA-seq alignment. By comparing this assembly with a previous assembly (GCA_000149555.1), we showed that numerous gaps were closed and three large inversions on the short arms of Chr3, Chr10 and Chr11 were corrected in this new assembly (Fig. 3a). Furthermore, unplaced scaffolds in GCA_000149555.1 are now anchored to correct chromosome positions (Fig. 3b). The gapless assembly contained a total of 890 kb new sequences including 25 kb to 231 kb per chromosomes which were absent in GCA_000149555.1 chromosomes (Fig. 3c).

Features and validation of telomeres and centromeres of Fusarium verticillioides strain 7600. (a) Dotplot of F. verticillioides centromere sequences assembled in this study visualized using GePard. (b,c) IGV (integrative genomics viewer) visualization of centromere regions (dashed red box) where PacBio HiFi and RNA-seq reads are mapped, from chromosomes 1 and 2, respectively. (d,e) IGV visualization of two telomere regions of chromosome 10 where PacBio HiFi and RNA-seq reads are mapped.

Fusarium verticillioides strain 7600 gap-free genome assembly represents a major improvement over the previous version. (a) Comparison of F. verticillioides genome characteristics between the previous version (GCA_000149555.1) and the gap-free assembly in this study. (b) Dotplot displaying the alignment of unplaced sequence of the previous genome against the gap-free chromosomes assembled in this study, indicating successful chromosome anchor of these sequences. (c) Lollipop plot summarizing the length of newly assembled sequences per chromosome compared to the previous version of the genome.
Lastly, we analyzed the three-dimensional genome of F. verticillioides based on the Hi-C sequencing data, generated from fungal mycelia collected from culture. With a total of 53.8 Gb (1272.8X coverage) Hi-C data containing 95.8% valid interaction pairs after initial quality control (Fig. 4a), from which we identified 60 TADs (topological associated domains) -like structures and five chromosome loops under 10 kb resolution (Table 4; Supplementary Table 1; Fig. 4b,c). Various candidate protein-coding genes were localized within the TADs-like and loop structures (Table 4; Supplementary Table 1; Fig. 4d). This gap-free genome assembly and updated annotation of F. verticillioides are excellent resources to study mechanisms of fungal genome evolution, mycotoxin production and pathogenesis on plant and human host.

Characteristics of the three-dimensional genome of Fusarium verticillioides strain 7600. (a) Donut chart summarizing the results of Hi-C data quality control performed by HiC-Pro. (b,c) Three-dimensional genomic feature (Hi-C matrix, A/B compartment, TADs(topological associated domains)-like structures and chromatin loops) for chromosome 3 and 4. (d) Boxplot summarizing the number of genes co-localized within TADs-like regions on each chromosome.
Methods
Fungal culture, DNA preparation and PacBio HiFi sequencing
F. verticillioides strain 7600 was routinely maintained on PDA (potato dextrose agar) slant and stored in −80 °C freezer. F. verticillioides 7600 mycelia and spores harvested from two-day old PDB (potato dextrose broth) culture in 150 rpm shaker at 25 °C were used to isolate high molecular weight DNA using CTAB (cetyltrimethylammonium bromide) method11. A total of 15 µg purified genomic DNA were used to construct a standard PacBio SMRTbell library using PacBio SMRT Express Template Prep Kit 2.0 (Pacific Biosciences, CA). The sequencing was performed using a PacBio Sequel II instrument at Biomarker Technologies Corporation (QingDao, China).
Hi-C sequencing and analysis
Hi-C library construction of F. verticillioides was prepared from cross-linked chromatins of fungal mycelia using a standard Hi-C protocol12. The constructed Hi-C sequencing library was sequenced by a test run and examined for valid interaction read pair ratios using HiCPro (v.3.1.0)13 before going through high coverage sequencing. The library was sequenced by Illumina NovaSeq. 6000 to yield 10.5 Gb (249.7 coverage) paired-end reads. The valid interaction pairs of Hi-C sequencing reads were used to anchor all contigs using Juicer (v.1.5)9, followed by using a 3D-DNA correction pipeline10 and manually correction with Juicebox (v.1.11.08)14. compartment A/B were analyzed using HiTC (v.1.40.1)15 and Cworld-dekker (https://github.com/dekkerlab/cworld-dekker), TADs-like structures and chromosome loop were identified by Juicer (v.1.5)9. Three-dimensional structure visualization of the whole genome using pyGenomeTracks (v.3.7)16.
Genome assembly
To optimize the genome assembly strategy and take into account the differences of assembly algorithm between software, we used Hifiasm (v.0.16.1)5, HiCanu (v.1.4)6 (parameters: -assemble -pacbio-hifi oeaErrorRate = 0.001), Flye (v.2.9)7 and NextDenovo (v.2.5.0) (https://github.com/Nextomics/NextDenovo, with parameters: minimap2_options_cns = -x ava-hifi), to assemble, respectively, and then sorted the number of contigs of different assemblers in ascending order. Based on four assemblies we used quickmerge (v.0.3) (https://github.com/mahulchak/quickmerge) to produce a merged genome assembly, and finally used Juicebox14 to manually adjust misassemblies.
RNA sequencing and analysis
Total RNA was extracted from the mycelia of F. verticillioides using Trizol (Thermal Fisher) agents following manufacturer recommendation protocol. The RNA Nano 6000 Assay Kit of Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA) was used to evaluate the total RNA integrity. The total RNA used for library preparation first enriched the mRNA with polyA tail through Oligo (dT) magnetic beads. The mRNA was then subjected to sequencing library construction using Illumina True-seq transcriptome kit (Illumina, CA) with an insert size of 370bp–420bp, and sequenced by an Illumina Novaseq. 6000 platform at Biomarker Technologies Corporation (QingDao, China) to generate 150 bp paired-end reads. RNA-seq data was checked for quality using fastp (v.0.23.2)17, mapped to F. verticillioides genome assembly using hisat2 (v.2.1.0)18, followed by calculating mapping ratios by samtools (v.1.15)19.
Identification of gene model and prediction of repeat sequences and non-coding RNA
For repetitive sequences, we firstly use de novo prediction and similarity aligmment to annotate it via RepeatModeler (v. 1.0.11)20 (parameters: -database -engine ncbi -pa) and softmasked genome by RepeatMasker (v. 4.1.2.p1)21. RepeatMasker’s perl script (rmOutToGFF3.pl) converts various types of repeat sequence annotation results into a common generic feature format (GFF) version 3. Gene model prediction combined with the following three aspects of evidence: (a) ab initio prediction, (b) homologous protein, (c)RNA-seq evidence. During the ab initio prediction, we firstly trained the GeneMark-ET model for five rounds using BRAKER2 (v.2.1.6)22 (parameters:–species = Fv–fungus–softmasking–genome–bam–prot_seq–prg = gth–gff3–rounds = 5), whose process employed GeneMark-ET23, NCBI BLAST24, DIAMOND25 and GenomeThreader26. We then trained the SNAP27 semi-HMM model for two rounds using MAKER28 (parameters: est2genome = 1, protein2genome = 1, pred_flank = 100, alt_splice = 1, correct_est_fusion = 1). AUGUSTUS29 used the built-in Fusarium genome feature model. To provide homologous protein evidences for gene prediction, we downloaded the protein data of this species (anchored chromosomes) from the public database, including 7600 (NCBI Assembly ID: GCA_ 000149555.1), BRIP53590 (NCBI Assembly ID: GCA_003316995.2), BRIP53590 (NCBI Assembly ID: GCA_003317015.2) and BRIP14953 (NCBI Assembly ID: GCA_003316975.2). For transcriptome data, RNA-seq reads from the vegetative phase were firstly aligned to our genome assembly through hisat218 for BRAKER2 (v.2.1.6)22. Then, we performed reference-based assembly and de novo assembly of transcriptomes by Scallop (v0.10.5)30 and Trinity (v.2.8.4)31 (parameters:–min_kmer_cov 3–normalize_max_read_cov 100), respectively. Transcripts obtained by two methods are de-redundant with CD-HIT (v.4.6)32 (parameters:-I -c 0.99 -T 50 -M 100000 -o). The above three evidences are integrated by MAKER (v.3.01.03)28 to predict the final gene model. Rfam/Infernal (v.1.1.4)33 (parameters: cmscan–cut_ga–rfam–nohmmonly–fmt 2 –clanin –tblout) and tRNAscan-SE (v. 2.0.9)34 (parameters: -E -X 20 –f -m -b -j–detail) are used to infer genome-wide non-coding RNAs. To compare the previous (NCBI: GCA_000149555.1) and our genome assembly, we performed the genome annotation on two genome assemblies by using the same software, parameters, and protein data.
Data Records
The raw PacBio HiFi sequencing data, Hi-C data and RNA-seq data have been deposited in the National Center for Biotechnology Information (NCBI) under the BioProject (PRJNA868307)35 with accession number of SRR2100352136, SRR2100352037, SRR2100351938, respectively. The gap-free genome assembly is deposited under the same BioProject at NCBI (GCA_027571605.1) and also in Genome Warehouse of National Genomics Data Center (https://ngdc.cncb.ac.cn/) at China National Center for Bioinformation under the accession number of GWHBQEB0000000039. Genome annotations including protein-coding regions, repeat sequence and ncRNA annotation files have been submitted to the online open access repository Figshare40.
Technical Validation
Manual adjustment of misjoin and detection of potentially contaminated sequences
To get a nearly complete and error-free nuclear genome, we first manually corrected the assembly using Hi-C read alignment within the Juicebox14. We then aligned the species’ mitochondrial genome to our assembly by megaBLAST24, which found no errors. Finally, we also used megaBLAST24 to aligned our genome assembly against a common database (ftp://ftp.ncbi.nlm.nih.gov/pub/kitts/contam_in_euks.fa.gz) to identify potentially contaminated sequences sequencing adaptor sequence (ftp://ftp.ncbi.nlm.nih.gov/pub/kitts/adaptors_for_screening_euks.fa) and nucleotide sequence database (remote mode), which again found no contamination.
Evaluation of the genome assembly
The genome assembly was validated by two independent methods. Firstly, HiFi reads were mapped to the assembly using Winnowmap2 (v.2.03)41 (parameters: -W repetitive_k15.txt -t 104 -ax map-pb) and the quality value (QV) was assessed using Merqury (v.1.3)42 (parameters: k = 18 count). Second, the BUSCO (Benchmarking Universal Single-Copy Ortholog)43 analysis was conducted to reflect the completeness of genome assembly. The final F. verticillioies gap-free genome assembly has a QV of 88.8, completeness of 99.7% and BUSCO score of 99%, suggesting the high accuracy and completeness of the assembly, respectively (Table 2).
Validation of the genome assembly
The resolved fungal telomere and centromere regions have been well covered by PacBio HiFi reads that span these complex regions (Fig. 2) by IGV (v.2.4.10)44. This assembly has reduced the length of gaps from 90,816 in previous version to 0, and captured eleven centromeres and nineteen telomeres (TTAGGG) except missing three telomeres via trf (v. 4.09.1)45 (parameters: 2 7 7 80 10 90 2000 -d -m -l 2) from assemblies and raw sequences, one each at the end of Chr2 and Chr4 (Fig. 3). There is a one-to-one correspondence between the old and new versions of the genome with 14,260 coding region genes via liftoff (v.1.6.3)46 and BEDtools (v.2.30.0)47 (parameters: intersect -wa -wb -f 1.0), which account for 99.5% of the old version genome and 93.6% of this study genome. Compared to previous version (NCBI: GCA_000149555.1), our assembly has corrected three major inversions (Fig. 3) located at the short arm of Chr3, Chr10 and Chr11 visualized via GenomeSyn48 plot.
Code availability
All software used in this study are in the public domain, with parameters being clearly described in Methods and this section. If no detail parameters were mentioned for the software, default parameters were used as suggested by developer.
References
Missmer, S. A. et al. Exposure to fumonisins and the occurrence of neural tube defects along the Texas-Mexico border. Environ Health Perspect 114, 237–241, https://doi.org/10.1289/ehp.8221 (2006).
Muhammed, M. et al. Fusarium infection: report of 26 cases and review of 97 cases from the literature. Medicine (Baltimore) 92, 305–316, https://doi.org/10.1097/MD.0000000000000008 (2013).
Nucci, M. & Anaissie, E. Fusarium infections in immunocompromised patients. Clin Microbiol Rev 20, 695–704, https://doi.org/10.1128/CMR.00014-07 (2007).
Ma, L. J. et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373, https://doi.org/10.1038/nature08850 (2010).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res 30, 1291–1305, https://doi.org/10.1101/gr.263566.120 (2020).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nature biotechnology 37, 540–546 (2019).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2020).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Allen, G. C., Flores-Vergara, M. A., Krasynanski, S., Kumar, S. & Thompson, W. F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat Protoc 1, 2320–2325, https://doi.org/10.1038/nprot.2006.384 (2006).
Belton, J. M. & Dekker, J. Hi-C in Budding Yeast. Cold Spring Harb Protoc 2015, 649–661, https://doi.org/10.1101/pdb.prot085209 (2015).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259, https://doi.org/10.1186/s13059-015-0831-x (2015).
Durand, N. C. et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Servant, N. et al. HiTC: exploration of high-throughput ‘C’ experiments. Bioinformatics 28, 2843–2844, https://doi.org/10.1093/bioinformatics/bts521 (2012).
Lopez-Delisle, L. et al. pyGenomeTracks: reproducible plots for multivariate genomic data sets. Bioinformatics (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 5, 4.10. 11–14.10. 14 (2004).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom Bioinform 3, lqaa108, https://doi.org/10.1093/nargab/lqaa108 (2021).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic acids research 42, e119–e119 (2014).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Gremme, G. Computational Gene Structure Prediction, (2013).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59, https://doi.org/10.1186/1471-2105-5-59 (2004).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491, https://doi.org/10.1186/1471-2105-12-491 (2011).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Shao, M. & Kingsford, C. Accurate assembly of transcripts through phase-preserving graph decomposition. Nature biotechnology 35, 1167–1169 (2017).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology 29, 644–652 (2011).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res 49, 9077–9096, https://doi.org/10.1093/nar/gkab688 (2021).
Yao, G. This study aimed to obtain high quality genomic sequence of Fusarium verticillioides. BioProject https://identifiers.org/ncbi/bioproject:PRJNA868307 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21003521 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21003520 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21003519 (2022).
Yao, G. Fusarium verticillioides 7600, whole genome sequencing project. NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/30265/show (2022).
Yao, G. The annotated file for Fusarium verticillioides strain 7600. Figshare https://doi.org/10.6084/m9.figshare.20465889.v6 (2022).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat Methods 19, 705–710, https://doi.org/10.1038/s41592-022-01457-8 (2022).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol Biol Evol 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Robinson, J. T. et al. Integrative genomics viewer. Nat Biotechnol 29, 24–26, https://doi.org/10.1038/nbt.1754 (2011).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Shumate, A. & Salzberg, S. L. Liftoff: accurate mapping of gene annotations. Bioinformatics 37, 1639–1643 (2021).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, https://doi.org/10.1093/bioinformatics/btq033 (2010).
Zhou, Z. W. et al. GenomeSyn: A bioinformatics tool for visualizing genome synteny and structural variations. J Genet Genomics S1673–8527(1622), 00104–00107, https://doi.org/10.1016/j.jgg.2022.03.013 (2022).
Acknowledgements
This project was supported by the National Natural Science Foundation of China (31970317) and the Fundamental Research Fund of Peking University Institute of Advanced Agricultural Sciences. LG is also supported by Taishan Scholars Program. We would like to thank the Bioinformatics Platform at Peking University Institute of Advanced Agricultural Sciences for providing the high-performance computing resources.
Author information
Authors and Affiliations
Contributions
L.G. conceived and supervised the study. H.W. collected the samples, conducted DNA extraction and conducted sequencing. G.Y., W.C., J.S., X.W. and T.M. performed bioinformatic analysis, prepared figures and tables. G.Y., L.Z. and L.G. wrote and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yao, G., Chen, W., Sun, J. et al. Gapless genome assembly of Fusarium verticillioides, a filamentous fungus threatening plant and human health. Sci Data 10, 229 (2023). https://doi.org/10.1038/s41597-023-02145-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02145-8
- Springer Nature Limited
This article is cited by
-
Technology-enabled great leap in deciphering plant genomes
Nature Plants (2024)