
Abstract
Listeria monocytogenes (Lm) is a ubiquitous bacterium that causes listeriosis, a serious foodborne illness. In the nature-to-human transmission route, Lm can prosper in various ecological niches. Soil and decaying organic matter are its primary reservoirs. Certain clonal complexes (CCs) are over-represented in food production and represent a challenge to food safety. To gain new understanding of Lm adaptation mechanisms in food, the genetic background of strains found in animals and environment should be investigated in comparison to that of food strains. Twenty-one partners, including food, environment, veterinary and public health laboratories, constructed a dataset of 1484 genomes originating from Lm strains collected in 19 European countries. This dataset encompasses a large number of CCs occurring worldwide, covers many diverse habitats and is balanced between ecological compartments and geographic regions. The dataset presented here will contribute to improve our understanding of Lm ecology and should aid in the surveillance of Lm. This dataset provides a basis for the discovery of the genetic traits underlying Lm adaptation to different ecological niches.
Measurement(s) | whole genome sequencing |
Technology Type(s) | Illumina Sequencing |
Factor Type(s) | Multi-locus sequence types • Geographic location • Animal associated environment isolates • Food product and food production environment isolates |
Sample Characteristic - Organism | Listeria monocytogenes |
Sample Characteristic - Environment | Farm • Ruminant • Agricultural soil • Wild animals • food processing building • dairy food product • meat or meat product (from mammal) (us cfr) • chicken meat food product • fish food product • vegetable or vegetable product (us cfr) |
Sample Characteristic - Location | Europe |
Similar content being viewed by others


Background & Summary
Listeria monocytogenes (Lm) is a facultative intracellular pathogen responsible for listeriosis, a serious disease affecting both humans and animals. Lm is a ubiquitous bacterium that is found in various ecological niches, including the natural and farm environments1,2. In particular, soil is a primary ecological niche of Lm and may thus be important in its transmission from natural/farm environment to food and food-processing environment (FPE)1,2. Farm animals, in particular ruminants, are also an additional important reservoir for Lm and contribute to contamination of the farm environment through fecal shedding3,4. In addition, Lm can persist for a long time in soil and the farm environment. Increasing amounts of information are also available on the prevalence of Lm in wildlife, showing that various animal species (e.g., deer, wild boars, bears, foxes, monkeys, rodents, hedgehogs, snails, slugs and birds) can act as a vehicles for this pathogen5,6,7,8,9,10,11. These findings point to an ecological role of wildlife as a reservoir of Lm and its potential importance in Lm infection cycle.
Lm is genetically heterogeneous species divided into four phylogenetic lineages, of which lineages I and II are the most frequently encountered. Multilocus sequence typing (MLST) classifies Lm into clonal complexes (CCs) and sequence types (STs), which are systematically used to describe its population structure12,13,14. Certain epidemiological clones account for the majority of outbreaks and sporadic cases in humans15 and animals16, worldwide13,17. The CCs that are commonly found in food and FPE, such as the most common CC9 and CC121, but also CC1, CC2, CC4, CC5, CC6, CC8 and CC3718, pose a serious challenge in food industry15,18,19. Moreover, they can persist in FPE for several years20,21,22,23,24. Remarkably, CC9 and CC121 are rarely reported in animals or natural/farm environments18,25.
In order to improve surveillance and the management of health risks associated with Lm, a deeper understanding of the genetic make-up of strains adapted to food and FPE is required. As part of the Horizon 2020 “One Health” European Joint Programme, the 3-year research project “LISTADAPT” (Adaptive traits of Listeria monocytogenes to its diverse ecological niches - https://onehealthejp.eu/jrp-listadapt/) aimed to identify the genetic mutations and mobile genetic elements underlying the adaptation of Lm to different ecological niches. With this objective in mind, strains were collected from i) farm environment and animals and ii) natural environment and wild animals to study their genetic make-up and to compare this background with that of strains isolated from food products and FPE. This work was made possible due to the LISTADAPT consortium which included (i) seven national reference laboratories (NRLs) for surveillance of Lm in food, animals and the environment (AT, CZ, DK, FR, IT, NO and SE) and (ii) three research laboratories at INRAE (the French National Research Institute for Agriculture, Food and Environment). Out of the seven NRLs, two are also national public health laboratories (AT and CZ) that are in charge of the surveillance of clinical strains isolated in outbreaks and sporadic cases. In addition, 14 institutes from 12 countries participated as external partners providing isolates.
In this data descriptor, we present a dataset of 1484 high-quality draft genomes originating from Lm strains isolated in 19 European countries within the framework of the LISTADAPT project. The constructed dataset cover a wide genetic diversity of Lm since it includes about 79 different CCs and singleton STs including the most prevalent CCs in Europe15 and worldwide13,17. The strains were collected from natural environment (wild animals and natural environment), primary production (farm environment and farm animals with or without listeriosis symptoms) until FPE and food products.
The constructed dataset provides a better understanding of the Lm transmission routes from the farm/natural environment to food and FPE and improves our understanding of its ecology. The dataset may also help to assess the importance of animal and food strains for human infection. Moreover, it can be used by the scientific community (i) to improve our understanding of the Lm population structure and the Lm evolutionary history, (ii) to facilitate the detection of the emerging Lm clones and (iii) to identify genetic traits related to the adaptation of Lm to particular ecological niches (ecophysiology). Such genetic traits could be used in the development of molecular assays for screening of food/FPE, animal and soil reservoirs.
Methods
Construction of the LISTADAPT dataset (n = 1484)
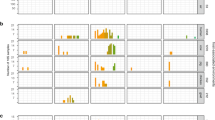
In order to build a dataset of Lm draft genomes suitable for investigating the adaptive traits of Lm to diverse ecological niches, we gathered a curated collection of Lm draft genomes. Strains isolated over the period 2010–2020 were preferred, regardless of their origin of isolation. We considered two geographic levels, (i) the 27 EU countries including Norway and Switzerland, heterogenous in size, population, climate, ecology and economical activities and (ii) based on country borders four European regions roughly equal in terms of surface area without consideration for other criteria (South-West, Central-South, Eastern and Northern). We included strains that were distributed evenly among these four European regions. The strain were gathered from already available strain collections and extensive sampling campaigns (Fig. 1). The LISTADAPT dataset was divided into two main ecological compartments: (i) C1 compartment, which included strains from animals and the natural/farm environment (n = 756), and (ii) C2 compartment, which included strains from food (n = 728) (Table 1).

Distribution of the LISTADAPT collection of Listeria monocytogenes strains (n = 1484) by time, geographic region and origin of isolation. (a) and (b) show the distribution of food strains by geographic region and food type, respectively. (c) and (d) show the distribution of environmental strains by geographic region and subcompartment, respectively.
Strains selected from the initial collection of the LISTADAPT consortium
At the beginning of the LISTADAPT project, the consortium had access to a collection of about 8000 food and animal Lm strains obtained from collaborative projects or national surveillance. Most of these strains were isolated from food, whereas the remainder were isolated from animals (C1 compartment: animal and environmental strains) with a substantial under-representation of certain animal species. This compartment mainly included strains isolated from animals showing listeriosis-related symptoms. Few strains were available from asymptomatic animals, soil and the agricultural environment, originating from three European countries (France, Italy and the Czech Republic).
Animal and environmental strains included in the collection during the LISTADAPT project (n = 756)
We collected isolates from animals showing listeriosis associated symptoms, asymptomatic animals, soil and the environment, in a large number of countries across Europe. These strains were isolated between 1978 and 2019. Regarding environmental niches, the consortium selected strains from continental environments remote to cities, large rivers and estuaries or marine environment to avoid the selection of human or food strains released in the environment, detailed strain information were provided in Figshare File 126. However, the six strains described by Szymczak et al.27 (Table 2) were isolated from city outskirts parks in Poland, distant from the city center. Similarly, the 47 strains from birds (mainly seagulls) (Hellström et al.)10 were isolated from localities from on the outskirts of Helsinki, Finland (Table 2).
Strains obtained from existing microbial collections (n = 648)
To increase the size and representativeness of the Lm genome dataset the LISTADAPT consortium performed an extensive review of all recent collections of published and unpublished Lm strains and then contacted researchers in charge of these collections. Finally, 14 external partners, food and veterinary laboratories and research institutes, all dealing with Lm hazards in Europe, collaborated with the LISTADAPT consortium (Tables 2 and 3).
The initial collection included more strains from animals with listeriosis-associated clinical symptoms than without symptoms. In order to reduce the number of strains originating from animals with listeriosis while maintaining maximum diversity of the dataset, we adopted an original method to select the strains based on metadata (e.g., type of sample, geographic location, time of isolation, molecular typing data such as PFGE profiles, animal species and geographic sampling location). This method relies on Gower’s coefficient (GC), which is a dissimilarity measure: the “distance” between two units is the sum of all the variable-specific distances (associated with metadata categories). The GC metric enables the combination of numeric and categorical data and enables applying weights to each variable, effectively altering the importance of each metadata category (e.g., geographical region as a more important category than year of isolation). The three steps are: (i) calculating the dissimilarity matrix based on Gower’s distance (ii) clustering the dissimilarity matrix with hierarchical clustering (agglomerative bottom-up approach of clustering) and (iii) assessing clusters with the “Silhouette” method. The silhouette plot displays a measure of how close each point in one cluster is to the points in the neighboring clusters. An R script available at https://github.com/lguillier/LISTADAPT/tree/master/metadata2assocation was used to perform the selection of strains based on this method. This script takes as input a Comma Separated Values (CSV) file that includes strain ID and metadata information, then outputs a CSV file of selected strains.
In the present study, we constructed a large dataset comprising 301 animal and environmental Lm strains from six European countries and published collections (Table 2), as well as 347 animal and environmental Lm strains from 12 European countries that were obtained from non-published collections (Table 3).
Strains collected from sampling campaigns (n = 108)
Soil, farm, and wild animal samples were collected in nine European countries (Table 4). For the collection of soil samples, the LISTADAPT project members raised awareness and organised crowd-sampling campaigns. All the soil samples were collected from agricultural or wild areas according to a common procedure provided to the samplers based on the existing recommendations reported in the literature2,28,29,30. The integration of feedback from samplers enabled a continuous improvement of the sampling protocol. The sampling campaigns were conducted in 17 areas in seven EU member states, Norway and Switzerland (Figs. 1 and 2, Table 4), namely AT, CH, CZ, FR, IT, NO, SE, SI and SK, resulting in the isolation of 58 Lm strains. Out of the 1752 available sampling records, the overall prevalence was 3%. We confirm in the present study the low prevalence of Lm in soil reported in the literature (below 1% and up to 6% depending on soil type)2,29. Soil strains from AT, FR, SI and SE were isolated by employing a two-step specific enrichment: the first enrichment was performed with modified Listeria Enrichment Broth for 24 h at 30 °C, followed by enrichment in University of Vermont Medium (UVM) enrichment broth for 48 h at 30 °C. Detection of Listeria spp. and Lm was then achieved by specific SYBR Green real-time PCR targeting prs2 and inlA genes, respectively. The samples positive for the presence of Listeria spp. and/or Lm were spread on RAPID’L.Mono agar plates (BioRAD, France). After 24 h incubation at 37 °C, colonies characteristic of Lm and other Listeria species were picked, purified and stored at –80 °C in Tryptone Soya Broth supplemented with 25% (v/v) glycerol. Strains from CH, CZ, IT and SK were isolated with the EN ISO 11290-1:2017 protocol (Horizontal method for the detection and enumeration of Lm and of Listeria spp.).

Microreact screenshot representing the distribution of the whole LISTADAPT dataset (n = 1484) by geographic region (a) and time (b). The k-mer-based phylogenomic clustering of the complete dataset is shown in (c). Interactive access to strain metadata and MLST types is available through Microreact44, a recently developed online tool for visualizing and sharing spacio-temporal and genetic distributions of strains (Fig. 2, accession link: https://microreact.org/project/8YtGBqEqhosJtysXTVY79M-figure-2-distribution-of-the-whole-listadapt-dataset-n1484-by-geographic-region-time-and-genetic-diversity). The dataset interactive map was generated using either the exact GPS coordinate, regional GPS coordinate or national GPS coordinate according to the level of details available for each strain. An annual timescale was used. The core genome MLST (Moura et al.) tree was generated from the draft genome assemblies using pairwise categorical difference and single linkage method in BioNumerics. The tree revealed three main clades corresponding to Lm phylogenetic lineages. Each clade included several clusters corresponding to MLST types (CC and singleton ST). Circles in shade of blue show food product isolates (clear blue: fish product, greeblue: dairy products, blue: composite dishes, deep blue: meat products). Circles in shade of orange show animal and environment isolates (beige: soil & farm environment, golden: wild animal, deep orange: farm animals). Circles size is proportional to the number of strains included.
Regarding the subcompartments of farm and wild animal, 50 Lm strains were isolated from sampling campaigns. Three campaigns targeting shelled gastropods sampled in IT, SK and CH resulted in the isolation of six strains (Figs. 1 and 2, Table 4). Sampling campaigns were also carried out for wild deer and reindeer feces in Southern Norway, and from cattle, roe deer, wild boar, wolf, bear and fox feces in the Abruzzo and Molise regions of Italy (Fig. 1, Table 4). Of the 2577 samples collected from vertebrates during the campaign conducted in IT and NO 41 isolates were detected, with an overall prevalence of 1.6%.
Food strains included in the collection during the LISTADAPT project (n = 728)
The food strains (C2 compartment) were classified according to the five main categories of risk food matrices for Lm defined by the European Food Safety Authority (EFSA)31: dairy products (n = 119), fish and fishery products (n = 165), meat products (n = 246), vegetables and fruits (n = 95), and composite dishes (food products combining several food categories) (n = 103). Six NRL project partners (AGES, ANSES, DTU, IZSAM, SLV and VRI) were instructed to target a maximum of 30 strains per food category from their strain collections, preferring strains isolated in the last 10 years. This time period was extended to the under-represented categories (vegetables and fruits); the final dataset included strains originating from the 2002–2020 period. We excluded raw materials from the selection based on the assumption that they could be contaminated by strains originating from farms or animals. The 728 strains from C2 compartment were isolated along the food chain, from food processing plants to food retail in several EU countries (Table 1), detailed strain information were provided in Figshare File 126
Complete LISTADAPT dataset (n = 1484)
The final LISTADAPT strain dataset that we constructed in collaboration with external partners was balanced with regard to the two main compartments: C1 (animals/environment, n = 756) and C2 (food/FPE, n = 728) (Table 1). The geographic distribution covered 19 of the 27 EU countries plus Norway and Switzerland (Figs. 1 and 2).
Although the C1 compartment (n = 756) covered a 41-year period (1978–2019), most of the strains (75%) were isolated since 2010. This panel covered all successive years between 2009 and 2019 in at least three European regions (Fig. 1c). Between 2008 and 2019, except for the year 2013, the C1 compartment covered all successive years in the following three categories of subcompartments: farm animals, wild animals and natural/farm environment (Fig. 1d).
Although the C2 compartment (n = 728) covered an 18-year period (2002–2020), most of the strains (78%) were recent, i.e. having been isolated between 2013 and 2019 (Fig. 1b). This panel covered all successive years between 2013 and 2019, as well as the five major categories in at least three European regions (Fig. 2b).
Strains from C1 compartment were isolated from more countries (n = 18) than strains from C2 compartment (n = 6). Finally, the majority (1135 of 1484 strains, 76%) of strains from both compartments originated from the period 2011–2019 (Fig. 1a,c).
Overall, the 1484 strains clustered into 137 MLST STs, which belonged to 54 CCs and 25 singleton STs (Fig. 3). For 22 strains, the allele profile was unknown (novel ST) or incomplete (When six out of seven MLST alleles were present, a CC was assigned when possible).

Distribution of the LISTADAPT dataset of Listeria monocytogenes genomes by multilocus sequence typing clonal complex (CC) or singleton sequence type (ST).
Standard strain nomenclature
In order to facilitate data sharing between partners, we adopted a standard nomenclature for strain identification (ID). This nomenclature was used as metadata codification to allow for fast identification of the geographic region and detailed isolation source of the strains (e.g., wild animal, food product or farm environment). In more detail, the LISTADAPT code has between 10 and 15 characters; the first two letters (level 1) correspond to the country code (ISO 3166-1-alpha-2 code), which is followed by a code detailing the origin of the strain and the sample type (level 2 to 4, depending on the nature of the source). Briefly, level 2 details the type of sample (e.g., animal species, environment and food categories) and level 3 details the nature of the sample (e.g., type of animal sample, type of food and nature of environmental sample). The level 4 gives additional information about the sample (e.g. type of preparation for aliments and health status of the animals). The code ends with a sequential number for each country, generated when the strain was added to the collection. For example: the strain DE-RDE-CP-13 was isolated in Germany (DE) from a roe deer (RDE) as a clinical strain (CP) and it was the 13th strain isolated from Germany included in the dataset. The Supplementary Table S2 provides a detailed overview of the employed LISTADAPT codification.
Whole Genome Sequencing and genomes data analysis
The next generation sequencing (NGS) paired-reads (2 × 150 bp) were generated during the project with Illumina platforms. Four LISTADAPT partners (AGES, IZSAM, ANSES and DTU) mainly performed the sequencing. Figshare File 126 lists the sequencing technology and the center which performed the library preparation and produced the sequences.
The genomes were all de novo assembled and annotated with a harmonized in-house workflow named ARTwork (Assembly of reads and typing workflow)32 used in the ANSES Laboratory for Food Safety. In addition to de novo assembly, the ARTwork pipeline also performs genome annotation using Prokka33. This whole genome sequencing (WGS) workflow has been described in detail in previous publications32,34,35,36, including the integrated bioinformatics tools and their corresponding versions, enabling repeatability and comparability of the results2 (Table 5). Assembled genome files were made publicly available in FASTA format through Figshare37.
Quality control of WGS data
Poor-quality reads or assemblies as well as contaminations can significantly affect gene prediction and cluster analyses38,39. Different WGS metrics and quality criteria were thus employed in the ARTwork pipeline to ensure high-quality WGS data. Reads with an estimated depth of coverage < 30 × (as estimated by BBmap40) as well as contigs and scaffolds with a length of < 200 bp were excluded (n = 22). Draft genomes with a total length outside the range of 2.7–3.3 Mb and with a total number of scaffolds > 200 (n = 46) were also excluded. In addition, inter- and intra-species contamination of reads was determined using the recently developed ConFindr software (v0.5.1)41. Since recently demonstrated, inter-and-intra species contamination of 10 single nucleotide variants (SNVs) assessed by ConFindr in the conserved core genes does not significantly impact cluster analysis39. We decided to exclude all genomes presenting SNVs lower than this cut-off (n = 12) as well as various read- or assembly-related errors (n = 34).
The employed WGS metrics and quality criteria of the complete LISTADAPT genome dataset are reported in Figshare File 126. In total, 114 sequenced genomes were of unsatisfactory quality after quality control and were thus excluded from the final dataset. After quality control of NGS and WGS data, the final LISTADAPT dataset included 1484 genomes.
Metadata and WGS data sharing
All metadata and WGS data collected herein were centralized and processed with standardized criteria for common nomenclature and NGS/WGS quality control before sharing between project partners. Reads normalized to 100 × coverage, draft assemblies (contigs and scaffolds) and annotated genomes (Genome Feature Format, GFF, and Genbank format, GBK) were also centralized at the MongoDB database located at ANSES (Maisons-Alfort Laboratory for Food Safety) providing quickly available, ready-to-use data.
Raw (non-normalized) reads for all the Lm strains sequenced in the LISTADAPT collection (n = 1484) were submitted to the NCBI Sequence Read Archive (SRA) for sharing with the LISTADAPT project’s partners. Raw (non-normalized) reads for 67 Lm food strains obtained from previous publications19,42 were submitted to the NCBI Sequence Read Archive (SRA) database and were linked to their existing accession numbers in Figshare File 126.
Data Records
All high-quality WGS data from this data descriptor are available for download at SRA/ENA public repository, including the sequences already available at the beginning of this study43. Assembly and annotation files are available through Figshare44. Complete metadata and quality check parameters are here reported in Figshare File 126.
Technical Validation
Redundant strains
The LISTADAPT dataset was analyzed by core-genome MLST (cgMLST) analysis, using BioNumerics (Table 5) according to a fixed cgMLST scheme consisting of 1748 Moura et al. loci45. All strains with genomes presenting less than < 7 allele differences (AD), isolated in the same year, as well as sharing the same source of isolation and sharing identical geographic location (same region or country) were considered as redundant. When the latter information was not available, the provider was used instead. Although year of isolation was unknown for four strains, they were marked as redundant because of similar cgMLST (<7 AD). Among the 1484 strains, 157 were identified as redundant. These strains were maintained in the dataset and marked accordingly (Figshare File 126)
Consistency analysis
The present study includes 648 strains from existing collections and 108 strains isolated in the framework of this study. The strains from historical collections were provided from 19 different laboratories. The management of large strain collections may lead to storage issue such as the isolation of two strains in the same tube. Furthermore, the sequencing of the strains involved several handling that may lead to human error.
For 380 of the 648 strains provided by partners, historical typing data were available. We established links between these typing data provided and the sequence obtained. These typing data were either, conventional serotyping data, molecular serotyping or MLST obtained by individual allele sequencing or mapping from PFGE. For conventional serotype the correspondence with the MLST type obtained from WGS was established following correspondence based on Ragon et al.12. The correspondence with molecular serotyping was established based on Hyden et al.46 mapping system using the Software SeqSphere (Table 5). For the strains isolated in Belgium (Table 3) the correspondence with PFGE was applied by our partner, based on the methodology described in Félix et al.18. For the strains isolated in Finland (Tables 2 and 3), the correspondence with PFGE was applied by our partners according to their in-house mapping methodology. The observed discordances were investigated with the partners. The concerned strains were re-sequenced if needed and discarded when unresolved. All results were reported in the Figshare File 126.
Code availability
The ARTwork pipeline, described in the WGS quality control section is publicly available at https://github.com/afelten-Anses/ARtWORK. The employed bioinformatics tools and their versions are specified in Table 5.
References
Piveteau, P., Depret, G., Pivato, B., Garmyn, D. & Hartmann, A. Changes in Gene Expression during Adaptation of Listeria monocytogenes to the Soil Environment. PLOS ONE 6, e24881 (2011).
Vivant, A. L., Garmyn, D. & Piveteau, P. Listeria monocytogenes, a down-to-earth pathogen. Front Cell Infect Microbiol 3, 87 (2013).
Buncic, S. et al. Microbial pathogen control in the beef chain: recent research advances. Meat Sci 97, 288–97 (2014).
Hurtado, A., Ocejo, M. & Oporto, B. Salmonella spp. and Listeria monocytogenes shedding in domestic ruminants and characterization of potentially pathogenic strains. Vet Microbiol 210, 71–76 (2017).
Yoshida, T., Sugimoto, T., Sato, M. & Hirai, K. Incidence of Listeria monocytogenes in wild animals in Japan. J Vet Med Sci 62, 673–5 (2000).
Weindl, L. et al. Listeria monocytogenes in Different Specimens from Healthy Red Deer and Wild Boars. Foodborne Pathog Dis 13, 391–7 (2016).
Parsons, C. et al. Listeria monocytogenes at the human-wildlife interface: black bears (Ursus americanus) as potential vehicles for Listeria. Microb Biotechnol 13, 706–721 (2020).
Lyautey, E. et al. Distribution and characteristics of Listeria monocytogenes isolates from surface waters of the South Nation River watershed, Ontario, Canada. Appl Environ Microbiol 73, 5401–10 (2007).
Hydeskov, H. B. et al. Listeria Monocytogenes Infection of Free-Living Western European Hedgehogs (Erinaceus Europaeus). J Zoo Wildl Med 50, 183–189 (2019).
Hellstrom, S., Kiviniemi, K., Autio, T. & Korkeala, H. Listeria monocytogenes is common in wild birds in Helsinki region and genotypes are frequently similar with those found along the food chain. J Appl Microbiol 104, 883–8 (2008).
Gismervik, K. et al. Invading slugs (Arion vulgaris) can be vectors for Listeria monocytogenes. J Appl Microbiol 118, 809–16 (2015).
Ragon, M. et al. A new perspective on Listeria monocytogenes evolution. PLoS Pathog 4, e1000146 (2008).
Chenal-Francisque, V. et al. Worldwide distribution of major clones of Listeria monocytogenes. Emerg Infect Dis 17, 1110–2 (2011).
Haase, J. K. et al. The ubiquitous nature of Listeria monocytogenes clones: a large-scale Multilocus Sequence Typing study. Environ Microbiol 16, 405–16 (2014).
Painset, A. et al. LiSEQ - whole-genome sequencing of a cross-sectional survey of Listeria monocytogenes in ready-to-eat foods and human clinical cases in Europe. Microb Genom 5(2019).
Dreyer, M. et al. Listeria monocytogenes sequence type 1 is predominant in ruminant rhombencephalitis. Sci Rep 6, 36419 (2016).
Cantinelli, T. et al. “Epidemic clones” of Listeria monocytogenes are widespread and ancient clonal groups. J Clin Microbiol 51, 3770–9 (2013).
Felix, B. et al. Population Genetic Structure of Listeria monocytogenes Strains Isolated From the Pig and Pork Production Chain in France. Front Microbiol 9, 684 (2018).
Henri, C. et al. Population Genetic Structure of Listeria monocytogenes Strains as Determined by Pulsed-Field Gel Electrophoresis and Multilocus Sequence Typing. Appl Environ Microbiol 82, 5720–8 (2016).
Holch, A. et al. Genome sequencing identifies two nearly unchanged strains of persistent Listeria monocytogenes isolated at two different fish processing plants sampled 6 years apart. Applied and environmental microbiology 79, 2944–2951 (2013).
Rychli, K. et al. Comparative genomics of human and non-human Listeria monocytogenes sequence type 121 strains. PLOS ONE 12, e0176857 (2017).
Stoller, A., Stevens, M. J. A., Stephan, R. & Guldimann, C. Characteristics of Listeria Monocytogenes Strains Persisting in a Meat Processing Facility over a 4-Year Period. Pathogens 8, 32 (2019).
Ortiz, S., Lopez, V. & Martinez-Suarez, J. V. Control of Listeria monocytogenes contamination in an Iberian pork processing plant and selection of benzalkonium chloride-resistant strains. Food Microbiol 39, 81–8 (2014).
Pasquali, F. et al. Listeria monocytogenes Sequence Types 121 and 14 Repeatedly Isolated Within One Year of Sampling in a Rabbit Meat Processing Plant: Persistence and Ecophysiology. Frontiers in microbiology 9, 596–596 (2018).
Papic, B., Pate, M., Felix, B. & Kusar, D. Genetic diversity of Listeria monocytogenes strains in ruminant abortion and rhombencephalitis cases in comparison with the natural environment. BMC Microbiol 19, 299 (2019).
Felix, B. Complete dataset, epidemiological informations, genomic quality assessment data and raw reads accession number. figshare https://doi.org/10.6084/m9.figshare.17696738 (2022).
Szymczak, B., Szymczak, M., Sawicki, W. & Dabrowski, W. Anthropogenic impact on the presence of L. monocytogenes in soil, fruits, and vegetables. Folia Microbiol (Praha) 59, 23–9 (2014).
Dowe, M. J., Jackson, E. D., Mori, J. G. & Bell, C. R. Listeria monocytogenes Survival in Soil and Incidence in Agricultural Soils (dagger). J Food Prot 60, 1201–1207 (1997).
Linke, K. et al. Reservoirs of listeria species in three environmental ecosystems. Appl Environ Microbiol 80, 5583–92 (2014).
Weller, D., Wiedmann, M. & Strawn, L. K. Spatial and Temporal Factors Associated with an Increased Prevalence of Listeria monocytogenes in Spinach Fields in New York State. Appl Environ Microbiol 81, 6059–69 (2015).
Nielsen, E.M. et al. Closing gaps for performing a risk assessment on Listeria monocytogenes in ready‐to‐eat (RTE) foods: activity 3, the comparison of isolates from different compartments along the food chain, and from humans using whole genome sequencing (WGS) analysis. EFSA Supporting Publications 14 (2017).
Vila Nova, M. et al. Genetic and metabolic signatures of Salmonella enterica subsp. enterica associated with animal sources at the pangenomic scale. BMC Genomics 20, 814 (2019).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–9 (2014).
Palma, F. et al. Dynamics of mobile genetic elements of Listeria monocytogenes persisting in ready-to-eat seafood processing plants in France. BMC Genomics 21, 130 (2020).
Radomski, N. et al. A Simple and Robust Statistical Method to Define Genetic Relatedness of Samples Related to Outbreaks at the Genomic Scale - Application to Retrospective Salmonella Foodborne Outbreak Investigations. Frontiers in microbiology 10, 2413–2413 (2019).
Felten, A. et al. First gene-ontology enrichment analysis based on bacterial coregenome variants: insights into adaptations of Salmonella serovars to mammalian- and avian-hosts. BMC Microbiology 17, 222 (2017).
Li, W. figshare https://doi.org/10.6084/m9.figshare.6025748 (2018).
Sims, D., Sudbery, I., Ilott, N. E., Heger, A. & Ponting, C. P. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet 15, 121–32 (2014).
Pightling, A. W., Pettengill, J. B., Wang, Y., Rand, H. & Strain, E. Within-species contamination of bacterial whole-genome sequence data has a greater influence on clustering analyses than between-species contamination. Genome biology 20, 286–286 (2019).
Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner https://www.osti.gov/servlets/purl/1241166 (2014).
Low, A. J., Koziol, A. G., Manninger, P. A., Blais, B. & Carrillo, C. D. ConFindr: rapid detection of intraspecies and cross-species contamination in bacterial whole-genome sequence data. PeerJ 7, e6995 (2019).
Fritsch, L. et al. Insights from genome-wide approaches to identify variants associated to phenotypes at pan-genome scale: Application to L. monocytogenes’ ability to grow in cold conditions. Int J Food Microbiol 291, 181–188 (2019).
Felix, B. NCBI Sequence Read Archive/European Nucleotide Archive. https://www.ebi.ac.uk/ena/browser/view/PRJEB38828 (2022). ListAdapt complete high-quality WGS data.
Felix, B. ListAdapt complete Listeria monocytogenes de novo assemblies. figshare https://doi.org/10.6084/m9.figshare.17315597 (2022).
Moura, A. et al. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat Microbiol 2, 16185 (2016).
Hyden, P. et al. Whole genome sequence-based serogrouping of Listeria monocytogenes isolates. J Biotechnol 235, 181–6 (2016).
Hellstrom, S. et al. Listeria monocytogenes contamination in pork can originate from farms. J Food Prot 73, 641–8 (2010).
Husu, J. R. Epidemiological studies on the occurrence of Listeria monocytogenes in the feces of dairy cattle. Zentralbl Veterinarmed B 37, 276–82 (1990).
Husu, J. R., Seppanen, J. T., Sivela, S. K. & Rauramaa, A. L. Contamination of raw milk by Listeria monocytogenes on dairy farms. Zentralbl Veterinarmed B 37, 268–75 (1990).
Ruusunen, M. et al. Pathogenic bacteria in Finnish bulk tank milk. Foodborne Pathog Dis 10, 99–106 (2013).
Esteban, J. I., Oporto, B., Aduriz, G., Juste, R. A. & Hurtado, A. Faecal shedding and strain diversity of Listeria monocytogenes in healthy ruminants and swine in Northern Spain. BMC Vet Res 5, 2 (2009).
Esteban, J. I., Oporto, B., Aduriz, G., Juste, R. A. & Hurtado, A. A survey of food-borne pathogens in free-range poultry farms. Int J Food Microbiol 123, 177–82 (2008).
Acknowledgements
This work was supported by the One Health European Joint Programme, European Union’s Horizon 2020 research and innovation programme (Grant Agreement No 773830). The authors would like to acknowledge Karol Romero and Karine Capitaine from ANSES, Laboratory for Food Safety, SEL Unit, for their technical help in the strain culture management. The authors also thank Thomas Berger, Jan-Erik Ingenhoff and René Imhof from Agroscope for collecting strains from soil and wild animals in Switzerland and for providing the associated genomes.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. Benjamin Félix contributed substantially to the study design, soil sampling, acquisition of strains and the corresponding genomes, data analysis as well as writing and editing of the manuscript. Yann Sévellec contributed extensively to the acquisition of strains and the corresponding genomes, data analysis, quality control of the WGS dataset as well as writing and editing the manuscript. Federica Palma contributed to the acquisition of WGS data, data analysis and quality control of the WGS dataset and to writing and revision of the manuscript. Arnaud Felten, Nicolas Radomski and Ludovic Mallet performed the assembly centralized in a meaningful database. Pierre Emmanuel Douarre contributed substantially to the technical validation of the dataset. Pascal Piveteau participated in the study design and collected strains from natural environment in France. Pascal Piveteau and Eliette Ascensio performed the detection of Lm for part of the soil samples collected by the partners. Christophe Soumet, Arnaud Bridier and Michel Hébraud contributed to the study design and the manuscript revision. Marina Torresi and Francesco Pomilio collected strains from natural environment and provided strains and genomes from animals and food in Italy. Cesare Cammà and Adriano di Pasquale were responsible for sequencing part of the strains. Taran Skjerdal collected strains from natural environment and provided strains from wild animals and food in Norway. Ariane Pietzka and Werner Ruppitsch performed soil sampling and provided WGS data of strains isolated from food in Austria. Renáta Karpíšková collected and provided strains from soil, ruminants and food isolated in the Czech Republic. Tereza Gelbíčová contributed to the selection and characterization of strains, DNA extraction in the Czech Republic and partly to data sequencing. Monica Ricao collected strains from soil and provided strains from wild animals and food isolated in Sweden. Bojan Papić collected strains from soil, provided strains from ruminants and natural environment in Slovenia and partly contributed to data sequencing. Bart Wullings provided strains and genomes from ruminants previously isolated from the Netherlands. Hana Bulawova provided strains from wild animals and ruminants isolated in Czech Republic. Hanna Castro, Miia Lindström and Hannu Korkeala collected and provided strains from wild and farm animals from Finland. Žanete Šteingolde and Toomas Kramarenko provided strains from farm animals isolated from Latvia and Estonia. Lenka Cabanova provided strains from ruminants and wild animals isolated from Slovakia. Barbara Szymczak provided strains from soil isolated from Poland. Verena Oswaldi provided strains from farm animals isolated from Germany. Manfred Gareis provided strains from wild animals isolated from Germany. Anne-Mette Seyfarth provided genomes from food strains isolated from Denmark. Ana Hurtado provided strains from farm animals isolated from Spain. Thomas Berger and the team from Agroscope collected strains from soil and wild animals, and provided the genomes of these strains isolated from Switzerland. Jean-Charles Leblanc contributed to the revision of the manuscript and the technical validation of the dataset. Laurent Guillier participated in coordination of the study, selection of strains for sequencing and in drafting the manuscript. Sophie Roussel designed and coordinated the study and contributed to the writing and editing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Félix, B., Sevellec, Y., Palma, F. et al. A European-wide dataset to uncover adaptive traits of Listeria monocytogenes to diverse ecological niches. Sci Data 9, 190 (2022). https://doi.org/10.1038/s41597-022-01278-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01278-6
- Springer Nature Limited