
Abstract
Single-cell RNA sequencing (scRNA-seq) is developing rapidly, and investigators seeking to use this technology are left with a variety of options for both experimental platform and bioinformatics methods. There is an urgent need for scRNA-seq reference datasets for benchmarking of different scRNA-seq platforms and bioinformatics methods. To be broadly applicable, these should be generated from renewable, well characterized reference samples and processed in multiple centers across different platforms. Here we present a benchmark scRNA-seq dataset that includes 20 scRNA-seq datasets acquired either as mixtures or as individual samples from two biologically distinct cell lines for which a large amount of multi-platform whole genome sequencing data are also available. These scRNA-seq datasets were generated from multiple popular platforms across four sequencing centers. We believe the datasets we describe here will provide a resource that meets this need by allowing evaluation of various bioinformatics methods for scRNA-seq analyses, including but not limited to data preprocessing, imputation, normalization, clustering, batch correction, and differential analysis.
Measurement(s) | single-cell gene expression analysis |
Technology Type(s) | RNA sequencing |
Sample Characteristic - Organism | Homo sapiens |
Sample Characteristic - Environment | cell line |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.13403753
Similar content being viewed by others


Background & Summary
A variety of scRNA-seq technologies and protocols have been developed for biomedical research1,2,3,4,5,6,7. These technologies can be divided into two broad categories: full-length and 3′ end counting-based. The 3′ end counting-based methods allow the incorporation of unique molecular identifiers (UMIs) to improve quantification of mRNA molecules; whereas full-length methods generally provide greater sensitivity of gene detection and ability to identify changes across the length of a transcript, such as alternative splicing, novel transcripts, and mutations, etc. Large differences exist across different protocols and platforms in specificity, sensitivity, throughput, chemistry of library construction, and bioinformatics8,9,10,11,12, as well as cost. As described in our associated Nature Biotechnology paper13, prior studies have attempted to address various aspects relating to scRNA-seq benchmarking9,11,12. However, technical factors (technology platform, inter-laboratory differences in cell handling, and library construction) cannot be distinguished from purely biological variability if only mixtures of cells are used. For example, it might be difficult to identify the effect of different technology platforms if studies considered only mixtures of multiple cell types across different platforms; as we pointed out in our Nature Biotechnology paper13, Scanorama failed to integrate data from different technology platforms. This was not noticed previously. By also distributing samples of both cell lines to different centers, where they were subsequently cultured before analysis, we were able to additionally evaluate the sort of experimental variability likely to be encountered in real-world collaborations. Therefore, currently there is no systematic multi-center study that evaluated the influence of technology platform, sample composition, and bioinformatic methods (including preprocessing, normalization, and batch-effect correction) using publicly available standard reference samples and datasets consisting of both mixed and non-mixed biologically distinct samples.
Recently, we benchmarked scRNA-seq performance across several popular instrumentation platforms at multiple centers, also focusing on the effects of bioinformatic processing; including preprocessing, normalization, and batch-effect correction13. As stated in this paper, our benchmark study has produced well-characterized reference materials (reference samples, datasets) and methods, which will have similar value for the single-cell sequencing community as the Zook et al. study14, carried out by the Genome in a Bottle Consortium (GIAB), did for genome sequencing. The findings from our study offer practical guidance for optimizing and benchmarking a platform or experimental protocol, and for selecting appropriate bioinformatics methods when designing scRNA-seq experiments. We analyzed two well-characterized, but biologically distinct reference cell lines, for which a large amount of multiplatform whole-genome and whole-exome sequencing data are available15: a human breast cancer cell line (HCC1395; sample A) and a B lymphocyte cell line (HCC1395BL; sample B) derived from the same donor. A total of 20 scRNA-seq datasets were generated from the two cell lines, processed either separately or as mixtures of different ratios of both cell lines, using four scRNA-seq platforms (10x Genomics Chromium, Fluidigm C1, Fluidigm C1 HT, and Takara Bio’s ICELL8 system) at four centers: Loma Linda University (LLU), US National Cancer Institute (NCI), US Food and Drug Administration (FDA), and Takara Bio USA (TBU). We evaluated seven preprocessing pipelines for raw scRNA-seq fastq data, eight normalization methods16,17,18,19,20,21, and seven batch correction methods22,23,24,25,26. Our study showed that although pre-processing and normalization contributed to variability in gene detection and cell classification, batch effects were quite large, and the ability to assign cell types correctly across platforms and sites was dependent on the bioinformatic pipelines, particularly the batch correction algorithms used. In many scenarios, Seurat v327, Harmony26, BBKNN25, and fastMNN22 all corrected the batch effects fairly well for scRNA-seq data derived from either biologically identical or dissimilar samples across platforms and sites. However, when samples containing large fractions of biologically distinct cell types were compared, Seurat v3 over-corrected the batch-effect and misclassified the cell types (i.e., breast cancer cells and B lymphocytes clustered together), while limma and ComBat failed to remove batch effects. The datasets we present here can help researchers select the scRNA-seq protocol and bioinformatic method best suited to the samples to be analyzed. In addition, they can be used to benchmark current or newly developed scRNA-seq protocols and evaluate various existing and emerging bioinformatics methods for scRNA-seq data analysis.
Methods
Detailed methods were described in our associated paper13. The following is a brief summary adapted from the Online Methods.
Study design
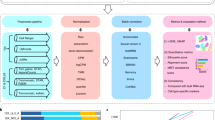
Fig. 1 shows our overall study design. A total of 20 scRNA-seq datasets were generated, including fourteen 3′ end counting-based and six full-length datasets, which were generated using two well-characterized reference cell lines: a human breast cancer cell line (sample A) and a matched control ‘normal’ B lymphocyte line (sample B) derived from the same donor. The fourteen 3′ end counting-based datasets were generated at three different centers (LLU, NCI, and FDA), and the datasets were referred to as follows: 10X_LLU, 10X_NCI, 10X_NCI_M (modified shorter sequencing protocol), and C1_FDA_HT. The six full-length datasets were generated at two centers (LLU and TBU), and the datasets were referred to as: C1_LLU and ICELL8 (includes both single-end/SE and paired-end/PE). In the case of the 10x Genomics (abbreviated 10x subsequently) data sets, mixtures of samples A and B were processed in addition to individual samples processed separately. All other data sets were generated from samples A and B separately. For simplicity, we will use the labels in Fig. 2 to represent the 20 datasets throughout our analysis.

Study design.

UMAPs before (a) and after batch correction using (b) Harmony, (c) BBKNN, and (d) Seurat v3.
Cell culture
We obtained the human breast cancer cell line (HCC1395, sample A) and the matched ‘normal’ B lymphocyte cell line (HCC1395BL, sample B) from ATCC (American Type Culture Collection, Manassas, VA, USA). The two cell lines were derived from the same human subject (43 years old, female). HCC1395 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS). HCC1395BL cells were cultured in Iscove’s Modified Dulbecco’s Medium supplemented with 20% FBS.
Full-length single-cell RNA-seq using the C1 fluidigm system
Single cell suspensions were loaded on a medium-sized (10-17 µm) RNA-seq integrated fluidic circuit (IFC) at a concentration of 200 cells/µl. Full-length cDNAs were generated using the Fluidigm C1 system at the LLU Center for Genomics using the SMART-Seq v4 Ultra Low Input RNA kit (Takara Bio) according to the manufacturer’s protocol. Libraries were prepared using the modified Illumina Nextera XT DNA library preparation protocol. 80 libraries were generated from HCC1395 cells (sample A) and 66 libraries were generated from HCC1395BL cells (sample B). Library pools were sequenced at the LLU Center for Genomics on an Illumina HiSeq 4000 sequencer,150×2 bp, paired-end sequencing.
3’-end single-cell RNA-seq using C1 Fluidigm high-throughput (HT) system
High-throughput single-cell 3′ end cDNA libraries were generated according to the manufacturer’s instructions at the FDA’s Center for Biologics Evaluation and Research. Briefly, single cells were loaded on an HT IFC at a concentration of 400 cells/µl (Nexcelom Cellometer Auto T4). After cell lysis, the captured mRNA was barcoded during the reverse transcription step with a barcoded primer, and the tagmentation step was done following the Nextera XT DNA library preparation guide. Lastly, sequencing adapters and Nextera indices were applied during library preparation. Only the 3′ end of the transcript was enriched following PCR amplification. 203 libraries were generated from HCC1395 cells (sample A) and 241 libraries were generated from HCC1395BL cells (sample B). Library pools were sequenced at the FDA/CBER Core Facility on an Illumina HiSeq 2500, 75×2 bp, paired-end sequencing.
Single-cell RNA-seq using Takara Bio ICELL8 platform
A bulk cell suspension of either cancer or B cells was fluorescently labeled and diluted to ~1 cell in 35 nl. Each cell type suspension was dispensed from a 384-well source plate into individually addressable wells in a 5,184 nano-well, 250 nl volume ICELL8 chip (SMARTer™ ICELL8® 250 v Chip, Takara Bio USA, CA, USA) using a SMARTer™ ICELL8® Single-Cell System (Takara). Wells containing individual live cells were identified by imaging using CellSelect software to generate a well-selection map (filter file), which was then used to enable individual addressing of the chosen wells for addition of cDNA synthesis and library preparation reagents as detailed in the following sections. All on-chip liquid handling was performed with the SMARTer™ ICELL8® Single-Cell System. Full-length cDNA synthesis, P5/P7 index addition and tagmentation were done on-chip. Following round 1 PCR, amplicons were collected, pooled by centrifugation of the chip, and purified using Ampure beads. This was followed by round 2 PCR amplification off-chip. All steps were performed per manufacturer’s instructions. The library quality was determined using a Qubit fluorometer (Thermo Fisher), a 2100 Bioanalyzer, and a corresponding High Sensitivity DNA Kit (Agilent). The ICELL scRNA-seq libraries were sequenced both at the Takara Bio USA site on an Illumina NextSeq 550, 75×2 bp, paired-end and at the LLU Center for Genomics on a HiSeq 4000, 150×1 bp, single-end sequencing.
Single-cell RNA-seq using the 10x Genomics platform
After filtering with a 30-micron MACS SmartStrainer (Miltenyi Biotec), single cells were resuspended in PBS (calcium and magnesium free) containing 0.04% weight/volume BSA, and further diluted to 300 cells/µl after cell count (Countess II FL, Life Technologies). For the 5% spike-in and 10% spike-in cell mixtures, 5% or 10% of HCC1395 breast cancer cells were mixed with either 95% or 90% of HCC1395BL cells. Library preparation was performed following the 3′ scRNA-seq10x Genomics platform protocol using v2 chemistry.
10x Genomics scRNA-seq library construction using fixed cells
We also constructed 10x scRNA-seq libraries using fixed cells at the NCI site. Briefly, for delayed captures, cells were fixed in methanol using a method described by Alles et al.28. The fixed samples underwent two different treatments. For the first sample (NCI_Mix5_F), the normal and cancer cells were harvested, washed, counted, and a 5% spike-in of breast cancer cells plus 95% normal B cells was prepared and mixed as described above. Approximately 130,000 cells were then processed for fixation. The cells were washed twice with 1X DPBS at 4 °C and resuspended gently in 100 µl 1X DPBS (ThermoFisher Scientific). 900 μl chilled methanol (100%) were then added drop by drop to the cells with gentle vortexing. Cells were then fixed on ice for 15 min, following which they were stored at 4 °C for 6 days. For rehydration, the fixed cells were pelleted by centrifugation at 3000 g for 10 min at 4 °C and washed twice with 1X DPBS containing 1% BSA and 0.4U/µl RNase inhibitor (Sigma Aldrich). The cells were then counted and the concentration was adjusted to be close to 1000 cells/µl. Approximately 8000 cells were loaded onto a single-cell chip for GEM generation using the 10x Genomics Chromium controller. 3′ mRNA-seq gene expression libraries were prepared using the Chromium Single Cell 3′ Library & Gel Bead Kit v2 (10x Genomics) according to the manufacturer’s guidelines.
For the second sample (NCI_Mix5_F2), breast cancer cells and normal B cells (approximately 4 million each) were harvested and fixed as described above. The cells were initially washed with 1X DPBS and resuspended in 10% 1X DPBS and 90% chilled methanol, as described above. Cells were then fixed on ice for 15 mins, following which they were stored at 4 °C for 24 hrs. For rehydration, the fixed cells were washed with 1X DPBS containing 1% BSA and 0.4U/µl RNase inhibitor and counted. Approximately 8000 cells were loaded onto a single-cell chip for GEM generation using the 10x Genomics Chromium controller. 3′mRNA-seq gene expression libraries were prepared using the Chromium Single Cell 3′ Library & Gel Bead Kit v2 (10x Genomics) according to the manufacturer’s guidelines.
All the 10x Genomics scRNA-seq libraries constructed at the LLU were sequenced on an Illumina NextSeq 550 and a HiSeq 4000 with the standard sequencing protocol of 26 + 98 bp read length at the LLU Center for Genomics, whereas the libraries constructed at the NCI site were both sequenced on an Illumina NextSeq 500 with a modified sequencing protocol of 26 + 57 bp read length at the NCI Sequencing Facility and on an Illumina HiSeq 4000 or a NextSeq 550 using the standard sequencing protocol of 26 + 98 bp read length at the LLU Center for Genomics.
Bulk cell RNA-seq
We isolated total RNA from bulk HCC1395 (cancer) and HCC1395BL (B cells) using the miRNeasy Mini kit (Qiagen), and constructed RNA-seq libraries using the NuGEN Ovation universal RNA-seq kit at LLU according to the manufacturer’s instructions. All the libraries were quantified using Qubit 3.0 (Life Technologies) and quality was checked on a TapeStation 2200 (Agilent Technologies). The bulk-cell RNA-seq libraries were sequenced both on an Illumina NextSeq 550, 75×2 bp, paired-end; and on a HiSeq 4000, 100×2 bp, paired-end at the LLU Center for Genomics.
Reference genome
The reference genome and transcriptome were downloaded from the 10x Genomics website as refdata-cellranger-GRCh38-1.2.0.tar.gz, which corresponds to the GRCh38 genome and Ensembl v84 transcriptome. All the bioinformatics data analyses were carried out based on the above reference genome and transcriptome.
Preprocessing of 10x Genomics scRNA-seq data
For UMI based 10x Genomics samples, four pre-processing pipelines, Cell Ranger1 (v2.0.0), Cell Ranger (v3.1.0), UMI-tools29 (v1.0.0), and zUMIs30 (v2.4.5) were used to process the raw fastq data and generate gene count matrices. In the Cell Ranger pipeline, ‘cellranger count’ was used with all default parameter settings. In the umi-tools and zUMIs pipelines, reads were filtered out if Phred sequence quality of either the cell barcode or UMI bases were <10. In UMI-tools, ‘umi_tools whitelist’ with default parameter settings was used to generate a list of cell barcodes for downstream analysis. ‘umi_tools extract’ was used to extract the cell barcodes and filter the reads (options: --quality-filter-threshold = 10 --filter-cell-barcode). STAR31 (v2.5.4b) was used for alignment to generate BAM files containing the unique mapped reads (option: outFilterMultimapNmax 1) for gene counting. featureCounts32 (v1.6.1) was used to assign reads to genes and generate a BAM file (option: -R BAM). ‘samtools (v1.3) sort’ and ‘samtools index’ were used to generate sorted and indexed BAM files. Finally, ‘umi_tools count’ (options: --per-gene --gene-tag = XT --per-cell --wide-format-cell-counts) was used for the sorted BAM files to generate gene count per cell matrices.
Preprocessing of non-UMI scRNA-seq data from C1 and Takara Bio ICELL8 platforms
For non-UMI based samples, three pre-processing pipelines were used to process the raw fastq data and generate gene count matrices. The pipelines included trimming and filtering, alignment, and gene counting. In the trimming and filtering process, one of the three tools [Trimmomatic33 (v0.35), trim_galore (v0.4.1), or cutadapt34 (v1.9.1)] was used to process the raw fastq data. Bases with quality less than 10 were trimmed from 5′ and 3′ ends of reads. Reads less than 20 bases were excluded from further analysis. STAR with default parameter settings was used for alignment to generate BAM files. Three gene counting tools, featureCounts, RSEM35 (v1.3.0), and kallisto36 (v0.43.1) were used to generate gene counts per cell. All default parameter settings were used except the following: In RSEM, option ‘--single-cell-prior’ was used to estimate gene expression levels for scRNA-seq data; Option of ‘--paired-end’ was used if the data were paired-end fastqs; In kallisto, options ‘-l 500’ and ‘-s 120’ were used to represent estimated average fragment length and standard deviation of fragment length if the data were single-end fastqs. For simplicity, we used featureCounts, RSEM, and kallisto to refer to the three pre-processing pipelines later.
Cell filtering and quality control metrics
We used gene count per cell matrices from Cell Ranger v3.1 and featureCounts pipelines in the downstream analyses for 10x and non-10x data, respectively. The following strategies were used to filter dead cells and doublets. (1) Cells were removed from analysis if they expressed less than 200 genes. We also removed genes expressed in less than 3 cells. (2) The total numbers of UMIs and genes for each cell were counted. The upper bound was calculated as mean plus two standard deviations (SD) and the lower bound as mean minus two SD for both the total UMIs and genes. Cells with total UMIs or genes outside of the upper and lower bounds were removed. (3) Cells were removed if greater than 10% reads mapped to mitochondrial genes.
scRNA-seq data batch effect correction
We used the filtered gene count per cell matrices from the Cell Ranger v3.1 and featureCounts pipelines as input to perform batch correction. Seurat (v3.0.3) was applied to each dataset. The datasets were then log transformed and scaled. The top 2,000 highly variable genes (HVGs) were selected in each dataset with the function FindVariableGenes. The processed data and HVGs were used as input to perform batch correction using Harmony, BBKNN, and Seurat v3. The Uniform Manifold Approximation and Projection (UMAP)37 plots were generated from the batch-corrected low-dimensional embedding matrices. The uncorrected 20 scRNA-seq datasets showed strong batch effects in UMAP plots (Fig. 2a), clustering by individual dataset instead of by cell type. When Harmony method was applied to the combined data, two clusters corresponding to the different cell types were clearly apparent (Fig. 2b). BBKNN batch correction also generated two clusters representing each cell type (Fig. 2c). However, Seurat v3 over-corrected and did not generate separate clusters for the two cell lines.
Estimation of copy number variation and clustering analysis
Copy number variation (CNV) is highly associated with the development and progression of many cancers. Recently developed scRNA-seq CNV inference methods38,39 enable the assessment of both RNA expression and genomic copy number information from the same cell using the transcriptomic data to study the genetic heterogeneity at the single-cell level. This is a significant advance because current methods for obtaining both DNA-seq and RNA-seq data from the same single cell are still not only technically challenging, but also expensive40. Here, we applied one of the published CNV inference methods to our dataset to examine the consistency of our data across different platforms and sites for CNV inference analysis. To estimate CNV of sample A cells (cancer cell line), we performed CNV analysis by inferCNV (https://github.com/broadinstitute/inferCNV, v1.1.3) on 13 datasets. These datasets included different batches of sample A cells either alone or mixed with cells from sample B. The 10x datasets were down sampled to 1,000 cells per dataset for CNV analysis, which generated a total of 10,353 cells. In addition, the 10X_LLU_B dataset was down sampled to 1,000 cells and used as a control. In the CNV analysis, a de-noising filtering step and a Hidden Markov Model (HMM)-based CNV prediction step were enabled with a cut-off of 0.1 for gene selection. For hierarchical clustering-based tree partitioning, parameters including ‘tumor_subcluster_partition_method = ‘qnorm”, ‘hclust_method = ‘ward.D2”, and ‘tumor_subcluster_pval = 0.05’ were used. Our CNV analysis showed a clear separation by cell type instead of by different dataset or platform. The results indicated good consistency across different platforms and datasets, which all captured similar CNVs in the tumor cells (Fig. 3a). Meanwhile, we applied Harmony batch correction and Seurat clustering to the same data and detected two major clusters as well (Fig. 3b). The CNV analysis and expression-based clustering analysis were highly consistent (Fig. 3c).

InferCNV analysis compared with expression-based clustering. (a) Estimation of copy number variants by inferCNV across 7 datasets including cancer cells (HCC1395) and 6 spike-in datasets containing both HCC1395 (breast cancer) and HCC1395BL (B lymphocytes). 10X_LLU_B was used as a control (heatmap not shown). All datasets were down sampled to 1,000 cells. Top color bars indicate different chromosome regions. Left color bars indicate different datasets. The color intensities of the heatmap correspond to the residual expression values by inferCNV, with red or blue indicating higher or lower values compared with those of control cells. (b) UMAP of Harmony-corrected expression data. Dark red and blue indicate the cell type identity of either HCC1395 or HCC1395BL. (c) A dendrogram of inferCNV clusters (left) and heatmap comparison (right) of cell labels generated by inferCNV and Seurat. The order of dendrogram leaves was generated by inferCNV as indicated in panel (a). Heatmap colors indicate the top two groups of cells in the dendrogram tree and cell clusters identified by Seurat.
Data Records
All sequence data have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Reads Archive (SRA) with accession ID: SRP199641 (BioProject: PRJNA504037)41. Dataset 142 provides detailed meta information of the deposited sequence data. The processed gene count matrices have been uploaded to Figshare42.
Technical Validation
QC assessment on the effect of preprocess pipelines for 10x and non-10x data
For the 10x scRNA-seq data, we evaluated four pre-processing pipelines: Cell Ranger 2.0, Cell Ranger 3.1 (10x Genomics), UMI-tools, and zUMIs, and examined the consistency between the four pipelines regarding the number of cells identified, the number of genes detected per cell, and percentage of sample A cells in spike-in mixtures (Fig. 4a–d). In most of the datasets (except 10X_LLU_A), Cell Ranger 3.1 and zUMIs always called the largest and second largest number of cells. For most of the datasets from sample A only or sample B only, Cell Ranger 2.0 and UMI-tools called consistent numbers of cells. For spike-in datasets, especially for cells fixed in methanol, the numbers of cells called were variable. The percentages of sample A cells in Fig. 4d were also inconsistent in the samples with fixed cells. These observations suggested that methanol fixation may reduce the quality of data, causing inconsistent cell calling by different pipelines. For the 10x single-cell protocol, we compared the standard sequencing protocol (26 + 98 bp) with the modified sequencing protocol (26 + 57 bp) using the same scRNA-seq libraries. The two sequencing protocols yielded consistent cell calling and percentage of spike-in cancer cells, suggesting that sequence length and sequencing instrument do not substantially impact the data quality. Fig. 4b shows the number of genes expressed per cell. Because Cell Ranger 3.1 modified the algorithm to call more cells with low RNA content, it always generated a lower number of genes expressed per cell because it called the largest number of cells compared with other pipelines. Fig. 4c shows the correlation of expressed genes of the same cells between any two preprocessing pipelines. Overall, the correlations between any two pipelines were consistently high across all the cells. However, we observed a relatively lower correlation in the datasets between the NCI modified sequencing protocol and the standard sequencing protocols, which was due to short sequencing reads in the modified protocol.

Effect of preprocessing pipelines. (a) Barplot of the number of cells identified by four pipelines using 10x datasets. (b,e) Boxplot of the number of genes detected per cell processed using four (10x datasets) or three (non-10xdatasets) pipelines, respectively. (c,f) Boxplot of Pearson’s correlations of consensus expressed genes per cell between any two pipelines in 10x and non-10x datasets, respectively. (d) Barplot of percentage of breast cancer cells detected in spike-in mixtures processed using four pipelines in 10x datasets.
For the non-droplet scRNA-seq data, consistent numbers of genes expressed per cell were observed using the three different pre-processing pipelines (Fig. 4e). Kallisto identified more genes per cell in the full-length protocols (C1_LLU and ICELL8) and fewer genes per cell in the 3′-end counting-based protocol (C1-FDA_HT). For ICELL8, we compared the paired-end (PE) read with single-end (SE) read data using the same scRNA-seq libraries; and found that the PE or SE sequencing did not affect the quality of the data from the same library. Our correlation analysis between any two preprocessing pipelines (Fig. 4f) showed consistently high correlations between full-length data (C1_LLU and ICELL8), but lower correlations between tag-based data, especially when comparing kallisto with the other two pipelines. This might be due to different alignment strategies (genome-based alignment vs. pseudoalignment) used in the three pipelines.
Cell quality assessment across 20 scRNA-seq datasets
We used three metrics: (1) average number of genes per cell; (2) average number of UMIs/read counts per cell; (3) average mitochondrial gene percentage per cell to evaluate the quality of the 20 scRNA-seq datasets by comparing high quality cells with filtered low-quality cells (Fig. 5a–c, see cell filtering in method section for details; Supplementary File 1 provides more detailed quality assessment results). In high quality cells, the average number of genes detected per cell was greater than 3,000 genes; only the C1 platform (C1_LLU, C1_FDA_HT) detected genes above this value in low quality cells (Fig. 5a). This may be due to relatively small numbers of cells captured with the C1 platform, leading to high read counts per cell. Samples analyzed using the same method showed similar average numbers of genes per cell and average numbers of UMIs/read counts per cell and were clustered together (Fig. 5a,b). Due to the sequencing depth differences between full-length and 3′ end counting-based methods, C1_LLU and ICELL8 showed higher values of metrics 1 and 2 in high quality cells than those in 10x and C1_FDA_HT. Furthermore, we found that no more than 3.85% of the cells had mitochondrial gene percentages greater than 10% across the combined 20 datasets. In high quality cells, the mitochondrial gene percentages were below 10% in all 20 datasets.

Evaluation of cell quality. (a) Average number of genes detected per cell; (b) Average number of UMIs/read counts (droplet/non-droplet) per cell; (c) Average mitochondrial gene percentage per cell. Auxiliary lines represented by grey dash lines are applied to (a–c) and several datasets are labeled for better visualization. # indicates the number of genes or read counts. % indicates percentage of mitochondrial genes.
Consistency of gene expression across 20 scRNA-seq datasets
For the 20 scRNA-seq data sets, we performed correlation analyses to evaluate the consistency of gene expression data using bulk RNA-seq data as a reference. We first labelled cells in the spike-in datasets as sample A and sample B cells, respectively. The subset data containing either A or B cells in the spike-in datasets were then used to perform correlation analyses (a total of 13 datasets for A and B cells, respectively). The top 2,000 HVGs of the 13 scRNA-seq datasets were used in the analyses. The gene expression mean and variance were calculated across all cells in each scRNA-seq dataset across three replicates of the bulk RNA-seq dataset. The bulk RNA-seq data sets showed good correlation (r ≥ 0.78) of gene expression mean with all 13 datasets in both A and B cells (Fig. 6a,d). The correlation of gene expression variance between the bulk RNA-seq datasets and scRNA-seq datasets suggested cell-to-cell diversity in scRNA-seq (Fig. 6b,e). High consistency was observed across the 10x and non-10x datasets in both gene expression mean and variance. The C1_LLU datasets showed the highest correlation of gene expression mean with bulk RNA-seq data in both A and B cells, likely because these datasets had the highest sequencing depth (an average of over 4 million reads per cell). We also found that the data between standard sequencing and modified sequencing protocols had high correlation (r ≈ 1). Our results showed high consistency of gene expression across all 20 scRNA-seq datasets.

Consistency across platforms and datasets. Pairwise Pearson correlation of the expression mean and variance of the top 2,000 highly variable genes (HVG) between bulk RNA-seq and scRNA-seq datasets for HCC1395 (a,b) and HCC1395BL (d,e). (c,f) represent the cell composition of each scRNA-seq dataset. The HVG expression in bulk RNA-seq data represents the average gene expression from 3 biological replicates. The HVG expression in scRNA-seq data represents the average gene expression across all cells in each dataset.
Code availability
All code used in processing the scRNA-seq data and in drawing the figures are available on Github at the following link: https://github.com/oxwang/SciData_scRNAseq.
References
Zheng, G. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Hashimshony, T. et al. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 17, 77 (2016).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Gao, R. et al. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nat. Commun. 8, 228 (2017).
Hashimshony, T., Wagner, F., Sher, N. & Yanai, I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Reports 2, 666–673 (2012).
Ramsköld, D. et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 30, 777-782 (2012).
Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013).
Ziegenhain, C. et al. Comparative analysis of single-cell RNA sequencing methods. Mol. Cell 65, 631–643. e4 (2017).
Tian, L. et al. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat. Methods 16, 479–487 (2019).
Zhang, X. et al. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol. Cell 73, 130–142.e5 (2019).
Tran, H. T. N. et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12 (2020).
Mereu, E., Lafzi, A., Moutinho, C. et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat. Biotechnol. 38, 747–755 (2020).
Chen, W. et al. A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples. Nat. Biotechnol. https://doi.org/10.1038/s41587-020-00748-9 (2020).
Zook, J. M. et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol. 32, 246–251 (2014).
Xiao, W. et al. Towards best practice in cancer mutation detection with whole-genome and whole-exome sequencing. Nat. Biotechnol. (in press).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Risso, D., Ngai, J., Speed, T. P. & Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896-902 (2014).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 1–15 (2019).
Lun, A. T., Bach, K. & Marioni, J. C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016).
Bacher, R. et al. SCnorm: robust normalization of single-cell RNA-seq data. Nat. Methods 14, 584-586 (2017).
Yip, S. H., Wang, P., Kocher, J.-P. A., Sham, P. C. & Wang, J. Linnorm: improved statistical analysis for single cell RNA-seq expression data. Nucleic Acids Res. 45, e179 (2017).
Haghverdi, L., Lun, A. T., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 37, 685–691 (2019).
Polański, K. et al. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965 (2020).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902. e1821 (2019).
Alles, J. et al. Cell fixation and preservation for droplet-based single-cell transcriptomics. BMC Biol. 15, 44 (2017).
Smith, T., Heger, A. & Sudbery, I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Parekh, S., Ziegenhain, C., Vieth, B., Enard, W. & Hellmann, I. zUMIs-a fast and flexible pipeline to process RNA sequencing data with UMIs. Gigascience 7, giy059 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 17, 10–12 (2011).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Bray, N., Pimentel, H., Melsted, P. et al. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016)
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2019).
Harmanci, A. S., Harmanci, A. O. & Zhou, X. CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nat. Commun. 11, 89 (2020).
Fan, J. et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 28, 1217–1227 (2018).
Macaulay, I. C. et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat. Methods 12, 519–522 (2015).
Yang, Z. & Wang, C. NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP199641 (2020).
Chen, X. et al. Gene count matrices from a scRNA-seq benchmark study. figshare https://doi.org/10.6084/m9.figshare.c.5213468 (2020).
Acknowledgements
The authors would like to thank Ms. Diana Ho of the LLU Center for Genomics for her great administrative support, particularly in coordinating the weekly Zoom conference calls and assistance in preparation of meeting minutes for the SEQC-2 single-cell sequencing project. The authors would like to thank Dr. Zhong Chen at LLU and Wells Wu of the FDA/CBER core facility for their technical assistance. The genomic work carried out at the LLU Center for Genomics was funded in part by the National Institutes of Health (NIH) grant S10OD019960 (CW), the American Heart Association (AHA) grant 18IPA34170301 (CW), the Ardmore Institute of Health grant 2150141 (CW) and Dr. Charles A. Sims’ gift to LLU Center for Genomics.
Author information
Authors and Affiliations
Contributions
C.W. conceived and designed the study. X.C., Z.Y. and W.C. drafted the manuscript. M.M.J., A.F., W.X. and C.W. edited the manuscript. W.C., A.F., B.T., M.M.J., V.F. performed single cell captures, scRNA-seq library construction and sequencing. X.C. and Z.Y. performed bioinformatics data analyses. All authors reviewed the manuscript. C.W. finalized and submitted the manuscript.
Corresponding author
Ethics declarations
Competing interests
Andrew Farmer is an employee of Takara Bio USA, Inc. All other authors claim noconflicts of interest. The views presented in this article do not necessarily reflect current or future opinion or policy of the US Food and Drug Administration. Any mention of commercial products is for clarification and not intended as an endorsement.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Chen, X., Yang, Z., Chen, W. et al. A multi-center cross-platform single-cell RNA sequencing reference dataset. Sci Data 8, 39 (2021). https://doi.org/10.1038/s41597-021-00809-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-021-00809-x
- Springer Nature Limited
This article is cited by
-
Vocal emotion perception in schizophrenia and its diagnostic significance
BMC Psychiatry (2023)
-
Towards accurate and reliable resolution of structural variants for clinical diagnosis
Genome Biology (2022)
-
Establishing community reference samples, data and call sets for benchmarking cancer mutation detection using whole-genome sequencing
Nature Biotechnology (2021)
-
Toward best practice in cancer mutation detection with whole-genome and whole-exome sequencing
Nature Biotechnology (2021)