

Abstract
The preinitiation complex (PIC) assembles on promoters of protein-coding genes to position RNA polymerase II (Pol II) for transcription initiation. Previous structural studies revealed the PIC on different promoters, but did not address how the PIC assembles within chromatin. In the yeast Saccharomyces cerevisiae, PIC assembly occurs adjacent to the +1 nucleosome that is located downstream of the core promoter. Here we present cryo-EM structures of the yeast PIC bound to promoter DNA and the +1 nucleosome located at three different positions. The general transcription factor TFIIH engages with the incoming downstream nucleosome and its translocase subunit Ssl2 (XPB in human TFIIH) drives the rotation of the +1 nucleosome leading to partial detachment of nucleosomal DNA and intimate interactions between TFIIH and the nucleosome. The structures provide insights into how transcription initiation can be influenced by the +1 nucleosome and may explain why the transcription start site is often located roughly 60 base pairs upstream of the dyad of the +1 nucleosome in yeast.
Similar content being viewed by others

Main
Previous studies provided structures of yeast and human preinitiation complexes (PICs) on various promoters1,2,3,4,5,6,7,8,9. However, promoters are flanked by nucleosomes within the chromatin environment in vivo10,11 and thus PIC structures must also be determined in the presence of nucleosomes. In the yeast Saccharomyces cerevisiae, PIC assembly occurs adjacent to the +1 nucleosome12,13, which resides at the downstream end of the core promoter. The +1 nucleosome is often well-positioned14,15 and thought to be involved in PIC assembly13, although the underlying mechanisms are unclear16. The +1 nucleosome is associated with the PIC at most Pol II promoters17. Correct positioning of the +1 nucleosome positively influences binding of the TATA box-binding protein (TBP), selection of the transcription start site (TSS) and transcription activity18,19,20,21 in vivo.
Here we use a combination of biochemistry and cryo-electron microscopy (cryo-EM) to provide structural insights into the PIC in the context of the +1 nucleosome. We show that TFIIH can engage with the +1 nucleosome in different ways and provide evidence that there is a preferred mode of TFIIH-nucleosome interaction that relies on multiple contacts. Finally, we use the PIC–nucleosome structures and modeling to provide a molecular explanation for long-standing observations on the preferred relative position of the TSS and the location of the +1 nucleosome. Our work thus provides the basis for a detailed analysis of structural and functional PIC-nucleosome interactions at gene promoters.
Results
Formation of PIC–nucleosome complex
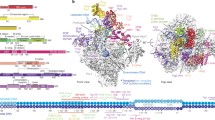
To investigate PIC assembly in the presence of the +1 nucleosome, we formed a PIC from the yeast S. cerevisiae on a promoter flanked by a +1 nucleosome (Fig. 1a,b). At most yeast promoters, the +1 nucleosome covers the TSS, which is typically located roughly 10–15 base pairs (bp) downstream of the proximal border of the nucleosome15,20,22. To mimic this natural arrangement, we prepared a His4 promoter template with a Widom-601-derived nucleosome positioning sequence that places the +1 nucleosome at a position in which the TSS is 10 bp downstream of the proximal border of the nucleosome (Fig. 1a). We reconstituted a nucleosome on this DNA and used the obtained nucleosomal template for in vitro assembly of the PIC4 (Extended Data Fig. 1a and Methods).

a, Schematic of template DNA. The distances between the TATA box, TSS and nucleosome dyad are indicated. The expected lengths of template and transcript are noted at the bottom right. b, Schematic of in vitro transcription assay. For details, see the Methods. c, The +1 nucleosome impairs promoter-dependent transcription in vitro. Assays were performed with template DNA without (DNA) or with +1 nucleosome (Nuc). Representative original scan of the urea–PAGE analysis that yielded results presented on the right. RNA transcripts were analyzed by urea-denaturing PAGE and the full-length product was quantified (dashed rectangle). Experiments were performed at least three times. Bars correspond to the mean of three independent experiments; error bars represent the s.d. FL, full-length transcript; ST, shorter transcripts.
The +1 nucleosome represses transcription
To evaluate the effect of the +1 nucleosome on transcription activity, we performed promoter-dependent in vitro transcription assays9 (Fig. 1b and Methods). We found that the presence of the +1 nucleosome strongly reduced RNA synthesis in this assay (Fig. 1c). The amount of full-length RNA product was reduced to 20% and shorter RNA transcripts were produced, as expected from Pol II stalling within the nucleosome23 (Fig. 1c). To test whether the reduction of RNA synthesis is caused by the high stability of a nucleosome obtained on a Widom-601 derived positioning sequence, we repeated the assay with a template containing the natural His4 promoter sequence. We observed that the level of RNA synthesis was again repressed, albeit to a lesser extent than for the original template containing the nucleosome positioning sequence (Extended Data Fig. 1b). These results indicate that the degree of transcription reduction is related to the stability of the nucleosome while any type of nucleosome causes a decrease in RNA production.
Cryo-EM structure determination
We then determined cryo-EM structures of the reconstituted PIC–nucleosome complex (Methods) in the absence (complex A) or presence (complex B) of nucleoside triphosphates (NTPs) under the conditions of our transcription assay. Classification of the data identified a subset of particles that contained the complete complex (Extended Data Figs. 2 and 3). Cryo-EM densities for TFIIH and the nucleosome were further improved by focused refinement. For complex A we obtained a reconstruction at an overall resolution of 3.3 Å, with local resolutions of 2.9 Å for Pol II, 3.2 Å for the nucleosome and 3.7 Å for TFIIH (Extended Data Figs. 2 and 4 and Supplementary Video 1). Complex B was resolved at an overall resolution of 4.0 Å, with local resolutions of 3.4 Å for Pol II, 3.6 Å for the nucleosome and 3.9 Å for TFIIH (Extended Data Figs. 3 and 4 and Supplementary Video 1). The structures were obtained with the use of atomic models of the PIC9 and the nucleosome24 and subsequent manual modeling, leading to good stereochemistry (Table 1).
PIC structures are largely unchanged
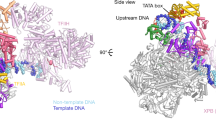
In both cryo-EM structures, the overall conformation of the PIC resembles that in the absence of the nucleosome4,9, except for a minor rotation of TFIIH with respect to the rest of the PIC (Fig. 2 and Extended Data Fig. 5a). The complex adopts the previously described closed promoter state with distorted DNA25 and shows the TFIIH ATPase Ssl2 in the pretranslocation state. The DNA is located above the active center cleft and the initially melting DNA region is flanked by the Rpb1 clamp head loop and the TFIIF charged region as observed before9 (Fig. 2a,b). In contrast to the closed promoter state observed here, previous cryo-EM studies of the yeast PIC revealed a large portion of PIC particles in the open promoter state4,9, suggesting that the +1 nucleosome counteracts DNA opening and that impaired DNA opening is responsible for the observed suppression of transcription in the presence of the +1 nucleosome.

a, Two views of a ribbon model of complex A with template and nontemplate DNA shown as dark and light blue spheres, respectively. b, Two views of a ribbon model of complex B with template and nontemplate DNA shown as dark and light blue spheres, respectively.
Rotation of the +1 nucleosome
Comparison of the structures of complexes A and B shows that the +1 nucleosome is rotated by roughly 75° in the presence of NTPs (Fig. 3a). This rotation is apparently caused by the translocase activity of TFIIH subunit Ssl2 that hydrolyzes ATP to propel downstream DNA into the PIC. Such ATPase action is predicted to cause a rotation of the nucleosome by 30–40° with respect to the PIC for each translocated DNA base pair. The observed roughly 75° rotation would thus correspond to a DNA translocation of 2 bp toward the active center of Pol II, and this is reflected by an observed bending of the DNA duplex into the cleft of Pol II around the initially melted region (Extended Data Fig. 5b). The transition from complex A to complex B also led to a further detachment of nucleosomal DNA from the histone octamer (Fig. 3b). In the absence of NTPs (complex A), only one turn of terminal nucleosomal DNA (SHL–7 to SHL–6) is detached from the histone octamer surface, whereas in the presence of NTPs (complex B), two turns of nucleosomal DNA are detached (SHL–7 to SHL–5).

a, Rotation of the +1 nucleosome as observed by comparison of complex A (light colors, without NTPs) and complex B (full colors, with NTPs). b, Detachment of promoter-proximal, terminal nucleosomal DNA from the histone octamer. Terminal nucleosomal DNA is displaced by roughly 20° and 60° in complexes A and B, respectively, with respect to the canonical nucleosomal DNA path (orange). The nucleosome dyad is indicated in black.
PIC–nucleosome contacts
The rotation of the incoming nucleosome leads to a more intimate association of the PIC and the +1 nucleosome. In the structure of complex A, TFIIH contacts the +1 nucleosome only via its Tfb2-Tfb5 dimerization domain (Fig. 4a and Supplementary Video 1). In complex B, however, TFIIH forms four contact sites with the nucleosome (Fig. 4b and Supplementary Video 1). TFIIH subunits Ssl2 (XPB in human), Tfb2 (p52), Ssl1 (p44) and Tfb4 (p34) all contain charged loop residues that protrude toward the nucleosome in complex B (Fig. 4b). The Ssl2 ATPase domain engages with downstream DNA, whereas the Ssl2 N-terminal extension and clutch domains form a wedge between DNA and the nucleosome (Fig. 4b). This Ssl2 wedge stabilizes the two detached turns of nucleosomal DNA. Tfb2 possesses a lysine-containing loop in its HTH-3 domain (residues 258–270) that approaches the acidic patch of the nucleosome (Fig. 4b). Ssl1 uses a lysine-rich insertion in its RING domain (residues 414–421) to contact nucleosomal DNA around the dyad (Fig. 4b). Finally, Tfb4 uses an extension in its vWA domain (residues 90–105) to reach near the N-terminal region of histone H4 (Fig. 4b). These PIC-nucleosome interactions may counteract further rotation of the +1 nucleosome and impair TFIIH translocase action beyond this state.

a, TFIIH–nucleosome interface in complex A. Tfb2 residues K495, K506 and R507, and Tfb5 residues R3, R5 and K6 from the dimerization domain contact DNA around the nucleosome dyad. Except for K495 in Tfb2, these TFIIH residues are conserved in human TFIIH. b, TFIIH–nucleosome interface in complex B. Four TFIIH subunits that are implicated in nucleosome contacts are shown in different colors. The view is related to the front view in Fig. 2a. The first contact may involve Ssl2 residue D103 and H3 residue R52. The second contact may involve Tfb2 residue K262 and the acidic patch on histones H2A and H2B. The third contact may involve Ssl1 residues K414, K417 and K420 that contact DNA around the nucleosome dyad. The fourth contact may involve Tfb4 residue R104 and histone H4 residue D24. Except for R104 in Tfb4 and K414 and K417 in Ssl1, these TFIIH residues are conserved in human TFIIH.
Evidence for a preferred nucleosome orientation
We next asked whether the position of the nucleosome observed in complex B, and its interactions with TFIIH are specific to the DNA sequence used here or whether they may be of more generic nature. Yeast promoters vary with respect to the distance between their TATA box and their TSS in a range of roughly 40–120 bp (ref. 26), and thus the distance between their TATA box and the dyad of the +1 nucleosome can vary in a range of roughly 100–180 bp. To investigate any such distance effects, we prepared an additional PIC–nucleosome complex on an altered DNA template where the nucleosome positioning sequence was shifted downstream by 10 bp (complex C), resulting in a 130 bp distance between the TATA box and the nucleosome dyad, as compared to a 120 bp distance in complexes A and B (Fig. 5a).

a, Schematic of template DNA for complexes A/B and C. The distances between the TATA box and nucleosome dyad are indicated. b, Structure of PIC–nucleosome complex C compared to complex B. The left panel shows the front view of complex C and B. The right panel shows the similar orientation of the +1 nucleosome in each complex aligned on TFIIH.
We incubated complex C with NTPs and could obtain a cryo-EM structure at an overall resolution of 6.6 Å (Extended Data Fig. 6). Although the resolution prevented us from observing molecular details within the cryo-EM map, the overall orientation of the nucleosome with respect to the PIC in complex C resembled that in complex B (Fig. 5b). This observation indicates that TFIIH uses a common surface to contact the nucleosome in the state of complexes B and C even though the detailed PIC-nucleosome interaction is partially different, suggesting a preferred orientation between TFIIH and the nucleosome may exist when they collide even with different initial PIC–nucleosome distances and on different promoters. We speculate that the preferred orientation of the nucleosome with respect to the PIC observed in complexes B and C occurs within a common intermediate of the transcription initiation process at yeast promoters.
Rpb6 N-terminal tail (NTT) in the Pol II active center
In all three PIC–nucleosome structures, we obtained an ordered conformation of the NTT of Pol II subunit Rpb6 (Fig. 6a and Supplementary Video 1) as confirmed by crosslinking mass spectrometry (Extended Data Fig. 7a). Whereas the NTT is mobile in all previous structures of Pol II complexes, Rpb6 residues 12–35 are observed here in the active center cleft of the polymerase (Fig. 6a). The Rpb6 NTT contains several negatively charged residues that interact with positively charged residues in the cleft that are often conserved (Fig. 6a and Extended Data Fig. 7b).

a, Close-up views of Pol II active center in complex A. Pol II subunits are colored and labeled individually. Rpb6 NTT residues are shown as sticks and labeled as indicated. Electrostatic interactions and hydrogen bonds are shown as yellow dotted lines. b, Superposition of the core PIC containing the Rpb6 NTT with the structure of an ITC (PDB 4BBS)27 shows that binding of the NTT is incompatible with nucleic acid binding during transcription. The template strand, nontemplate strand and RNA transcript in the ITC structure are indicated.
Structural superposition shows that the NTT would clash with DNA and RNA in an initially transcribing complex (ITC)27 (Fig. 6b), indicating its position is incompatible with transcription. The NTT is dispensable for growth of budding yeast28 but its mutation causes temperature sensitivity in the fission yeast S. pombe29. We note that Pol I and Pol III also contain elements that can transiently occupy the active center (Extended Data Fig. 8). These elements are referred to as the expander or the DNA-mimicking loop of A190 in Pol I30,31, and as the C-terminal tail of RPC7 (C31 in yeast) in Pol III32. Further analysis of the function of the NTT awaits the development of a recombinant Pol II system that is currently not available.
Coactivators may be accommodated
Finally, we asked whether the coactivators Mediator and TFIID may be accommodated in our PIC–nucleosome structures. Superimposition of the yeast core Mediator-PIC structure4 indeed showed that Mediator could be appended to our structures without clashes (Extended Data Fig. 9a). In the resulting model, the Mediator hook domain approaches the +1 nucleosome up to a distance of roughly 40 Å (Extended Data Fig. 9a). We also superimposed our PIC–nucleosome structures onto two human PIC structures containing TFIID6,7 since no yeast TFIID-containing PIC structure is available. This showed that TFIID could in principle be accommodated in the PIC–nucleosome complex surface without clashes (Fig. 7 and Extended Data Fig. 9b). The putative TFIID position is consistent with reports that the double bromodomain of TFIID subunit TAF1 and the PHD finger domain of TAF3 can contribute to promoter recognition by binding modified histone tails33,34 (Extended Data Fig. 9b). Histone modifications might also be involved in regulating the assembly of PIC.

Discussion
Here, we report structures of Pol II PIC–nucleosome transcription complexes. The structures show that TFIIH directly interacts with the nucleosome in distinct conformations. They also indicate that a preferred orientation of the nucleosome with respect to the PIC is adopted that is characterized by multiple TFIIH–nucleosome contacts. Although the nucleosome may initially be found in different rotational states relative to the PIC, action of the Ssl2 translocase may lead to a preferred nucleosome orientation that allows for multiple TFIIH contacts as observed in complex B and C of this study. As TFIIH apparently has ceased to unravel nucleosomal DNA beyond SHL–5 in complex B, the translocase activity of TFIIH may not be sufficient to enable the PIC to pass through nucleosomal sites with strong histone-DNA interactions (such as SHL–5 and –1). Passage of PIC through the +1 nucleosome may thus rely on assistance from chromatin remodelers and chromatin modifying complexes.
Together with modeling, the observed structures with the stalled PIC on the nucleosome also provide a possible explanation for why the TSS is often located roughly 60 bp upstream of the dyad of the +1 nucleosome in yeast15. Before initiation of RNA chain synthesis, the yeast PIC scans downstream DNA for the TSS35. Modeling based on complex B indicates that scanning requires further progression of the PIC into the +1 nucleosome and detachment of three additional turns of DNA (Extended Data Fig. 10). This may be achieved by the ATP-dependent Ssl2 translocase whose activity is required for scanning36,37. We speculate that scanning may be impaired at the major histone-DNA interacting sites (for example, SHL–1) just upstream of the nucleosome dyad23,38, which then may trigger TSS usage and RNA chain initiation as suggested13. This model of nucleosome-defined TSS usage may explain why TSSs in yeast are generally located at a distance of roughly 60 bp from the dyad of the +1 nucleosome, even though the distance from the TSS to the TATA box varies in the range of roughly 40–120 bp (ref. 26). In our experimental system, the high stability of the nucleosome on the Widom-601 positioning sequence may have prevented scanning and led to a stable intermediate amenable to structure determination.
Methods
Preparation of PIC–nucleosome complex
S. cerevisiae 12-subunit Pol II, TBP, TFIIA, TFIIB, TFIIE, TFIIF and TFIIH were purified as described previously2,4,39. The DNA scaffolds containing the modified His4 promoter and Widom-601 sequence were synthesized by Integrated DNA Technologies, amplified and purified as described40. Xenopus laevis and S. cerevisiae histones were prepared and assembled into nucleosomes as described41,42. The sequences for the scaffold used in this study is listed with the Widom-601 sequence underlined. The TATA box and TSS are indicated in bold.
His4 promoter (209 bp):
5′-AGCACGCTGTGTATATAATAGCTATGGAACGTTCGATTCACCTCCGATGTGTGTTGTACATACATAAAAATATCATAGCTCTTCTGCGCTGTGTTATAGTAATACAATAGTTTACAAAATTTTTTTTCTGAATAATGGTTTTGCCGATTCTACCGTTAATTGATGATCTGGCCTCATGGAATAGTAAGAAGGAATACGTTTCACTTGTT-3′
Scaffold for complex A and B (209 bp):
5′-AGCACGCTGTGTATATAATAGCTATGGAACGTTCGATTCACCTCCGATGTGTGTTGTACATACATAAAAATATCATAGCTCTTCTGCGCTGTGTTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCGAT-3′
Scaffold for complex C (219 bp):
5′-AGCACGCTGTGTATATAATAGCTATGGAACGTTCGATTCACCTCCGATGTGTGTTGTACATACATAAAAATATCATAGCTCTTCTGCGCTGTGTTCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCGAT-3′
The PIC–nucleosome complex was assembled according to a previously reported protocol4, with minor modifications. Briefly, scaffold containing a reconstituted nucleosome was incubated with TBP, TFIIA and TFIIB, whereas Pol II and TFIIF were incubated for 10 min at 25 °C. These two preparations were then combined and incubated for another 5 min. TFIIE and preassembled 10-subunit TFIIH were added to the mixture simultaneously and incubated for 5 min. NTPs at a concentration of 400 μM were added and the assembly was incubated for 1 h at 25 °C. The PIC–nucleosome samples were subjected to GraFix ultracentrifugation43 at 137,600g (32,000 r.p.m. for SW60 rotor) for 16 h at 4 °C in a 15–40% (w/v) sucrose gradient with 0–0.1% glutaraldehyde crosslinker. Subsequently, the gradient solutions were fractionated and quenched with a mixture of 40 mM aspartate and 10 mM lysine for 10 min. Fractions were analyzed by native PAGE. Gels were stained with SYBR Gold (Invitrogen) and Coomassie brilliant blue. Peak fractions containing crosslinked PIC–nucleosome complex were dialyzed for 16 h in dialysis buffer (20 mM HEPES-Na pH 7.5, 50 mM KCl, 2 mM MgCl2, 1 mM TCEP) to remove sucrose. The dialyzed samples were concentrated to 0.2 mg ml−1 and used for grid preparation.
In vitro promoter-dependent transcription assay
Assays were performed as described9, with minor alterations. DNA scaffolds with or without nucleosome were prepared as described above. All scaffolds contained identical DNA sequences irrespective of the nucleosome component. Assembled scaffolds were stored in a low salt buffer (50 mM KCl, 5 mM K-HEPES pH 7.5, 0.025 mM EDTA). PIC was reconstituted on scaffold DNA essentially as reported9. All incubation steps were performed at 25 °C unless indicated otherwise. Per sample, 1.6 pM of TBP, 1.8 pM Pol II, 2.7 pM TFIIE and TFIIH, 9 pM TFIIF, 9 pM TFIIB and 18 pM TFIIA were used. Reactions were prepared in a sample volume of 23.8 µl with final assay conditions of 3 mM HEPES-K pH 7.9, 20 mM Tris-HCl pH 7.9, 60 mM KCl, 8 mM MgCl2, 2% (w/v) PVA, 3% (v/v) glycerol, 0.5 mM DTT, 0.5 mg ml−1 BSA and 20 units of RNase inhibitor. Samples were incubated for 45 min and transcription was started by adding 1.2 µl of 10 mM NTP solution and permitted to proceed for 60 min. Reactions were quenched with 100 µl of Stop buffer (300 mM NaCl, 10 mM Tris-HCl pH 7.5, 0.5 mM EDTA) and 14 µl of 10% SDS, followed by treatment with 4 µg of proteinase K (New England Biolabs) for 30 min at 37 °C. RNA products were isolated from the samples as described9, applied to urea gels (7 M urea, 1× TBE, 6% acrylamide:bis-acrylamide 19:1) and separated by denaturing gel electrophoresis (urea–PAGE) in 1× TBE buffer for 45 min at 180 V. Gels were stained for 30 min with SYBR Gold (Invitrogen) and RNA was visualized with a Typhoon 9500 FLA imager (GE Healthcare Life Sciences). The densities of the bands on the gels were quantitated with ImageJ.
Cryo-EM analysis and data processing
Four microliters of PIC–nucleosome samples were applied to glow-discharged UltrAuFoil 2/2 grids (Quantifoil). After incubation on grids for 10 s, samples were blotted for 4 s and vitrified by plunging into liquid ethane via a Vitrobot Mark IV (FEI) operated at 4 °C and 100% humidity. Cryo-EM data were collected on a Titan Krios G2 transmission electron microscope (FEI) operated at 300 keV, equipped with a K3 summit direct detector and a GIF quantum energy filter (Gatan). Automated data acquisition was performed using SerialEM software at a nominal magnification of ×81,000, corresponding to a physical 1.05 Å per pixel. Image stacks of 40 frames were collected in counting mode over 1.5 s at a defocus range from 0.8–2.0 μm. The dose rate was 27 e−/Å2 per second resulting in 1.02 e−/Å2 per frame. Totals of 26,764, 31,286 and 15,515 videos were collected for complexes A, B and C, respectively.
Image stacks were motion-corrected, contrast-transfer function corrected, dose-weighted and auto-picked using Warp44. Image processing was performed with RELION v.3.0.5 (ref. 45). Particles were extracted using a box size of 4002 or 3602 pixels, and normalized. Reference-free 2D classification was performed to remove poorly aligned particles. An ab initio model generated with cryoSPARC46 was used for subsequent 3D classification. All classes containing PIC–nucleosome density were combined and used for a global 3D refinement. To obtain an improved density map for cPIC, TFIIH and the nucleosome, particles were subjected to focused 3D classification without image alignment. All classes containing good cPIC density were subjected to contrast-transfer function refinement, Bayesian polishing and 3D refinement. Postprocessing of refined models was performed using automated B factor determination in RELION and reported resolutions were based on the gold-standard Fourier shell correlation 0.143 criterion. The density of TFIIH was further improved by applying signal subtraction and focused refinement. Local resolution estimates were obtained using the built-in local resolution estimation tool of RELION and previously estimated B factors.
Model building
The structural models were built into the density of the final reconstructions with the best local resolutions for PIC or the TFIIH–nucleosome complex. A nucleosome structure with 145 bp Widom-601 DNA (Protein Data Bank (PDB) 7OHC)24 and the structure of yeast PIC (PDB 7O73)9 were placed into the density maps by rigid-body fitting in Chimera47, followed by manually adjustment and connection of linker DNA. The assignment of Rpb6 NTT was guided by the densities of bulky side chains, the crosslinking mass spectrometry and secondary structure prediction. The models were subjected to alternating real-space refinement and manual adjustment using PHENIX48 and COOT49, resulting in very good stereochemistry as assessed by Molprobity50.
Crosslinking and mass spectrometry
Samples for mass spectrometry were prepared by sucrose gradient centrifugation as described above for cryo-EM sample preparation without glutaraldehyde. Fractions containing fully assembled complexes were pooled, subjected to chemical crosslinking using zero-length crosslinker EDC (100 mM) and NHS (100 mM) for 1 h at room temperature, and quench with 100 mM ammonium bicarbonate. The samples were adjusted to 8 M urea, 50 mM NH4HCO3, 10 mM DTT followed by an incubation for 30 min at 37 °C. Proteins were alkylated in the presence of 40 mM iodoacetamide for another 30 min at 37 °C in the dark and the reaction was quenched by 10 mM DTT for 5 min at 37 °C. The reaction volume was adjusted to reach a final concentration of 1 M urea and 50 mM NH4HCO3. Nucleic acids fragments within the PIC–nucleosome complex were digested for 30 min at 37 °C by the addition of 0.1 M MgCl2 to a final concentration of 1 mM in the reaction and 500 U of universal nuclease (Pierce, catalog no. 88702, 250 U µl−1). Trypsin digest was performed overnight at 37 °C with 5 μg of trypsin (Promega, V5111). Peptides were acidified with 4 µl of 100% formic acid, desalted on MicroSpin columns (Harvard Apparatus) following the manufacturer’s instructions and vacuum dried. Dried peptides were dissolved in 50 µl of 30% acetonitrile/0.1% TFA and peptide size exclusion (pSEC, Superdex Peptide 3.2/300 column) on an ÄKTA micro system (GE Healthcare) was performed to enrich for crosslinked peptides at a flow rate of 50 µl min−1. Fractions of 50 µl were collected. The first 21 fractions enriched in crosslinked peptides were vacuum dried and dissolved in 5% acetonitrile/0.05% TFA (v/v) for analysis by liquid chromatography with tandem mass spectrometry.
Crosslinked peptides derived from pSEC were analyzed as technical duplicates on Q Exactive HF-X hybrid quadrupole-orbitrap mass spectrometer (Thermo Scientific), coupled to a Dionex UltiMate 3000 UHPLC system (Thermo Scientific). The sample was separated on an in-house-packed C18 column (ReproSil-Pur 120 C18-AQ, 1.9 µm pore size, 75 µm inner diameter, 30 cm length, Dr. Maisch GmbH) at a flow rate of 300 nl min−1. Sample separation was performed over 60 min using a buffer system consisting of 0.1% (v/v) formic acid (buffer A) and 80% (v/v) acetonitrile, 0.08% (v/v) formic acid (buffer B). The main column was equilibrated with 5% B, followed by sample application and a wash with 5% B. Peptides were eluted by a linear gradient from 15–48% B or 20–50% B. The gradient was followed by a wash step at 95% B and re-equilibration at 5% B. Eluting peptides were analyzed in positive mode using a data-dependent top-30 acquisition methods. MS1 and MS2 resolution were set to 120,000 and 30,000 full width at half maximum, respectively. Precursors selected for MS2 were fragmented using 30% normalized, higher-energy collision induced dissociation fragmentation. Allowed charge states of selected precursors were +3 to +7. Further tandem mass spectrometry parameters were set as follows: isolation width, 1.4 m/z; dynamic exclusion, 10 s and maximum injection time (MS1/MS2), 60/200 ms.
For identification of crosslinked peptides, raw files were analyzed by pLink (v.2.3.5), pFind group51 using EDC as crosslinker and trypsin/P as digestion enzyme with maximal three missed cleavage sites. The search was conducted against a customized protein database containing all proteins within the complex (Supplementary Table 1). Carbamidomethylation of cysteines was set as a fixed modification, oxidation of methionines and acetylation at protein N termini were set as a variable modification. Searches were conducted in combinatorial mode with a precursor mass tolerance of 10 ppm and a fragment ion mass tolerance of 20 ppm. The false discovery rate was set to 0.05 (separate mode). Spectra of both technical duplicates were combined and evaluated manually.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The electron density reconstructions and final models were deposited with the EM Data Bank (accession codes EMD-14927, 14928 and 14929) and with the PDB (accession codes PDB 7ZS9, 7ZSA and 7ZSB). All mass spectrometry raw files were deposited to the ProteomeXchange Consortium (https://www.proteomexchange.org/) via the PRIDE52 partner repository with the dataset identifier PRIDE: PXD029840. Source data are provided with this paper.
References
He, Y. et al. Near-atomic resolution visualization of human transcription promoter opening. Nature 533, 359–365 (2016).
Plaschka, C. et al. Transcription initiation complex structures elucidate DNA opening. Nature 533, 353–358 (2016).
Robinson, P. J. et al. Structure of a complete Mediator-RNA polymerase II pre-initiation complex. Cell 166, 1411–1422 e1416 (2016).
Schilbach, S. et al. Structures of transcription pre-initiation complex with TFIIH and Mediator. Nature 551, 204–209 (2017).
Aibara, S., Schilbach, S. & Cramer, P. Structures of mammalian RNA polymerase II pre-initiation complexes. Nature https://doi.org/10.1038/s41586-021-03554-8 (2021).
Chen, X. et al. Structural insights into preinitiation complex assembly on core promoters. Science https://doi.org/10.1126/science.aba8490 (2021).
Chen, X. et al. Structures of the human Mediator and Mediator-bound preinitiation complex. Science https://doi.org/10.1126/science.abg0635 (2021).
Rengachari, S., Schilbach, S., Aibara, S., Dienemann, C. & Cramer, P. Structure of human Mediator–RNA polymerase II pre-initiation complex. Nature https://doi.org/10.1038/s41586-021-03555-7 (2021).
Schilbach, S., Aibara, S., Dienemann, C., Grabbe, F. & Cramer, P. Structure of RNA polymerase II pre-initiation complex at 2.9 A defines initial DNA opening. Cell https://doi.org/10.1016/j.cell.2021.05.012 (2021).
Bernstein, B. E., Liu, C. L., Humphrey, E. L., Perlstein, E. O. & Schreiber, S. L. Global nucleosome occupancy in yeast. Genome Biol. 5, R62 (2004).
Lee, C. K., Shibata, Y., Rao, B., Strahl, B. D. & Lieb, J. D. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat. Genet. 36, 900–905 (2004).
Jiang, C. & Pugh, B. F. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 10, 161–172 (2009).
Rhee, H. S. & Pugh, B. F. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483, 295–301 (2012).
Yuan, G. C. et al. Genome-scale identification of nucleosome positions in S. cerevisiae. Science 309, 626–630 (2005).
Albert, I. et al. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446, 572–576 (2007).
Lai, W. K. M. & Pugh, B. F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol. 18, 548–562 (2017).
Rossi, M. J. et al. A high-resolution protein architecture of the budding yeast genome. Nature 592, 309–314 (2021).
Kubik, S. et al. Sequence-directed action of RSC remodeler and general regulatory factors modulates +1 nucleosome position to facilitate transcription. Mol. Cell 71, 89–102 e105 (2018).
Kubik, S. et al. Opposing chromatin remodelers control transcription initiation frequency and start site selection. Nat. Struct. Mol. Biol. 26, 744–754 (2019).
Challal, D. et al. General regulatory factors control the fidelity of transcription by restricting non-coding and ectopic initiation. Mol. Cell 72, 955–969 e957 (2018).
Klein-Brill, A., Joseph-Strauss, D., Appleboim, A. & Friedman, N. Dynamics of chromatin and transcription during transient depletion of the rsc chromatin remodeling complex. Cell Rep. 26, 279–292 e275 (2019).
Whitehouse, I., Rando, O. J., Delrow, J. & Tsukiyama, T. Chromatin remodelling at promoters suppresses antisense transcription. Nature 450, 1031–1035 (2007).
Kujirai, T. et al. Structural basis of the nucleosome transition during RNA polymerase II passage. Science 362, 595–598 (2018).
Wang, H., Xiong, L. & Cramer, P. Structures and implications of TBP-nucleosome complexes. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.2108859118 (2021).
Dienemann, C., Schwalb, B., Schilbach, S. & Cramer, P. Promoter distortion and opening in the RNA Polymerase II cleft. Mol. Cell 73, 97–106 e104 (2019).
Struhl, K. Molecular mechanisms of transcriptional regulation in yeast. Annu. Rev. Biochem. 58, 1051–1077 (1989).
Sainsbury, S., Niesser, J. & Cramer, P. Structure and function of the initially transcribing RNA polymerase II-TFIIB complex. Nature 493, 437–440 (2013).
Nouraini, S., Archambault, J. & Friesen, J. D. Rpo26p, a subunit common to yeast RNA polymerases, is essential for the assembly of RNA polymerases I and II and for the stability of the largest subunits of these enzymes. Mol. Cell. Biol. 16, 5985–5996 (1996).
Ishiguro, A., Nogi, Y., Hisatake, K., Muramatsu, M. & Ishihama, A. The Rpb6 subunit of fission yeast RNA polymerase II is a contact target of the transcription elongation factor TFIIS. Mol. Cell. Biol. 20, 1263–1270 (2000).
Fernandez-Tornero, C. et al. Crystal structure of the 14-subunit RNA polymerase I. Nature 502, 644–649 (2013).
Engel, C., Sainsbury, S., Cheung, A. C., Kostrewa, D. & Cramer, P. RNA polymerase I structure and transcription regulation. Nature 502, 650–655 (2013).
Wang, Q. et al. Structural insights into transcriptional regulation of human RNA polymerase III. Nat. Struct. Mol. Biol. 28, 220–227 (2021).
Jacobson, R. H., Ladurner, A. G., King, D. S. & Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 288, 1422–1425 (2000).
Vermeulen, M. et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 (2007).
Hahn, S. & Young, E. T. Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189, 705–736 (2011).
Fishburn, J., Galburt, E. & Hahn, S. Transcription start site scanning and the requirement for ATP during transcription initiation by RNA polymerase II. J. Biol. Chem. 291, 13040–13047 (2016).
Qiu, C. et al. Universal promoter scanning by Pol II during transcription initiation in Saccharomyces cerevisiae. Genome Biol. 21, 132 (2020).
Ehara, H. et al. Structural insight into nucleosome transcription by RNA polymerase II with elongation factors. Science 363, 744–747 (2019).
Sydow, J. F. et al. Structural basis of transcription: mismatch-specific fidelity mechanisms and paused RNA polymerase II with frayed RNA. Mol. Cell 34, 710–721 (2009).
Wang, H., Farnung, L., Dienemann, C. & Cramer, P. Structure of H3K36-methylated nucleosome-PWWP complex reveals multivalent cross-gyre binding. Nat. Struct. Mol. Biol. 27, 8–13 (2020).
Dyer, P. N. et al. Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 375, 23–44 (2004).
Kingston, I. J., Yung, J. S. & Singleton, M. R. Biophysical characterization of the centromere-specific nucleosome from budding yeast. J. Biol. Chem. 286, 4021–4026 (2011).
Kastner, B. et al. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5, 53–55 (2008).
Tegunov, D. & Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife https://doi.org/10.7554/eLife.42166 (2018)
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290 (2017).
Pettersen, E. F. et al. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. D. Struct. Biol. 74, 531–544 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D. 66, 486–501 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 12–21 (2010).
Chen, Z. L. et al. A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat. Commun. 10, 3404 (2019).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Acknowledgements
We thank C. Dienemann and U. Steuerwald for advice and for maintaining the EM facility. We thank F. Grabbe for purification of the PIC components and E. Oberbeckmann and L. Xiong for discussions. H.W. was supported by an EMBO Long-Term-Fellowship (grant no. ALTF 650-2017). H.U. was supported by the Deutsche Forschungsgemeinschaft (grant no. SFB860). P.C. was supported by the Deutsche Forschungsgemeinschaft (grant no. SFB860, EXC 2067/1-390729940) and the European Research Council Advanced Investigator grant CHROMATRANS (grant agreement no. 882357).
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
H.W. and P.C. conceived the study. H.W. designed and carried out all experiments and data analysis, unless stated otherwise. S.S. performed transcription experiments and provided advice on cryo-EM processing. M.N. and H.U. carried out mass spectrometry analysis. P.C. supervised research. H.W. and P.C. interpreted the data and wrote the manuscript, with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Structural and Molecular Biology thanks Patrick Schultz and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Carolina Perdigoto, in collaboration with the Nature Structural & Molecular Biology team. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Transcription assay using His4 promoter DNA.
a. Reconstitution of nucleosome with His4 promoter DNAs. Template DNAs (DNA) and reconstituted nucleosomes (Nuc) were analyzed by 1% agarose gel. M: single strand RNA marker with size for each band on the right. b. The +1 nucleosome impairs promoter-dependent transcription in vitro. Assays were performed with template DNA without (DNA) or with +1 nucleosome (Nuc). RNA transcripts were analyzed by urea-denaturing PAGE and the full-length product was quantified (dashed rectangle). This experiment was repeated 3 times independently with similar results. Bars correspond to the mean of three independent experiments; error bars represent the s.d.
Extended Data Fig. 2 Cryo-EM structure determination and analysis of PIC-nucleosome complex A.
a. Exemplary cryo-EM micrograph. A scale bar is provided. In total 26,764 micrographs were collected with similar results. b. Particle sorting and classification tree. Regions corresponding to Pol II, general transcription factors (GTFs) and template DNA are colored as in Fig. 2a, histones are colored in light yellow. Maps deposited to EMDB are indicated with grey background and outlined in black. The subpopulation of TBP-nucleosome complex is not described in this study since a similar TBP-NCP structure has been published before24. c. Representative 2D class averages of sorted particles used for final reconstruction.
Extended Data Fig. 3 Cryo-EM structure determination and analysis of PIC-nucleosome complex B.
a. Exemplary cryo-EM micrograph. A scale bar is provided. In total 31,286 micrographs were collected with similar results. b. Particle sorting and classification tree. Regions corresponding to Pol II, general transcription factors (GTFs) and template DNA are colored as in Fig. 2b, histones are colored in light yellow. Maps deposited to EMDB are indicated with grey background and outlined in black. The subpopulation of TBP-nucleosome complex is not described in this study since a similar TBP-NCP structure has been published before24. c. Representative 2D class averages of sorted particles used for final reconstruction.
Extended Data Fig. 4 Quality of cryo-EM reconstructions of complexes A and B.
a. PIC-nucleosome complex A and B reconstructions colored according to local resolution43 for the core PIC (left), TFIIH (middle) and the +1 nucleosome (right). The color bars from blue to red indicate the local resolution range in Å. Angular distribution diagrams for particles in the final reconstructions are shown on the right. Color shading from blue to yellow correlates with the number of particles at a specific orientation as indicated. b. Fourier shell correlation (FSC) between the half maps of the reconstruction. The average resolution is estimated at the FSC 0.143 cut-off criterion (dashed line). c. Electron densities (blue mesh) for various parts as indicated. d. Model-to-map FSC correlation between the final model and reconstruction. The depicted correlation curves were calculated from the FSC between the derived model and the reconstruction. The resolutions at the FSC 0.5 cut-off criterion (dashed line) are denoted.
Extended Data Fig. 5 Structural changes in the PIC-nucleosome complexes.
a. Superimposition of PIC-nucleosome complex A and B on previous PIC complex (PDB code: 7O73)9. b. Superimposition of PIC-nucleosome complex A and B aligned on Pol II. The red dashed box encloses the DNA around initially melting region. The black arrows indicate the directions of movement of the DNA towards the active center of Pol II.
Extended Data Fig. 6 Cryo-EM structure determination and analysis of PIC-nucleosome complex C.
a. Exemplary cryo-EM micrograph. A scale bar is provided. In total 15,515 micrographs were collected with similar results. b. Particle sorting and classification tree. Regions corresponding to Pol II, general transcription factors (GTFs) and template DNA are colored as in Fig. 2a, histones are colored in light yellow. Maps deposited to EMDB are indicated with grey background and outlined in black. The subpopulation of TBP-nucleosome complex is not described in this study since a similar TBP-NCP structure has been published before24. c. Representative 2D class averages of sorted particles used for final reconstruction.
Extended Data Fig. 7 Rbp6 NTT occupies the active center cleft of Pol II.
a. Crosslinks between Rpb6 NTT and other Pol II subunits. The close-up view shows three observed EDC crosslinks (black dashed lines). b. Sequence alignment of Rpb6 orthologs. The NTT regions are boxed. Acidic residues of Rpb6 NTT that are involved in interaction with the Pol II cleft are indicated in bold.
Extended Data Fig. 8 Comparison of structural elements occupying the cleft of Pol I, II, and III.
a. Pol I and Pol III contain elements that can occupy the active center cleft at a location corresponding to that observed for the Rpb6 NTT in Pol II (Extended Data Fig. 4). These elements are referred to as the expander or DNA-mimicking loop of A190 in yeast Pol I (PDB code: 4C3H)31, and as the C-terminal tail of RPC7 (C31 in yeast) in human Pol III (PDB code: 7D59)32. b. Superposition of cleft elements in Pol I, Pol II and Pol III. The bridge helix and the catalytic magnesium ion in the active site are indicated.
Extended Data Fig. 9 Mediator and TFIID are compatible with the PIC-nucleosome structure.
a. Yeast core Mediator (cMed) was placed onto our PIC-nucleosome structure by superimposing the core PIC in the yeast cMed-PIC structure (PDB code: 5OQM)4. b. TFIID was placed onto the PIC-nucleosome structure by superimposing the core PIC in the human TFIID-containing PIC structure (PDB code: 7EGB)6 since no yeast TFIID-containing PIC structure is available. The unmodelled double bromodomain of TAF1 and the PHD finger domain of TAF3 are depicted as ovals. The unmodelled histone tails in the structure are depicted as threads with histone modifications symbolized as spheres.
Extended Data Fig. 10 Model for TSS scanning.
a. The PIC-nucleosome complex structure presented here corresponds to the state before scanning starts (pre-scanning state). The Pol II active center and the TSS are indicated. The lower panel shows a schematic representation. Distances between the TATA box and selected DNA elements are denoted. b. Model of the PIC-nucleosome complex in the post-scanning state. The modelled structure was generated using structures of the yeast PIC with open DNA (PDB code: 7O75)9 and of Pol II with a partially unraveled nucleosome (PDB code: 6A5T)23. The partially unraveled nucleosome with 5 turns of nucleosomal DNA detached from the histone octamer position was aligned with the PIC-nucleosome structure based on DNA and manually adjusted to avoid clashes between the nucleosome and TFIIH.
Supplementary information
Supplementary Table 1
PIC–nucleosome EDC crosslinking data.
Supplementary Video 1
Overview of PIC–nucleosome structures.
Source data
Source Data Fig. 1
Unprocessed gel.
Source Data Extended Data Fig. 1
Unprocessed gels.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Schilbach, S., Ninov, M. et al. Structures of transcription preinitiation complex engaged with the +1 nucleosome. Nat Struct Mol Biol 30, 226–232 (2023). https://doi.org/10.1038/s41594-022-00865-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-022-00865-w
- Springer Nature America, Inc.
This article is cited by
-
Nucleosomal DNA has topological memory
Nature Communications (2024)
-
Quantitative analysis of transcription start site selection reveals control by DNA sequence, RNA polymerase II activity and NTP levels
Nature Structural & Molecular Biology (2024)
-
When transcription initiation meets chromatin
Nature Structural & Molecular Biology (2023)