

Abstract
Amyloid-β (Aβ) is thought to be neuronally derived in Alzheimer’s disease (AD). However, transcripts of amyloid precursor protein (APP) and amyloidogenic enzymes are equally abundant in oligodendrocytes (OLs). By cell-type-specific deletion of Bace1 in a humanized knock-in AD model, APPNLGF, we demonstrate that OLs and neurons contribute to Aβ plaque burden. For rapid plaque seeding, excitatory projection neurons must provide a threshold level of Aβ. Ultimately, our findings are relevant for AD prevention and therapeutic strategies.
Similar content being viewed by others
Main
In Alzheimer’s disease (AD), amyloid-β (Aβ) production has primarily been attributed to excitatory neurons (ExNs)1, despite emerging evidence that other cell types might contribute to Aβ production2,3. Cultured oligodendrocytes (OLs) are capable of generating detectable levels of Aβ in vitro4,5,6. Because OL lineage cells are abundantly present in both gray matter and white matter (WM), and myelin alterations have been implicated in AD7,8,9, we asked whether OLs directly contribute to Aβ plaque burden in vivo.
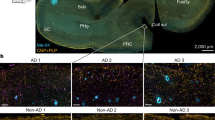
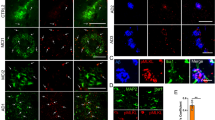
We first interrogated multiple sequencing datasets of wild-type (WT) mouse9,10,11 and healthy control human12,13,14 nervous tissue for expression of amyloidogenic pathway genes (APP, BACE1, PSEN1 and PSEN2) (Fig. 1 and Extended Data Fig. 1a,b). Depending on the sequencing technology and tissue input, positive cell rates of amyloidogenic transcripts varied, but expression levels were similar between neurons and OLs (Extended Data Fig. 1c,d). We validated the expression of amyloid precursor protein (APP) in murine OLs in vitro and in vivo (Extended Data Fig. 2a,b), alongside human OLs via immunolabeling (Extended Data Fig. 2c). By in situ hybridization (ISH) in human cortical tissue, we found that approximately 50% of all gray matter OLs express considerable levels of APP and BACE1 mRNA in both AD cases and controls (Extended Data Fig. 2d–f). Thus, both mouse and human OLs express the essential components for Aβ generation.

a, UMAP visualization of cell types from a 6-month-old mouse brain snRNA-seq dataset9. b, Feature plots showcasing expression of key amyloidogenic genes (App, Bace1, Psen1 and Psen2) across all cell types in WT mouse brains. c, Expression level half violin plots of key amyloidogenic genes in neurons and OLs of mouse brains normalized by the SCTransform method, highlighting the similar expression of all genes between neurons and OLs. d, Expression level half violin plots of key amyloidogenic genes in neurons and OLs normalized by the SCTransform normalization method from additional mouse datasets9,10,11. e, Mouse nervous tissue inputs for sequencing from each study are shown. f, UMAP visualization of cell types from a human brain snRNA-seq dataset12. g, Feature plots showcasing expression of key amyloidogenic genes (APP, BACE1, PSEN1 and PSEN2) across all cell types in control human brains. h, Expression level half violin plots of key amyloidogenic genes in neurons and OLs of human brains with the SCTransform normalization method, highlighting the similar expression of these genes between neurons and OLs. i, Expression level half violin plots of key amyloidogenic genes in neurons and OLs normalized by the SCTransform normalization method from additional human datasets13,14. j, Positive rate bar plots of APP processing genes in mouse and human nervous tissue inputs. k, Human nervous tissue inputs for sequencing from each study are shown. c,d,h,i, Half violins represent aggregated expression levels of respective genes from each cell type, and data points refer to individual expression levels from single cells or nuclei normalized by SCTransform. The results published here are based on data obtained from the Gene Expression Omnibus and the AD Knowledge Portal. mo, months; UMAP, uniform manifold approximation and projection.
Next, we created novel AD mouse lines to assess Aβ contribution from OLs and ExNs separately (Fig. 2a). For this, we employed APPNLGF knock-in mice that express a humanized and triple-mutated APP in the endogenous App locus to circumvent transgenic mouse artifacts. These mice were crossed with Bace1fl/fl mice to conditionally knock out Bace1 (Bace1 cKO), the rate-limiting enzyme in Aβ generation, using cell-type-specific Cre drivers, namely, Cnp-Cre for OLs and Nex-Cre for dorsal telencephalic ExNs. We termed the resultant triple-mutant mice OL-Bace1cKO;AD and ExN-Bace1cKO;AD, respectively, and compared them to non-Cre controls termed Control;AD.

a, Mouse breeding setup to investigate the OL-specific and ExN-specific contributions to Aβ burden. b–f, LSM data of plaque burden (Congo red) comparing 6-month-old OL-Bace1cKO;AD mice to age-matched and sex-matched littermate controls. g–k, LSM data of plaque burden (Congo red) comparing 6-month-old ExN-Bace1cKO;AD mice to age-matched and sex-matched littermate controls. b–k, Color-region allocation is as follows: white, isocortex; blue, hippocampus; yellow, alveus; pastel blue, inferior colliculus; pastel yellow, thalamus. b,g, LSM 3D visualization of control and cKO hemibrains. c,h, Brain region-segmented plaques of control and cKO hemibrains. d,i, LSM 2D single plane of control and cKO hemibrains. Arrowheads point to plaques with colors indicating specific regions. e,j, LSM 3D renders of representative cortical Aβ plaques of control and cKO hemibrains. f,k, Quantification of LSM data between controls (n = 5 per sex) and cKOs (n = 5 per sex). Normalization of cKO data points to sex-matched controls was performed. Circles represent controls, and triangles represent cKOs. Filled shapes represent male mice, and hollowed shapes represent female mice. For each parameter, unpaired, two-tailed Student’s t-test was performed (P values indicated in graphs with significance highlighted in bold) comparing cKOs to controls. Bars represent means with s.e.m., and individual data points are displayed. Raw data are available in Supplementary Tables 1 and 2.
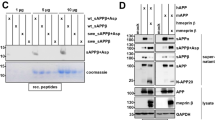
We assessed Cnp-Cre specificity using a stop-flox tdTomato reporter mouse line as transient neuronal Cnp-Cre activity has been detected to varying degrees15,16. In concordance with recent findings17, only a very low percentage of cortical (0.756% ± 0.057%) and hippocampal (0.468% ± 0.111%) neurons were tdTomato+ (Extended Data Fig. 3). We then validated the cell-type-specific Bace1 transcript reduction using ISH, whereby Bace1 transcripts were massively reduced in the intended target cell type (Extended Data Fig. 4). Notably, we confirmed that ExN Bace1 transcript levels were unaffected in OL-Bace1cKO;AD animals (Extended Data Fig. 4a–d,h–m). We next attempted to demonstrate BACE1 protein expression in OLs and to validate successful knockdown in OLs on a protein level in OL cultures from Control;AD and OL-Bace1cKO;AD mice (Extended Data Fig. 5a,b). However, we failed to validate Bace1 cKO via immunocytochemistry using the 3D5 antibody. We, therefore, investigated the specificity and sensitivity of this antibody in tissue sections by comparing WT animals to constitutive Bace1 KO animals. We only observed a loss of 3D5 labeling in the mossy fibers in Bace1 KO (Extended Data Fig. 5c), yet no somatic BACE1 was observed (Extended Data Fig. 5d). As immunohistochemical validation of Bace1 KO was not feasible, we sorted OLs from Control;AD and OL-Bace1cKO;AD mice and showed abolishment of BACE1 in cKO tissue via immunoblotting by which BACE1 can be readily detected (Extended Data Fig. 5e). We also validated the specificity of the APP antibody Y188 by investigating constitutive APP KO tissue, which revealed loss of both neuronal and OL APP (Extended Data Fig. 5f).
We then turned to western blot analysis to validate APP processing alterations (Extended Data Fig. 5g,h). Full-length APP (FL-APP) levels were 40% lower in control APPNLGF lysates compared to WT brains. As expected, both Bace1 cKOs in ExNs and OLs resulted in a region-dependent depletion of BACE1, reflecting local differences in neuron-to-OL ratio. Of note, WM tracts harbor a substantial amount of axoplasm containing neuronally expressed BACE1 (ref. 18), explaining the reduction seen in the WM of ExN-Bace1cKO;AD mice. Cell-type-specific losses of BACE1 diminished β C-terminal fragments (β-CTFs) in the cKOs and restored FL-APP to nearly baseline WT amounts. Levels of presenilin-1 (PSEN1) remained unchanged.
Next, we used light sheet microscopy (LSM) for in toto imaging of amyloid plaques in OL-Bace1cKO;AD and ExN-Bace1cKO;AD mouse hemibrains at 6 months in both sexes (Extended Data Fig. 6). We analyzed cortex and hippocampus for gray matter and the alveus as a representative WM tract, alongside the thalamus and inferior colliculus as regions that do not show Nex-Cre recombination.
OL-Bace1cKO;AD mice accumulated approximately 30% fewer plaques when compared to respective controls in both sexes (Fig. 2b–f). The decrease in plaque amount and plaque size was greatest in the alveus. Microgliosis was proportional to Aβ plaque pathology (Extended Data Fig. 7). Surprisingly, plaque burden in ExN-Bace1cKO;AD mice was reduced by 95–98% compared to controls (Fig. 2g–k), which was much more than anticipated given our findings in OL-Bace1cKO;AD. Accordingly, plaque sizes were smaller, and microgliosis was markedly reduced (Extended Data Fig. 7). Moreover, ExN-Bace1cKO;AD mice also showed a striking reduction in the amount of thalamic plaques. The unchanged levels of Bace1 transcript in the thalamus of ExN-Bace1cKO;AD mice (Extended Data Fig. 8a,b) indicate that a large amount of subcortical Aβ must be derived from cortico-thalamic axonal projections. Indeed, the inferior colliculus, receiving limited cortical input (Extended Data Fig. 8c) primarily from the auditory cortex19, was spared from plaque attenuation in ExN-Bace1cKO;AD mice. In fact, immunolabeling of 5×FAD brain slices with the human APP-specific antibody, 1D1, revealed that human APP reactivity in neuronal soma is almost restricted to the cortex (Extended Data Fig. 8d) and hippocampus, confirming the Thy1.2 promoter domain. This further indicates that a subset of dorsal telencephalic neurons is the predominant source of local and distal Aβ plaques.
It was nonetheless puzzling that plaque burden was reduced by more than 95% in ExN-Bace1cKO;AD animals, as we had expected that the residual plaque burden would reflect the contribution of OLs (30%). Aβ fibrillation and plaque formation follow sigmoidal growth kinetics20,21, and a threshold level of Aβ accumulation is essential for plaque seeding to occur. This threshold level apparently cannot be reached without neuronal Aβ. Fittingly, compared to homozygous APPNLGF mice, heterozygotes did not develop 50% plaque burden but, rather, less than 10% (Extended Data Fig. 8e–g). This highlights the nonlinear relationship among APP processing, Aβ production and plaque load. Indeed, analysis of 12-month-old ExN-Bace1cKO;AD mice revealed considerable plaque deposition (Extended Data Fig. 9a,b), hinting that, with enough time, plaques can still be formed by Aβ from non-ExN sources.
Lastly, we performed a sensitive electrochemiluminescence assay for different Aβ species (Aβ38, Aβ40 and Aβ42) to determine total Aβ levels. As inputs, we analyzed soluble and insoluble (representing Aβ primarily bound in plaques) fractions of microdissected cortex for gray matter and corpus callosum (CC) for WM (Fig. 3a,b). OL-Bace1cKO;AD brains contained less insoluble and soluble Aβ42 compared to controls, especially in the WM. ExN-Bace1cKO;AD brains were almost devoid of insoluble Aβ, but a moderate amount (14.925% ± 0.066%) of soluble Aβ42 was detected in ExN-Bace1cKO;AD cortical tissue. Additionally, the residual amount of WM Aβ42 was higher in ExN-Bace1cKO;AD brains (27.604% ± 0.072%). In short, although plaque amount was marginally low in the ExN-Bace1cKO;AD brains, an adequate amount of soluble Aβ was still generated by non-ExN sources of Aβ, including OLs and potentially other cell types.

a,b, Aβ electrochemiluminescence immunoassay data of insoluble (SDS-soluble, left) and soluble (Tris-NaCl-soluble, right) lysates of microdissected cortical (a) and WM (b) tissues from control and cKO 6-month-old male mouse hemibrains (n = 7 per group). Triplex immunoassay measured Aβ38, Aβ40 and Aβ42 levels, and data points were normalized to Control;AD samples. Of note, SDS-soluble fractions from both regions mirrored LSM data, whereas Tris-NaCl-soluble fractions revealed a substantial amount of soluble Aβ still being produced, even in ExN-Bace1cKO;AD mice, signifying a residual Aβ production from other cells. Statistical analysis: one-way ANOVA with Tukey multiple comparison tests (P values indicated in graphs with significance highlighted in bold). Bars represent means with s.e.m., and individual data points are displayed. Raw unnormalized data are available in Supplementary Table 6. c, Working model of modulating cell-type-specific Aβ contributions. Selectively ablating Aβ from specific cell types results in steady-state rate change of Aβ production, causing exponentially slower plaque growth that follows a sigmoidal growth curve. ctrl, control; rel., relative.
To investigate if residual plaques found in ExN-Bace1cKO;AD hemibrains are primarily derived from OLs, we generated Cnp-Cre Nex-Cre Bace1fl/fl APPNLGF mice, hereby termed OL-ExN-Bace1cKO;AD. OL-ExN-Bace1cKO;AD developed almost no plaques in the cerebrum (Extended Data Fig. 9c–e). Moreover, OL-ExN-Bace1cKO;AD lysates showed an almost complete loss of Aβ42 when compared to ExN-Bace1cKO;AD samples (Extended Data Fig. 9f), further highlighting that OLs are a main source of Aβ production even in the absence of the ExN contribution.
Functionally, cKO of Bace1 did not result in any changes to neuronal nor axonal abundance (Extended Data Fig. 10a,b). We also recently showed that OL dysfunction drives neuronal amyloid deposition in AD mouse models9. To exclude this as a confounding factor, we compared myelin profiles in OL-Bace1cKO;AD and ExN-Bace1cKO;AD mice but found no changes (Extended Data Fig. 10c–g).
The high expression level of amyloidogenic pathway genes in OLs was contrasted by the smaller relative contribution to overall Aβ deposition. This could be explained by the differences in number, localization or size between neurons and OLs22. There is also the alternative possibility that Aβ processing is more efficient in neuronal compartments or that Aβ isoforms differ between cell types. Beyond their abundance and unique activity profile23, however, what makes ExNs so efficient at producing Aβ remains elusive. Identifying the mechanisms that slow down Aβ generation in OLs, despite the abundance of processing substrate and enzymes, could pave the way for novel therapies targeting Aβ generation.
In conclusion, we provide, to our knowledge, the first in vivo evidence that OLs, a glial cell type, are key players in AD—even in the context of establishing primary Aβ pathology. Notably, the 30% plaque reduction in OL-Bace1cKO;AD mice falls in the range of effect sizes achieved by aducanumab24 and the FDA-approved lecanemab25 antibody therapies. Potentially, selective targeting of BACE1 in OLs could spare the detriments of widespread BACE1 inhibition, especially given the adverse effects seen in BACE1 inhibitor clinical trials26,27,28. Additionally, we showed that ExN-derived Aβ is still required for rapid plaque seeding locally and distally (Fig. 3c). Ultimately, our non-linearity data are relevant when considering anti-Aβ therapeutic interventions, including BACE1 inhibitors, which, as suggested29,30, may have potential in preventing amyloidosis before threshold levels are reached.
Methods
Reanalysis of single-nucleus RNA sequencing and single-cell RNA sequencing data from mouse and human nervous system
Single-cell/single-nucleus RNA sequencing (snRNA-seq/scRNA-seq) datasets were collected and screened for expressions of APP, BACE1, PSEN1 and PSEN2 across major cell populations in the central nervous system (CNS). In total, four mouse datasets9,10,11 and three human datasets12,13,14 were used. The in-house generated data underwent alignment toward reference genome GRCm38/mm10 using the Cell Ranger toolkit (10x Genomics), where other external datasets were processed from raw count matrices. All data were processed with the R package Seurat (version 4.3.0)31 based on original study protocols. Cell type annotations were cross-checked with cluster-specific gene signatures. Afterward, major CNS cell populations, including excitatory neuron (Ext_Neuron), inhibitory neuron (Inh_Neuron), oligodendrocyte precursor cell (OPC), newly formed oligodendrocyte (NFOL), mature oligodendrocyte (MOL), astrocyte (AST), microglia (MG), endothelial cells (Endo) and pericyte, were subset for further screening for APP metabolism-related gene expressions. Each subset dataset underwent renormalization, high variable feature calculation and scaling using the SCTransform pipeline with default parameters. Gene expression levels are visualized in half violin plots using the R package raincloudplots (version 0.0.4)32. The positive expression rate of each gene was calculated upon more than one unique molecular identifier (UMI), and the relative proportion is visualized using the R package ggplot2 (version 3.4.4)33.
Mouse models, husbandry and genotyping
All animal experiments were conducted in concordance with German animal welfare practices and local authorities (documentation: 24_KAN_0021_CNCBFL, 24_KAN_0026_NXCBFL and 24_KAN_0024_FFDE). Mice were group-housed in the animal facility of the Max Planck Institute for Multidisciplinary Sciences (MPI-NAT), City Campus, with ad libitum food and regular cage maintenance. All mice were kept under a 12-h dark and 12-h light cycle in an ambient temperature of 21 °C and 45% humidity. All animals are characterized as unburdened, and only organ collection was performed. Mouse strains were kept on a C57BL/6N background. Both sexes were used and indicated in the respective figures. The following mouse strains were used: APPNLGF (ref. 34), Bace1fl/fl (ref. 35), CnpCre/+ hereby termed Cnp-Cre36, Nex-Cre37, stop-flox tdTomato38 and 5×FAD39. The crossbreeds generated and analyzed are as follows: Cnp-Cre Bace1fl/fl APPNLGF to assess OL-Aβ contribution (OL-Bace1cKO;AD); Nex-Cre Bace1fl/fl APPNLGF (ExN-Bace1cKO;AD) to assess ExN-Aβ contribution; and Cnp-Cre stop-flox tdTomato to validate Cnp-Cre specificity. Bace1−/− samples were provided by the laboratory of Robert J. Vassar. App−/− samples were provided by the laboratory of Ulrike Müller. Ages of animals analyzed are listed on the respective figures. Genotyping was carried out on ear clips from the marking process (see individual strain references for genotyping protocols). Re-genotyping was performed on a small tail biopsy gathered after mice were euthanized. Most experimental cohorts were defined by genotype, and littermate controls were analyzed.
Mouse tissue extraction
To acquire samples for imaging experiments, animals were euthanized and immediately flushed with cold PBS until the liver was decolorized. Extracted tissues underwent overnight immersive fixation in 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer before switching to PBS for long-term storage at 4 °C. For electron microscopy, perfusion was done with 4% PFA and 2.5% glutaraldehyde in 0.1 M phosphate buffer (K+S buffer) after flushing with PBS and stored at 4 °C long term in 1% PFA in PBS. For biochemical experiments, animals were cervically dislocated, and their brains were quickly extracted before a quick wash in PBS. Tissues were then placed into a custom 1-mm-spaced coronal brain matrix developed in-house (Workshop, MPI-NAT, City Campus) for manual, sequential sectioning with blades. Brain slices were transferred to a glass plate on ice before microdissection of cortices, CC and hippocampi. Dissected tissues were snap frozen and placed in −80 °C until further use.
Sample preparation and staining for LSM
Fixed tissues were processed for LSM imaging using a modified iDISCO protocol optimized for amyloid plaque staining, as previously reported9. Hemibrains were subjected to a sequence of dehydration, autofluorescence quenching, permeabilization and labeling with Congo red dye. After labeling, hemibrains were subjected to a final ascending methanol wash in PBS and overnight incubation in a 1:2 mixture of 100% methanol and dichloromethane (DCM). Finally, the samples were placed in 100% DCM for 1 h 40 min before clearing in ethyl cinnamate (ECI) for imaging.
In toto LSM imaging and analysis
Cleared hemibrains were imaged with an LSM setup (UltraMicroscope II, LaVision Biotec) with a corrected dipping cap at ×2 objective lens magnification. InspectorPro (version 7.124, LaVision Biotec) software was used to visualize the samples in the mosaic acquisition mode with the following settings: 5-µm light sheet thickness, 20% sheet width, 0.154 sheet numerical aperture, 4-µm z-step size, 2,150 × 2,150 pixels field of view, dynamic focus steps of 5, dual light sheet illumination and 100-ms camera exposure time. Red fluorescence of Congo red-stained hemibrains was recorded with 561-nm laser excitation at 80% laser power and a 585/40-nm emission filter. Image stacks were imported and stitched with Vision4D (version 3.2, Arivis). Regions of interest (ROIs) in this study include isocortex, hippocampus, alveus, inferior colliculus and thalamus. The ROIs were defined based on anatomical landmarks and labeled before segmentation of plaque burden. A machine learning pipeline to extrapolate three-dimensional (3D) shape recognition from two-dimensional (2D) inputs was generated by supplementing 200 desired objects (plaques) and backgrounds (non-plaque structures), respectively. Next, segment co-localization was performed to delineate plaques within specific ROIs. Upon acquiring plaques as voxel objects, noise was removed by deleting objects with voxel sizes 1–10 and plane counts 1–3. Lastly, object metadata and correlated features were exported as spreadsheets.
Paraffin sample preparation and immunohistochemical staining
Preparation of paraffinized samples and immunohistochemistry (IHC) were performed as previously described9. Details of antibodies used for IHC are listed in Supplementary Tables 7 and 8. Nuclei were stained with DAPI (Thermo Fisher Scientific, 300 nM) in PBS. Slides were washed in PBS twice for 5 min and mounted with Aqua-Poly/Mount medium (Polysciences). Finally, slides were left to dry overnight before imaging.
Vibratome sectioning and immunostaining
Fixed hemibrains were sectioned at 30-µm thickness with a VT 1000S microtome (Leica). Selected vibratome sections were placed into a 12-well plate with 1 ml of TBS in each well. The sections were rinsed three times in PBS for 5 min. For 3D5 immunolabeling, the sections were placed in a 0.1 M sodium citrate pH 9.0 buffer for antigen retrieval. Sections were then incubated in 16 mM glycine in TBS-0.1% Triton solution for 1 h, followed by washing with TBS three times for 5 min each. Blocking was performed in 5% donkey serum in TBS with 0.1% Triton X-100 for 1 h with shaking. Sections were washed twice for 10 min in 1% BSA in TBS with 0.1% Triton X-100 with shaking. Next, the designated primary antibodies (500 µl per well) were applied onto the sections in 1% BSA in TBS-0.1% Triton overnight at 4 °C with shaking. The following primary antibodies were used: anti-BACE1-3D5 (mouse, hybridoma culture supernatant, 1:250) and anti-BACE1-ab183612 (rabbit, Abcam, 1:250). The next day, sections were washed three times for 10 min each in 1%BSA/0.21% Triton-TBS with shaking. Fluorescent secondary antibodies (500 µl per well) were applied in 1% BSA in TBS-0.25% Triton for 2 h in the dark. The following secondary antibodies were used: anti-rabbit Alexa Fluor 555 (donkey/goat, Thermo Fisher Scientific, 1:1,000) and anti-mouse Alexa Fluor 555 (donkey/goat, Thermo Fisher Scientific, 1:1,000). Nuclei were stained with DAPI (Thermo Fisher Scientific, 300 nM) in PBS. Finally, the sections were washed three times for 15 min each in TBS in the dark with shaking. Sections were carefully retrieved from the 12-well plate and mounted with Aqua-Poly/Mount medium (Polysciences). Slides were left to dry overnight before imaging.
Human tissue collection
Human patient samples (control—one female, two male, age: 74 ± 2.83 years; AD—two female, two male, age: 72.75 ± 1.78 years) were obtained from Neurobiobank Munich with ethical approval from the Ethical Commitee at Ludwig-Maximilians University. Selection of patients was performed upon Braak staging with scores of patients with AD ranging from Braak 5 to 6 and control patient scores ranging from Braak 1 to 3. Postmortem interval of patients ranged between 26 h and 51 h. APOE genotype of all control patients is 3/3, and APOE genotypes of patients with AD are 3/3, 3/4 and 4/4. For ISH experiments, formalin-fixed paraffin-embedded tissue sections were used from human samples.
ISH
We employed the RNAscope Fluorescent Multiplex assay (ACDBio) for paraffin-embedded samples (5-µm mouse and 4-µm human sections) as previously described9. To ensure optimal hybridization, a hydrophobic barrier was created followed by protein digestion via incubation in RNAscope Protease Plus at 40 °C for 15 min (mouse) or RNAscope Protease IV for 20 min (human). The following probes were used: Mm-Mbp (451491-C1), Mm-Bace1-C2 (400721-C2), Mm-Slc17a7-C3 (416631-C3), Hs-MBP-C2 (411051-C2), Hs-APP-C1 (418321-C1) or Hs-BACE1-C3 (422541-C3) at 40 °C for 2 h. For triple visualization of mouse sections, the following fluorophores were applied: Opal 520, 570 and 690, at 40 °C for 30 min. For double visualization of human sections, TSA Vivid Fluorophores (570 and 650) were used. The slides were again washed and stained with DAPI (Thermo Fisher Scientific, 300 nM) for 10 min before mounting with Aqua-Poly/Mount medium (Polysciences).
Upon epifluorescence imaging, validation of Bace1 deletion in mouse cortical ExNs was performed manually due to the presence of ample satellite OLs in the cortex. A 500-μm-wide ROI spanning all cortical layers was drawn for each coronal brain slice, more specifically in the parietal or somatosensory cortex overlying the hippocampus. For nuclei expressing Slc17a7, Bace1 puncta were quantified to yield individual ExN Bace1 counts. The data were grouped into distinct cortical layers, which were delineated based on landmarks. Similar manual quantification was also performed on Mbp+ OLs in the fimbria. Validation of Bace1 deletion in hippocampal ExNs was carried out semi-automatically by creating a pipeline employing a nuclear detection plugin (StarDist) and expanding the captured nuclear ROIs by a pixel size of 10. Combined with particle analyzer and watershed binarization, Bace1 puncta were detected per hippocampal neuron. For human samples, images of the entire human brain sections were acquired with the PANNORAMIC Midi II Slide Scanner (3DHISTECH) with the ×20 objective and smaller selected regions with the ×40 objective. The human cortex was divided into three areas corresponding to layers 1 and 2 (L1–2), layers 3 and 4 (L3–4) and layers 5 and 6 (L5–6). For each area, 6–12 images of similar size were selected using CaseViewer (version 2.4, 3DHISTECH) and exported via Slide Converter (version 2.3.2, 3DHISTECH). Selected images were randomized using a Fiji filename-randomizer plugin, and counting was done using the Fiji CellCounter plugin.
In vitro OL culture
OPCs were isolated from p7 mouse brains using magnetic-activated cell sorting (MACS) and anti-NG2 MicroBeads (Miltenyi Biotec). Tissue dissection and cell sorting were performed under sterile conditions. The neural tissue dissociation kit was used according to the manufacturer’s protocol and as described40. Dissected brains were transferred into enzyme mix 1, followed by incubation at 37 °C for 15 min. Next, incubation with enzyme mix 2 was done at 37 °C for 20 min with manual dissociation. Tubes were centrifuged at 1,200 r.p.m. for 5 min, and the supernatant was decanted while the pellet was resuspended in DMEM with 1% horse serum. We then passed the cell suspension through a 70-μm and then a 40-μm strainer. The tubes were again centrifuged at 1,200 r.p.m. for 10 min, and the pellet was resuspended and incubated in warm OPC culture medium consisting of 100 ml of NeuroMACS media, 2 ml of MACS NeuroBrew21, 1 ml of penicillin–streptomycin and 1 ml of L-GlutaMAX at 37 °C for 2 h. Next, tubes were centrifuged at 1,200 r.p.m. and 4 °C for 10 min, followed by pellet resuspension and incubation in NG2 MicroBeads diluted in DMEM with 1% horse serum (10 μl of NG2 beads per 107 total cells) at 4 °C for 15 min. The cell suspension was again centrifuged at 1,200 r.p.m. and 4 °C for 10 min, and the supernatant was removed before pellet resuspension in 5 ml of DMEM with 1% horse serum. LS columns (Miltenyi Biotec) were first attached to a magnet before activating with DMEM containing 1% horse serum. The columns were washed three times with DMEM after addition of the cell suspension. The columns were finally detached from the magnet and flushed with 5 ml of DMEM containing 1% horse serum to collect bound cells. Upon detachment, samples were centrifuged at 1,200 r.p.m. for 5 min, and the pellet was resuspended in proliferation medium. OPCs were plated at a density of 1.2 × 105 cells per well on a 12-well plate in proliferation medium, before replacement with OPC differentiation medium at 4 days in vitro (DIV4). Cells were fixed at DIV8 with 4% PFA and washed with PBS three times for 5 min each.
Immunocytochemical staining
For immunocytochemical labeling, cells were permeabilized with cold 0.3% Triton X-100 in PBS and blocked with 10% goat serum and 0.03% Triton X-100 in PBS for 1 h. Primary antibodies were diluted in 1.5% horse serum in PBS and applied at 4 °C overnight. Coverslips were washed with PBS three times for 5 min and incubated in secondary antibodies diluted in PBS for 1 h. Details of antibodies used for immunocytochemistry are listed in Supplementary Tables 7 and 8. The samples were washed twice for 5 min before incubation with DAPI (Thermo Fisher Scientific, 300 nM) in PBS. Lastly, cells were washed briefly in PBS before mounting with Aqua-Poly/Mount for confocal imaging. All incubation steps were done at room temperature unless stated otherwise.
Epifluorescence and confocal microscopy
Epifluorescence imaging was carried out with parameters as previously described9. Resulting tiled images were stitched in ZEN. For confocal microscopy, images were partially acquired via ZEN software with a Zeiss LSM 800 Airyscan confocal microscope equipped with Plan-Apochromat ×63/1.4 oil DIC M27 objective. Alternatively, images were acquired via LasAF software with a Leica SP8 Lightning confocal microscope equipped with an argon laser and a tuneable white-light laser with ×63/1.4 glycerin objective. Both confocal microscopes are situated at the European Neuroscience Institute and at MPI-NAT, City Campus, respectively.
Analysis of 2D microscopy images
All 2D image analysis was performed on Fiji (version 1.53c)41. For validation of Cnp-Cre specificity, thresholding and particle analyzer were performed to segment and quantify neurons, OLs and RFP+ cells. Quantification of RFP+ OLs in the CC, however, was performed manually due to the dense amount of OLs. Quantification of 2D Aβ and microgliosis was done via thresholding and measurement of positive area. Microscopic analysis of OL numbers between controls and cKOs similarly started with ROI segmentation followed by thresholding and particle analyzer. As for OL numbers in WM tracts, manual quantification was again performed. Finally, percentage ROI area of the cortex and hippocampus occupied by myelinated structures was obtained upon thresholding, and mean intensity values of major WM tracts were measured.
Electron microscopy
Sample preparation for electron microscopy was performed based on an optimized protocol in the working group9. At least ten digital pictures were captured at ×4,000 magnification with a Zeiss EM900 for ultrastructural analysis. Electron micrographs of the caudal CC were analyzed with Fiji. Analysis of g-ratio was conducted as previously described9.
MACS of OLs
OLs were isolated from whole brains (excluding the olfactory bulb and the cerebellum) of 1-month-old mice using an adult brain dissociation kit (Miltenyi Biotec, 130-107-677). OLs were sorted via positive selection by labeling with OL-specific anti-O4 MicroBeads (Miltenyi Biotec, 130-096-670, 1:40). Sorted cells were eluted in 1× PBS containing cOmplete Mini protease inhibitor cocktail (Roche, one tablet per 10 ml of 1× PBS) and PhosSTOP (Roche, one tablet per 10 ml of 1× PBS) and were centrifuged at 13,000 r.p.m. for 5 min. Pellets were snap frozen for further protein analysis and stored at −80 °C. For western blotting, the pellets were resuspended in 28.5 µl of RIPA buffer taken from an aliquot of 10 ml containing one tablet of protease inhibitor and one tablet of phosphatase inhibitor. This was followed by sonication for 3 min in an ultrasonic bath. For loading, 30 µl of 2× Tris-tricine sample buffer (Invitrogen) was added, as was 1.5 µl of 2 M DTT.
Protein fractionation
Preparation of insoluble and soluble fractions from mouse brain tissue was carried out based on a modified protocol42. Tissue homogenization was carried out using a homogenizer (Precellys) on microdissected cortical and CC fractions in reaction tubes containing ceramic beads in cold lysis buffer (pH 8.0) (700 μl for cortex and 500 μl for CC). The following settings were used for the homogenization at 4 °C: 6,500g twice for 30 s. The homogenate was carefully transferred to a 1.5-ml reaction tube before spinning with a benchtop centrifuge (Eppendorf) at 17,000g and 4 °C for 20 min. The supernatant was collected and served as the soluble protein fraction, while the pellet was resuspended in 2% SDS (500 μl for cortex and 300 μl for CC). The solution was then sonicated on ice for 1 min until the pellet completely dissolved. To remove DNA, 1 μl of benzonase was added into the solution and incubated at room temperature for 5 min. The samples were again centrifuged at 17,000g and 4 °C for 20 min before transferring the supernatant to a fresh collection tube, serving as the insoluble fraction. Fractions were stored at −80 °C for further use.
Western blotting
Only the insoluble fraction was used to probe for APP processing machinery via western blotting. Protein concentration measurement as well as protocol for SDS-PAGE and western blotting with 10–20% gradient gels (Novex) were carried out as previously described9. Details of antibodies used for western blotting are listed in Supplementary Tables 7 and 8. For chemiluminescent blots, equal amounts of Western Lightning Plus ECL Oxidizing Reagent Plus and Enhanced Luminol Reagent Plus (PerkinElmer) were first mixed and then applied onto the membrane. To visualize protein with low abundance, SuperSignal West Femto Stable Peroxide and Luminol/Enhancer (Thermo Fisher Scientific) were instead used. Upon washing in TBS-T three times for 10 min each, membranes were fluorescently scanned using an Odyssey platform (LI-COR) or using a ChemoStar imager (Intas) for chemiluminescent visualization. For quantification, background was subtracted, and bands were analyzed using Fiji. Target protein content was normalized to the FastGreen bands of respective controls as indicated in the graphical representations.
Electrochemiluminescence Aβ immunoassay
To determine Aβ levels in specific brain regions, we turned to the V-PLEX Plus Aβ Peptide Panel 1 (6E10) kit (Meso Scale Discovery (MSD)) and conducted experiments based on instructions provided by the manufacturer. The kit allows multiplex measurement of Aβ38, Aβ40 and Aβ42 from single wells. First, 150 μl of Diluent 35 was added into each well for blocking before the plates were sealed and incubated with shaking at room temperature for 1 h. Each well was subsequently washed three times with 150 μl of wash buffer containing 0.05% Tween 20 in PBS (PBS-T). From a detection antibody solution containing 50× SULFO-TAG anti-Aβ 6E10 antibody diluted in Diluent 100, 25 μl was added into each well, followed by the addition of 25 μl of samples or calibrators per well. The plate was again sealed and incubated with shaking at room temperature for 2 h. Each well was again washed three times with 150 μl of PBS-T (0.05% Tween 20 in PBS) before the addition of 150 μl of 2× Read Buffer T. Lastly, plate measurement was carried out using the MSD QuickPlex SQ 120 reader. In all assays performed, two technical replicates of samples and calibrators were included.
Data analysis, statistics and figure preparation
All statistical analyses and preliminary graphs were performed with GraphPad Prism 8.0.2. Statistical tests were chosen based on tests for normality. Experimenters were blinded in the analysis of electron microscopy data. Due to the visible effects that Bace1 cKO has on the plaque load, blinding was not possible for various imaging analyses. No statistical methods were used to pre-determine sample sizes, but sample sizes for primary experiments (that is, quantitative LSM of cKOs and immunoassay) are similar to those shown in our previous publication9. The specific statistical analyses performed are listed in the respective figure legends. No animals or data points were excluded from this study. Brain connectivity images were adapted from the Allen Brain Atlas: Mouse Connectivity: Projection (https://connectivity.brain-map.org/)43. All figures were prepared with Adobe Illustrator 28.3.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The four mouse scRNA-seq/snRNA-seq datasets analyzed were obtained from Depp et al. (GSE178295 and GSE208683)9, Ximerakis et al. (GSE129788)10 and Zeisel et al. (SRP135960)11. The three human scRNA-seq/snRNA-seq datasets were obtained from Zhou et al. (accessed via the AD Knowledge Portal under study snRNAseqAD_TREM2)12, Jäkel et al. (GSE118257)13 and Lake et al. (GSE97942)14. Source data are provided with this paper.
Code availability
The code used for snRNA-seq is available on GitHub at https://github.com/TSun-tech/2023_Sasmita_OL_BACE.
References
Zhao, J. et al. β-secretase processing of the β-amyloid precursor protein in transgenic mice is efficient in neurons but inefficient in astrocytes. J. Biol. Chem. 271, 31407–31411 (1996).
Veeraraghavalu, K., Zhang, C., Zhang, X., Tanzi, R. E. & Sisodia, S. S. Age-dependent, non-cell-autonomous deposition of amyloid from synthesis of β-amyloid by cells other than excitatory neurons. J. Neurosci. 34, 3668–3673 (2014).
Rice, H. C. et al. Contribution of GABAergic interneurons to amyloid-β plaque pathology in an APP knock-in mouse model. Mol. Neurodegener. 15, 3 (2020).
Skaper, S. D., Evans, N. A., Rosin, C., Facci, L. & Richardson, J. C. Oligodendrocytes are a novel source of amyloid peptide generation. Neurochem. Res. 34, 2243–2250 (2009).
Walter, S. et al. The metalloprotease ADAMTS4 generates N-truncated Aβ4–x species and marks oligodendrocytes as a source of amyloidogenic peptides in Alzheimer’s disease. Acta Neuropathol. 137, 239–257 (2019).
Gazestani, V. et al. Early Alzheimer’s disease pathology in human cortex involves transient cell states. Cell 186, 4438–4453.e23 (2023).
Chen, J. F. et al. Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer’s disease. Neuron 109, 2292–2307 (2021).
Mathys, H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019).
Depp, C. et al. Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature 618, 349–357 (2023).
Ximerakis, M. et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 22, 1696–1708 (2019).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014 (2018).
Zhou, Y. et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 26, 131–142 (2020).
Jäkel, S. et al. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 566, 543–547 (2019).
Lake, B. B. et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 36, 70–80 (2018).
Tognatta, R. et al. Transient Cnp expression by early progenitors causes Cre-Lox-based reporter lines to map profoundly different fates. Glia 65, 342–359 (2017).
Jo, Y. R. et al. Potential neuron-autonomous Purkinje cell degeneration by 2′,3′-cyclic nucleotide 3′-phosphodiesterase promoter/Cre-mediated autophagy impairments. FASEB J. 35, e21225 (2021).
Lam, M. et al. CNS myelination requires VAMP2/3-mediated membrane expansion in oligodendrocytes. Nat. Commun. 13, 5583 (2022).
Hu, X., Hu, J., Dai, L., Trapp, B. & Yan, R. Axonal and Schwann cell BACE1 is equally required for remyelination of peripheral nerves. J. Neurosci. 35, 3806–3814 (2015).
Blackwell, J. M., Lesicko, A. M., Rao, W., De Biasi, M. & Geffen, M. N. Auditory cortex shapes sound responses in the inferior colliculus. eLife 9, e51890 (2020).
Lee, C. C., Nayak, A., Sethuraman, A., Belfort, G. & McRae, G. J. A three-stage kinetic model of amyloid fibrillation. Biophys. J. 92, 3448–3458 (2007).
Burgold, S., Filser, S., Dorostkar, M. M., Schmidt, B. & Herms, J. In vivo imaging reveals sigmoidal growth kinetic of β-amyloid plaques. Acta Neuropathol. Commun. 2, 30 (2014).
Herculano-Houzel, S. The glia/neuron ratio: how it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 62, 1377–1391 (2014).
Kamenetz, F. et al. APP processing and synaptic function. Neuron 37, 925–937 (2003).
Sevigny, J. et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537, 50–56 (2016).
Tucker, S. et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe mice. J. Alzheimers Dis. 43, 575–588 (2015).
Chatila, Z. K. et al. BACE1 regulates proliferation and neuronal differentiation of newborn cells in the adult hippocampus in mice. eNeuro 5, ENEURO.0067-18.2018 (2018).
Sur, C. et al. BACE inhibition causes rapid, regional and non-progressive volume reduction in Alzheimer’s disease brain. Brain 143, 3816–3826 (2020).
Wessels, A. M. et al. Cognitive outcomes in trials of two BACE inhibitors in Alzheimer’s disease. Alzheimers Dement. 16, 1483–1492 (2020).
Satir, T. M. et al. Partial reduction of amyloid β production by β-secretase inhibitors does not decrease synaptic transmission. Alzheimers Res. Ther. 12, 63 (2020).
Peters, F. et al. BACE1 inhibition more effectively suppresses initiation than progression of β-amyloid pathology. Acta Neuropathol. 135, 695–710 (2018).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Allen, M., Poggiali, D., Whitaker, K., Marshall, T. R. & Kievit, R. A. Raincloud plots: a multi-platform tool for robust data visualization. Wellcome Open Res. 4, 63 (2019).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2009).
Saito, T. et al. Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 17, 661–663 (2014).
Hu, X., Das, B., Hou, H., He, W. & Yan, R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J. Exp. Med. 215, 927–940 (2018).
Lappe-Siefke, C. et al. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 33, 366–374 (2003).
Goebbels, S. et al. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44, 611–621 (2006).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010).
Oakley, H. et al. Intraneuronal β-amyloid aggregates, neurodegeneration and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 (2006).
Weil, M. T. et al. Isolation and culture of oligodendrocytes. Methods Mol. Biol. 1936, 79–95 (2019).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Wirths, O. Extraction of soluble and insoluble protein fractions from mouse brains and spinal cords. Bio Protoc. 7, e2422 (2017).
Oh, S. W. et al. A mesoscale connectome of the mouse brain. Nature 508, 207–214 (2014).
Acknowledgements
We would like to thank all personnel of the animal facility of the Max Planck Institute for Multidisciplinary Sciences (MPI-NAT), City Campus; A. Fahrenholz for her technical contribution in sample preparation for imaging experiments; B. Sadowski for sample preparation for electron microscopy; members of the KAGS subgroup and the Department of Neurogenetics for scientific input; U. Muller and V. Bengelsdorff for the preparation and transport of APP KO tissue; S. Rossner for the 1D1 antibody recommendation; and M. Thalmann for his input on statistical analysis and data segmentation. Work pertaining to this manuscript was performed in the laboratory of K.-A.N. and was supported by the Deutsche Forschungsgemeinschaft (DFG, TRR274), the Dr. Myriam and Sheldon Adelson Medical Foundation (AMRF) and an ERC Advanced Grant (MyeliNANO). Work carried out in the laboratory of O.W. is supported by the DFG (WI3472/11-1). We would like to extend our gratitude to the team behind the Allen Brain Atlas: Mouse Connectivity: Projection. We also extend our gratitude to the Gene Expression Omnibus and the AD Knowledge Portal data platforms. The valuable data provided by these platforms owe their existence to the dedication of research volunteers and the collaborative efforts of contributing researchers.
Funding
Open access funding provided by Max Planck Society.
Author information
Authors and Affiliations
Contributions
A.O.S., C.D. and K.-A.N. conceptualized and designed the experiments. A.O.S. and T.N. planned and performed imaging and biochemical experiments. T. Sun arranged and analyzed snRNA-seq datasets. X.Y., S.M. and S.B.S. executed in vitro culture experiments. C.B. provided technical assistance, prepared samples for imaging and executed biochemical experiments. E.C.O. carried out biochemical experiments. Y.B.N. and G.S. planned and carried out cell sorting experiments for cKO validation. Z.W. and T.R. prepared samples and advised on electron microscopy experiments. B.B. assisted in developing an automated analysis pipeline for ISH quantification. B.B. and S.B.S. performed confocal imaging. L.E. and S.J. performed and analyzed ISH on human tissue. B.M. guided and assisted with the electrochemiluminescence assay. S.S. and S.M. assisted in data analysis. F.B. and K.O. performed all genotyping. L.S. and S.A.B. conducted pilot biochemical experiments. S.E. optimized in vitro culture experiments. K.R.S. and R.V. supplied constitutive BACE1 KO brains and the 3D5 antibody for imaging and biochemical experiments. S.G. provided and collected tissue from Cnp-Cre stop-flox tdTomato and Nex-Cre mice and provided valuable scientific input. T. Saito and T. Saido provided APPNLGF mice and valuable scientific input. G.C.-B. provided infrastructure for and assisted with computation of mouse snRNA-seq datasets. W.M. provided access to equipment and materials for electron microscopy. H.-W.K. and O.W. advised on the electrochemiluminescence assay and provided valuable scientific input. J.W. provided infrastructure for the electrochemiluminescence assay. S.J. provided and advised on human ISH data and provided valuable scientific input. R.Y. provided Bace1fl/fl mice and valuable scientific input. A.O.S. analyzed data and constructed figures. A.O.S., C.D. and K.-A.N. prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Neuroscience thanks Ben Emery and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Expression levels and positive rates of amyloidogenic pathway genes across all cell types in the nervous system.
(a) Expression level half violin plots of amyloidogenic pathway genes in all cell types of mouse nervous tissue inputs with SCTransform normalization method from all chosen mouse datasets9,26,27. (b) Expression level half violin plots of amyloidogenic pathway genes in all cell types of human nervous tissue inputs with SCTransform normalization from all chosen human datasets28,29,30. (a, b) Half violins represent aggregated expression levels of respective genes from each cell type and data points refer to individual expression levels from single cells or nuclei normalized by the SCTransform method. (c) Positive rate barplots of amyloidogenic pathway genes in all cell types of mouse nervous tissue inputs from all chosen mouse datasets9,26,27. (d) Positive rate barplots of amyloidogenic pathway genes in all cell types of human nervous tissue inputs with SCTransform normalization method from all analyzed human datasets28,29,30. The results published here are based on data obtained from GEO and the AD Knowledge Portal.
Extended Data Fig. 2 OLs exhibit protein expression of APP and an abundance of APP and BACE1 transcripts.
(a) A confocal image of an in vitro OL with APP present in the soma and cellular processes. Experiment was repeated thrice in separate in vitro cultures. (b) A confocal image of in vivo mouse cortical OLs with APP present in the perinuclear space. Experiment was repeated thrice in separate in vitro cultures. (c) A fluorescence microscopy image of human cortical OLs showing APP reactivity in the perinuclear space. Immunolabeling was performed once on all control human samples (n = 3). (d) ISH images of human cortical layers 5–6 with visible APP puncta in MBP+ cells of control (left) and AD (right) patients. Arrowheads point to APP-expressing OLs. (e) ISH images of human cortical layers 5-6 with visible BACE1 puncta in MBP+ cells of control (left) and AD (right) patients. Arrowheads point to BACE1-expressing OLs. (f) Quantification of nuclear count, MBP+ nuclei, MBP+ APP+ nuclei, and MBP+ BACE1+ nuclei in control (n = 3) and AD (n = 3–4) patients. One-way ANOVA was performed with Sidak multiple comparison tests (P values indicated in graphs with significance highlighted in bold) comparing AD patients to controls. Bars represent means with SEM, and individual data points are displayed.
Extended Data Fig. 3 Validation of Cre specificity in Cnp-Cre stop-flox tdTomato mice.
(a) Fluorescence microscopy sagittal overview of a Cnp-Cre stop-flox tdTomato mouse. (b-d) Closeup images of cortex, hippocampus, and CC of a Cnp-Cre stop-flox tdTomato mouse. Yellow arrowheads point to RFP+ OLs and the blue arrowhead points to a single RFP+ neuron in the cortex. (e) Barplots showing percentages of RFP+ OLs and neurons in specific brain regions. Mean percentage values are shown above each bar. Rounded average total number of cells considered for quantification is as follows: Cortex–OLs=283, cortex–neurons=9,232, hippocampus–OLs=66, hippocampus–neurons=3,203, CC–OLs=344, CC–neurons=1.
Extended Data Fig. 4 ISH validation of Bace1 cKO in OL-Bace1cKO;AD and ExN-Bace1cKO;AD.
(a-d) ISH validation of Bace1 cKO in cortical ExNs. (e-g) ISH validation of Bace1 cKO in fimbria OLs. (h-m) ISH validation of Bace1 cKO in hippocampal ExNs. (a) Fluorescence microscopy images of cortices showing reductions of Bace1 transcripts in ExNs of ExN-Bace1cKO;AD samples. (b) Violin plots showing individual mouse distribution of Bace1 puncta/cortical ExN nuclei. (c) Frequency distribution heatmap of individual mouse distribution of Bace1 puncta/cortical ExN nuclei. (d) Violin plots showing cortical layer-specific individual mouse distribution of Bace1 puncta/ExN nuclei. (e) Fluorescence microscopy images of hippocampal fimbriae showing reductions of Bace1 transcripts in OLs of OL-Bace1cKO;AD samples. (f) Violin plots showing individual mouse distribution of Bace1 puncta/fimbria OL nuclei. (g) Frequency distribution heatmap of individual mouse distribution of Bace1 puncta/fimbria OL nuclei. (h-j) Fluorescence microscopy images of hippocampal CA1, CA2/3, and DG respectively showing reductions of Bace1 transcripts only in ExNs of ExN-Bace1cKO;AD samples. (k) Violin plots showing individual mouse distribution of Bace1 puncta/hippocampal ExN nuclei. (l) Frequency distribution heatmap of individual mouse distribution of Bace1 puncta/hippocampal ExN nuclei. (m) Violin plots showing hippocampal region-specific individual mouse distribution of Bace1 puncta/ExN nuclei. For (b,d,f,k,m), comparison was made between control, OL-Bace1cKO;AD, and ExN-Bace1cKO;AD mice (n = 2 per group). Solid lines represent median and faded lines represent quartiles. For (c,g,l), n-numbers refer to amount of nuclei considered for each region and cell type analysis and make up the data cloud for violin plots shown in (b,d,f,k,m).
Extended Data Fig. 5 Validation of BACE1 and APP antibodies with KO materials.
(a) Confocal images of primary OLs harvested from Control;AD and OL-Bace1cKO;AD mice labeled with PLP (yellow), APP (blue), and 3D5 for BACE1 (white). The punctate 3D5 labeling is equally present in Control;AD and OL-Bace1cKO;AD, suggesting an unspecific BACE1 staining. Experiment was repeated twice in separate in vitro cultures. (b) Confocal images of primary OLs harvested from Control;AD and OL-Bace1cKO;AD mice labeled with PLP (yellow), APP (blue), and ab183612 for BACE1 (white). No BACE1 reactivity could be detected. Experiment was repeated twice in separate in vitro cultures. (c) Fluorescence microscopy images of mossy fibers of 9-month-old WT (left) and constitutive BACE1 KO (right) animals labeled with 3D5 (top) and ab183612 (bottom) for BACE1 (white). Loss of mossy fiber staining was only detected via 3D5 labeling. Immunolabeling was performed once on brain slices from different mice (n = 2 per group). (d) Confocal images of 9-month-old WT (top) and constitutive BACE1 KO (bottom) cortices labeled with DAPI (gray) and 3D5 for BACE1 (blue). Note the absence of any intracellular staining of 3D5 in both WT and BACE1 KO animals, hinting at the inability of the 3D5 antibody to detect BACE1 in cell somas. Immunolabeling was performed once on brain slices from different mice (n = 2 per group). (e) Immunoblot representative images of BACE1 and the loading control, actin, on sorted OLs from 1-month-old Control;AD and OL-Bace1cKO;AD (n = 1 per group), showing a proof of concept ablation of BACE1 in a cell-type-specific manner. (f) Fluorescence microscopy images of piriform cortices of 3-month-old WT (left) and constitutive APP KO (right) animals labeled with CAII (yellow) and Y188 for APP (blue) for BACE1 (white). APP reactivity, including the APP labeling in OL soma (insets), is abolished in the KO tissue. Immunolabeling was performed once on brain slices from different mice (n = 2 per group). (g-h) Cell-type-specific deletion of Bace1 alters APP processing. (g) Immunoblots and total protein content of microdissected cortical and WM tissues from 6-month-old male mice targeting key amyloidogenic proteins in lysates. (h) Immunoblot quantification showing APP processing in WT, control, ExN-Bace1cKO;AD and OL-Bace1cKO;AD (n = 4–6 per group) lysates. Top–cortical, bottom–WM. All immunoblots were normalized to WT relative protein amount except β-CTFs which were normalized to control AD relative protein amount. Data was statistically analyzed via one-way ANOVA was performed with Tukey multiple comparison tests (P values indicated in graphs with significance highlighted in bold). Bars represent means with SEM and individual data points displayed.
Extended Data Fig. 6 Female APPNLGF animals develop more Aβ plaque burden compared to age-matched male APPNLGF animals.
(a) LSM 3D visualization of female and male control APPNLGF hemibrains at 6 months of age. (b) Brain region-segmented plaques of female and male control APPNLGF hemibrains. Color-region allocation is as follows: White–isocortex, blue–hippocampus, yellow–alveus. (c) Fluorescence microscopy images of female and male control APPNLGF cortices. (d) Quantification of LSM data between female and male control APPNLGF hemibrains (n = 10 per sex). Male data points were normalized to female data. Filled shapes represent male and hollowed shapes represent female mice. For each parameter, unpaired, two-tailed Student’s t-test was performed (P values indicated in graphs) comparing males to females. Bars represent means with s.e.m. and individual data points are displayed. Raw unnormalized data is available in Supplementary Table 1. (e,f) Quantification of Aβ load and microgliosis in different brain regions of male and female control APPNLGF mice (n = 4 per sex). Unpaired, two-tailed Student’s t-test was performed for each regional quantification (P values indicated in graphs with significance highlighted in bold) comparing males to females. Bars represent means with SEM and individual data points displayed.
Extended Data Fig. 7 Microgliosis is proportional to Aβ burden.
(a) Coronal sections of female control, OL-Bace1cKO;AD and ExN-Bace1cKO;AD mouse hemibrains stained for microglia (Iba1) and Aβ (6E10). (b,c) Closeup images of cortex and WM of control and cKO mice showing moderate and marked reductions of both Aβ deposits and microgliosis in OL-Bace1cKO;AD and ExN-Bace1cKO;AD samples, respectively. Inherent changes in microgliosis could thus be excluded as microglia only appear reactive to plaques and not in regions devoid of them. (d,e) Quantification of Aβ load and microgliosis between controls and cKOs (n = 5 per group) spanning different regions. Microgliosis was shown to be directly linked to plaque load. One-way ANOVA was performed with Tukey multiple comparison tests (P values indicated in graphs with significance highlighted in bold). Bars represent means with SEM and individual data points displayed.
Extended Data Fig. 8 Thalamic plaques contain Aβ derived from cortical and hippocampal sources and plaque deposition is not directly proportional to APPNLGF gene dosage.
(a) Fluorescence microscopy images of thalami of control and ExN-Bace1cKO;AD samples with no apparent reduction in Bace1 transcripts in ExNs. (b) Violin plots showing individual mouse distribution of Bace1 puncta/thalamic nuclei. Solid lines represent medians and faded lines represent quartiles. (c) Isocortical regions containing neuronal projections into the thalamus (left) and the inferior colliculus (right). Inferior colliculus primarily receives cortical input from the auditory cortex. Images were adapted from the Allen Brain Atlas: Mouse Connectivity: Projection (https://connectivity.brain-map.org/). (d) Fluorescence microscopy images of 5xFAD mouse cortex (left) and thalamus (right) stained for Aβ42 (blue) and human-specific APP (yellow). White arrowheads point to neuronal somas in the cortex but not the thalamus of 5xFAD mice positive for human-specific APP. Immunolabeling was performed once on brain slices of 6-month-old 5xFAD mice (n = 4) (e-g) Plaque deposition is not directly proportional to APPNLGF gene dosage. (e) LSM 3D visualization of female WT, heterozygous, and homozygous APPNLGF hemibrains at 6 months of age. (f) Quantification of LSM data between female homozygous (n = 10) and heterozygous APPNLGF mice (n = 7). Heterozygous data points were normalized to homozygous data. Circles represent homozygous APPNLGF mice and half-filled circles represent heterozygous APPNLGF mice. For each parameter, unpaired, two-tailed Student’s t-test was performed (P values indicated in graphs) comparing the two groups. Bars represent means with SEM and individual data points displayed. Raw unnormalized data is available in Supplementary Table 3. (g) Immunoblot of 6-month-old male homozygous APPNLGF, heterozygous APPNLGF, and WT mice labeled for APP (n = 2 per group). Heterozygous mice express half of the APPNLGF gene dosage, resulting in a FL-APP level between WT and homozygous APPNLGF mice. This is accompanied by a concomitant accumulation of half β-CTFs in heterozygous lysates compared to the homozygous amount.
Extended Data Fig. 9 Considerable plaque deposition occurs in older ExN-Bace1cKO;AD mice and double Bace1 cKO in OLs and ExNs ablated cerebral Aβ burden.
(a) LSM 3D visualization of female ExN-Bace1cKO;AD mice at 3, 6, 9, and 12 months alongside a Control;AD mouse brain at 6 months stained with Congo Red. Bottom right insets represent 2D cortical planes with yellow arrowheads pointing to plaques. (b) Quantification of LSM total cortical plaque volume between ExN-Bace1cKO;AD at various time points (n = 3–5 per group). Normalization of ExN-Bace1cKO;AD data points to Control;AD data points was performed. Blue bars represent ExN-Bace1cKO;AD data points and the gray bar represents Control;AD. An unpaired, two-tailed Student’s t-test was performed (P values indicated in the graph with significance highlighted in bold) comparing 12-month-old ExN-Bace1cKO;AD and 6-month-old Control;AD data. Bars represent means with SEM and individual data points displayed. Raw unnormalized data is available in Supplementary Table 4. (c-f) Double Bace1 cKO in OLs and ExNs ablated cerebral Aβ burden. Light sheet microscopy data of plaque burden (Congo red) comparing 6-month-old OL-ExN-Bace1cKO;AD male mice to age- and sex-matched ExN-Bace1cKO;AD mice. Color-region allocation is as follows: White–isocortex, blue–hippocampus, yellow–alveus, pastel blue–inferior colliculus, pastel yellow–thalamus. (c) LSM 3D visualization of ExN-Bace1cKO;AD and OL-ExN-Bace1cKO;AD male mouse hemibrains. (d) Brain region-segmented plaques of ExN-Bace1cKO;AD and OL-ExN-Bace1cKO;AD male mouse hemibrains. (e) Quantification of LSM data between ExN-Bace1cKO;AD (n = 5) and OL-ExN-Bace1cKO;AD (n = 3) hemibrains. Normalization of OL-ExN-Bace1cKO;AD data points was performed to ExN-Bace1cKO;AD. Hollowed triangles represent ExN-Bace1cKO;AD and hollowed squares represent OL-ExN-Bace1cKO;AD. For each parameter, unpaired, two-tailed Student’s t-test was performed (P values indicated in graphs) comparing the two groups. Bars represent means with SEM and individual data points displayed. Raw unnormalized data is available in Supplementary Table 5. (f) Aβ42 electrochemiluminescence immunoassay data of insoluble (SDS-soluble, left) and soluble (Tris-NaCl-soluble, right) lysates of microdissected cortical (top) and WM (bottom) tissues from ExN-Bace1cKO;AD (n = 7) and OL-ExN-Bace1cKO;AD (n = 3) fractions of 6-month-old male mouse hemibrains. Data points were normalized to ExN-Bace1cKO;AD samples. The small amount of Aβ42 still produced in ExN-Bace1cKO;AD fractions is almost completely ablated in OL-ExN-Bace1cKO;AD fractions, highlighting the OL contribution to Aβ42 even in the absence of Aβ42 from ExNs. An unpaired, two-tailed Student’s t-test was performed (P values indicated in the graph with significance highlighted in bold) comparing the two groups. Bars represent means with SEM and individual data points displayed. Raw unnormalized data are available in Supplementary Table 6.
Extended Data Fig. 10 Bace1 cKO does not result in changes to neuronal numbers, axonal densities, and myelination profile.
(a) Closeups of fluorescence microscopy images of cortices of controls and cKOs highlighting unchanged density of neurons and axons in both cKO lines at a gross level. (b) Quantification of neuronal and axonal densities between 6-month-old female controls and cKOs (n = 5 per group) in the isocortex. (c-g) Bace1 cKO does not alter myelination profile. (c) Closeups of fluorescence microscopy images of cortices of controls and cKOs highlighting unchanged density of OLs and myelination in both cKO lines at a gross level. (d) Quantification of OL density between controls and cKOs (n = 5 per group) spanning different regions. (e) Quantification of myelin density between controls and cKOs (n = 5 per group) spanning different regions. As CC and alveus are densely myelinated tracts, mean intensity values were instead measured. (f) Representative electron micrographs of caudal corpus callosum (CC) of controls and cKOs at 6 months of age. (g) Analysis of myelin thickness via g-ratio measurement with single dots representing single myelinated axons quantified (Control;AD = 397, OL-Bace1cKO;AD = 417, ExN-Bace1cKO;AD = 394). Lines represent linear regressions of each group and shaded area indicates error bars. Myelinated axon counts and mean g-ratio comparisons from electron micrographs of controls and cKOs (n = 4 per group). For (b,d,e,g), one-way ANOVA was performed with Tukey multiple comparison tests (P values indicated in graphs with significance highlighted in bold). Bars represent means with SEM and individual data points displayed.
Supplementary information
Supplementary Fig. 1.
Raw immunoblots shown in this study.
Supplementary Tables 1–8
Supplementary Table 1. Raw LSM data of OL-Bace1cKO;AD analysis at 6 months (n = 5 per sex and genotype) from Fig. 2f. Supplementary Table 2. Raw LSM data of ExN-Bace1cKO;AD analysis at 6 months (n = 5 per sex and genotype) from Fig. 2k. Supplementary Table 3. Raw LSM data of female heterozygous APPNLGF analysis at 6 months (n = 7) from Fig. 8f. Supplementary Table 4. Raw LSM data of female ExN-Bace1cKO;AD analysis at 3 months, 9 months and 12 months (n = 3 per age group) from Fig. 9b. Supplementary Table 5. Raw LSM data of male OL-ExN-Bace1cKO;AD analysis at 6 months (n = 3 per group) from Fig. 9f. Supplementary Table 6. Raw Aβ immunoassay data pertaining to this study. Normalized Aβ immunoassay data of control, OL-Bace1cKO;AD and ExN-Bace1cKO;AD (n = 4 per group) at 6 months of age (Fig. 3). Cortical and WM data are both represented with adjacent columns containing Aβ38, Aβ40 and Aβ42 values. Top half represents measurements of the SDS-soluble fractions, and bottom half represents measurements of the Tris-NaCl-soluble fractions. All values below the detection range of the immunoassay were reported as 0. Supplementary Table 7. Details of primary antibodies used in this study. Supplementary Table 8. Details of secondary antibodies used in this study.
Source data
Source data Figs. 2 and 3 and Extended Data Figs. 1–10
Source data for Figs. 2 and 3 and Extended Data Figs. 1–10
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sasmita, A.O., Depp, C., Nazarenko, T. et al. Oligodendrocytes produce amyloid-β and contribute to plaque formation alongside neurons in Alzheimer’s disease model mice. Nat Neurosci (2024). https://doi.org/10.1038/s41593-024-01730-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41593-024-01730-3
- Springer Nature America, Inc.