

Abstract
The BEACON CRC study demonstrated that encorafenib (Enco)+cetuximab (Cetux)±binimetinib (Bini) significantly improved overall survival (OS) versus Cetux + chemotherapy in previously treated patients with BRAF-V600E-mutant mCRC, providing the basis for the approval of the Enco+Cetux regimen in the United States and the European Union. A greater understanding of biomarkers predictive of response to Enco+Cetux±Bini treatment is of clinical relevance. In this prespecified, exploratory biomarker analysis of the BEACON CRC study, we characterize genomic and transcriptomic correlates of clinical outcomes and acquired resistance mechanisms through integrated clinical and molecular analysis, including whole-exome and -transcriptome tissue sequencing and circulating tumor DNA genomic profiling. Tumors with higher immune signatures showed a trend towards increased OS benefit with Enco+Bini+Cetux. RAS, MAP2K1 and MET alterations were most commonly acquired with Enco+Cetux±Bini, and more frequent in patients with a high baseline cell-cycle gene signature; baseline TP53 mutation was associated with acquired MET amplification. Acquired mutations were subclonal and polyclonal, with evidence of increased tumor mutation rate with Enco+Cetux±Bini and mutational signatures (SBS17a/b). These findings support treatment with Enco+Cetux±Bini for patients with BRAF-V600E-mutant mCRC and provide insights into the biology of response and resistance to MAPK-pathway-targeted therapy. ClinicalTrials.gov registration: NCT02928224
Similar content being viewed by others


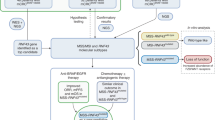
Main
BRAF-V600E mutations occur in approximately 10% of tumors in patients with mCRC and are associated with poor prognosis relative to wild-type BRAF tumors1,2. The BEACON CRC study (NCT02928224; Binimetinib, Encorafenib and Cetuximab Combined to Treat BRAF-Mutant Colorectal Cancer) demonstrated that the BRAF inhibitor Enco plus the anti-epidermal growth factor receptor (EGFR) antibody Cetux, with or without the MEK inhibitor Bini, improved OS, objective response rate and progression-free survival (PFS) compared with Cetux plus chemotherapy in patients with BRAF-V600E-mutant mCRC who had been previously treated3,4. A greater understanding of the BRAF-mutant (BM) biological landscape and prognostic and predictive determinants is important for optimizing therapy.
Transcriptional profiling of CRC tumors has identified subtypes that have similar biological characteristics. These include the consensus molecular subtypes (CMS) and BM subtypes5,6. These subtypes may have utility in predicting prognosis and treatment response in patients with CRC5,7. Previously treated patients with BRAF-V600E-mutant CRC have shown acquired resistance alterations in the MAPK pathway and amplifications in MET following treatment with BRAF inhibitor combinations8,9,10. To our knowledge, clinical associations with molecular subtypes and mechanisms of acquired resistance following treatment with a BRAF inhibitor plus an anti-EGFR antibody, with or without a MEK inhibitor, versus conventional cytotoxic chemotherapy plus an anti-EGFR antibody, have not been studied in large, randomized trials.
Through genomic and transcriptional profiling of tumor tissue and genomic profiling of plasma samples obtained from the BEACON CRC study cohort, we characterized the molecular landscape of BRAF-V600E-mutant mCRC treated with Enco+Cetux±Bini versus Cetux plus chemotherapy to understand the evolving biology of these tumors on treatment. The objective of this analysis was to identify molecular correlates of clinical outcomes and define resistance mechanisms acquired following treatment. The integrated tumor and plasma molecular profiling analyses performed in this study provide comprehensive data from the largest randomized study in BRAF-V600E-mutant CRC.
Results
BEACON CRC biomarker analysis set
Of the 665 patients enrolled in the full analysis set, 621 (93.4%) were included in the biomarker analysis set (Fig. 1a). To assess the molecular landscape of baseline tumors, we analyzed 503 patients (81.0%) by whole-exome sequencing (WES) and 441 (71.0%) by whole transcriptome sequencing (WTS). For assessment of the genomic landscape by circulating tumor DNA (ctDNA) profiling, we analyzed 544 patients (87.6%) at baseline and 320 (51.5%) with paired end-of-treatment (EoT) samples. Baseline biomarker analyses were performed in a representative patient population (Fig. 1b). The number of patients was balanced among the three treatment arms (Enco+Bini+Cetux, Enco+Cetux and control) for all biomarker analysis subsets (Supplementary Table 1).

a, Patient flow in analysis sets. aFull analysis set included patients who were enrolled to receive randomized treatment. bSafety analysis set included patients who received one or more dose of any trial drug and had one or more post-treatment safety assessment. cBiomarker analysis set included patients who had baseline tumors analyzed by WTS or WES or who were analyzed for ctDNA at baseline. dBiomarker analysis subsets were not mutually exclusive. b, Heatmap of patient characteristics and molecular profiling coverage in the biomarker analysis set, across treatment arms. CEA, carcinoembryonic antigen; CRP, C-reactive protein; ECOG PS, Eastern Cooperative Oncology Group performance status; tx, treatment; ULN, upper limit of normal.
Baseline molecular profiling of patients
Genomic profiling of baseline tumor tissue and ctDNA showed that BRAF-V600E was detected by WES and ctDNA in 476 of 503 (94.6%) and 492 of 544 patients (90.4%) analyzed, respectively (Fig. 2a, Extended Data Fig. 1 and Supplementary Table 2). Of the patients who had genomic profiling performed on both tumor and ctDNA samples, 368 of 404 (91.1%) had BRAF-V600E detected in both (Supplementary Table 3).

a, Oncoprint showing frequency of cancer gene mutations in baseline tumors profiled by the GuardantOMNI panel, as well as TMB and MSI status (top bars), across treatment arms determined using WES. BRAF alterations included V600E and other alterations. The MSS group also included patients who were MSI-low. b, Baseline tumor size (left panel) and baseline ctDNA maxVAF (right panel) by baseline ctDNA BRAF-V600E status. P values were based on the Wilcoxon rank-sum test (two-sided). The boxes show medians and IQR; the whiskers represent 1.5 × IQR. c, Kaplan–Meier plots of OS by ctDNA BRAF-V600E VAF status (split on median) in each treatment arm. P values (two-sided Wald test) were based on the Cox model without adjustment for baseline covariates. d, Distribution of BM1 and BM2 subtypes across CMS subgroups. aThe ‘not detected’ group included patients with no ctDNA detected. bPatients with no ctDNA detected were excluded from the analysis. cThe BM1 subtype is characterized by high KRAS/mTOR/AKT/4EBP1, epithelial–mesenchymal transition (EMT) and immune infiltration, whereas the BM2 subtype is characterized by cell-cycle checkpoint dysregulation5. dCMS1 is characterized by MSI and immune patterns and enrichment for BRAF mutations; CMS2 by chromosomal instability and WNT activation; CMS3 by metabolic pattern and enrichment for KRAS mutations and CMS4 by EMT, angiogenesis and stromal infiltration5.
Compared with patients who had detectable BRAF-V600E by ctDNA genomic profiling, those who had undetectable BRAF-V600E had smaller baseline tumor sizes (median (interquartile range; IQR) 40.00 mm (28.00–72.00) versus 66.00 mm (42.00–97.00); P = 0.001) and lower baseline ctDNA maximum variant allele frequency (maxVAF) (0.830% (0.350–4.200) versus 13.605% (2.595–35.680); P < 0.0001) (Fig. 2b). Consistent with this finding, when maxVAF was below the first quartile (2.01%), the percentage of patients with detectable BRAF-V600E was 78.4%; when maxVAF was in the third quartile (33.77%) and above, the percentage increased to >99% (P < 0.0001) (Extended Data Fig. 2a). maxVAF by ctDNA genomic profiling correlated with BRAF-V600E VAF (r = 0.93; P < 0.0001; Extended Data Fig. 2b). Patients with liver metastases at baseline had higher BRAF-V600E VAF than those who did not (P < 0.0001), whereas BRAF-V600E VAF did not differ between patients with three or more involved organs and those with fewer involved organs (P = 0.25) (Supplementary Fig. 1a,b).
Evaluation of BRAF-V600E VAF levels in ctDNA at baseline showed that, in all treatment arms, OS was longer in patients with low (≤median (7.385%)) BRAF-V600E VAF than in those with high (>median) BRAF-V600E VAF (P < 0.0001 for all arms) (Fig. 2c). maxVAF by ctDNA genomic profiling showed similar associations with OS in all treatment arms (Extended Data Fig. 3). Overall, results were similar with or without adjustments for baseline covariates (Extended Data Fig. 3).
The cancer-associated genes comutated most frequently in tumor tissue were TP53 (70%), APC (36%), SMAD4 (28%), FAT1 (27%), RNF43 (26%) and LRP1B (25%) (Fig. 2a). Microsatellite instability (MSI)-high (MSI-H) was detected in 11.1% of patients (Supplementary Table 2). The median tumor mutational burden (TMB) was 7.740 mutations (mut)/Mb, and 15.7% of patients had a TMB of ≥10 mut/Mb (Supplementary Table 2). ctDNA genomic profiling showed similar frequently mutated cancer-associated genes; MSI-H was detected in 8.8% of patients and median TMB was 11.480 mut/Mb across all arms (Extended Data Fig. 1 and Supplementary Table 2).
Tumors were classified into CMS and BM subtypes by transcriptomic analyses. Most patients analyzed had either CMS1 (214/441; 48.5%) or CMS4 (166/441; 37.6%) tumors; 33.1% (146/441) of patients were classified as having the BM1 subtype, which was observed predominately in those with CMS4 tumors (122/166; 73.5%) (Fig. 2d).
Associations of CRC subtypes and biomarkers with OS benefit
We examined the association between OS benefit and the CMS and BM subtypes in the BEACON CRC patient population. OS benefit with Enco+Cetux versus control was observed irrespective of BM subtype (Pinteraction = 0.0803) or CMS (Pinteraction = 0.2961), with a trend towards increased benefit in the BM2 and CMS1 subgroups (Extended Data Fig. 4 and Supplementary Fig. 2a (adjusted analyses)). Similarly, OS benefit with Enco+Bini+Cetux versus control was observed across the cohort with no differences according to BM subtype (Pinteraction = 0.7607) or CMS (Pinteraction = 0.8656) (Extended Data Fig. 4 and Supplementary Fig. 2a (adjusted analyses)).
We examined whether the mutational status of the most frequently comutated genes in CRC (APC, PIK3CA, RNF43 and TP53), MSI status or TMB level was associated with differences in OS in patients treated with either Enco+Bini+Cetux or Enco+Cetux compared with control (Fig. 3 and Supplementary Fig. 2b (adjusted analyses)). Patients with wild-type TP53 showed greater OS benefit with Enco+Cetux (hazard ratio (HR), 0.37 (95% confidence interval (CI), 0.224–0.627) for wild type and 0.76 (95% CI, 0.547–1.060) for mutant; Pinteraction = 0.0276), with a similar trend observed for the OS difference in the Enco+Bini+Cetux versus control arms (Pinteraction = 0.0712). We did not observe differences in OS benefit in subgroups defined by other specific gene mutations, MSI-H or TMB. Specifically, OS benefit was observed in patients treated with Enco+Cetux±Bini regardless of RNF43 mutational status (Enco+Cetux: unadjusted HR, 0.53 (95% CI, 0.381–0.733) for wild type and 0.93 (95% CI, 0.535–1.622) for mutant; Pinteraction = 0.0693 and Enco+Bini+Cetux: unadjusted HR, 0.55 (95% CI, 0.405–0.745) for wild type and 0.71 (95% CI, 0.394–1.263) for mutant; Pinteraction = 0.6269) (Fig. 3 and Supplementary Fig. 2b). Subgroup analyses showed similar OS findings by RNF43 mutational status in MSS patients (Supplementary Table 4).

Forest plots of HRs for OS with Enco+Cetux (top panel) or Enco+Bini+Cetux (bottom panel) versus control in subgroups defined by specific gene mutations, MSI status or TMB levels. The diamonds show HRs and the lines show 95% CI. P values for interaction (two-sided Wald test) were based on the Cox model without adjustment for baseline covariates. aThe MSS subgroup included patients with MSI-low; those with inconclusive results were excluded from the analysis.
Immune associations with OS benefit from Enco+Cetux±Bini
To evaluate gene expression profiles predictive of OS benefit from Enco+Bini+Cetux or Enco+Cetux, we used Cox models to test for interactions between univariable gene expression and treatment arm (Fig. 4a). The expression of genes linked to T-cell biology and immune processes (for example, TBX21, GZMA, TLR7, CD7)11,12,13,14 was strongly enriched in patients who had an OS benefit from Enco+Bini+Cetux, more modestly from control and in those who had decreased OS benefit from Enco+Cetux (all Pinteraction < 0.001 for Enco+Cetux versus Enco+Bini+Cetux; Pinteraction < 0.001 for Enco+Cetux versus control) (Fig. 4a and Extended Data Fig. 5a). Univariable tests of gene expression associations with OS in each arm (Fig. 4a), followed by gene set enrichment analysis (GSEA) against hallmark genesets, confirmed these observations (Fig. 4b and Extended Data Fig. 5b), revealing that immune processes (for example, interferon (IFN)-γ signaling, allograft rejection) were enriched most strongly in patients who had prolonged OS in the Enco+Bini+Cetux arm and in those who had shorter OS in the Enco+Cetux arm (Supplementary Fig. 3).

a, Associations between OS and immune gene expression (scaled) were determined by Cox proportional hazards models (two-sided Wald test). Genes in the top quartile of variance in expression (based on IQR and measured in log2(TPM)) were tested. b, GSEA hallmark genesets; highest average absolute normalized enrichment score (NES) across arms are shown; point size indicates Benjamini–Hochberg-adjusted P values from permutation testing. c, Kaplan–Meier plots of OS by cytolytic score (split on median). P values for interaction (two-sided Wald test) were based on Cox proportional hazard models with and without adjustment for baseline covariates.
To evaluate the association between a simplified metric of tumor immune status and OS, we applied a cytolytic score used widely to measure the immune status of the tumor microenvironment (mean of log GZMA and PRF1 expression)13 to characterize tumors as cytolytic high (>median of 3.11 log2(transcripts per million (TPM)) or cytolytic low (≤median). The cytolytic score was correlated with single-sample GSEA scores for the hallmark IFN-γ response (r = 0.73; P < 0.001), allograft rejection (r = 0.79; P < 0.001) and inflammatory response (r = 0.59; P < 0.001) signatures (Extended Data Fig. 6a), as well as sample scores for multiple metrics inferring CD8+ T-cell infiltration and other gene signatures identifying immune infiltration15 (all P < 0.001 and Extended Data Fig. 6b). Patients with cytolytic-low tumors had longer OS than those with cytolytic-high tumors in the Enco+Cetux arm (HR, 1.85 (95% CI, 1.203–2.847) for cytolytic-high versus cytolytic-low]); in contrast, Enco+Bini+Cetux-treated patients with cytolytic-high tumors had longer OS (HR, 0.56 (95% CI, 0.377–0.844) for cytolytic-high versus cytolytic-low); Pinteraction = 0.0012) (Fig. 4c and Supplementary Table 5). Similar trends were observed for PFS, albeit slightly less pronounced (HR, 1.62 (95% CI, 1.107–2.365) for cytolytic-high versus cytolytic-low in the Enco+Cetux arm and HR, 0.69 (95% CI, 0.476–0.997) in the Enco+Bini+Cetux arm; Pinteraction = 0.0088) (Extended Data Fig. 7a,b). Inferring immune cell type abundance using xCell16 affirmed an enrichment of lymphocytes (for example T cells, B cells, plasma cells and natural killer cells) in tumors with higher cytolytic scores (Extended Data Fig. 8).
BM1 tumors had higher cytolytic scores than BM2 tumors (P < 0.0001) (Supplementary Fig. 4, top panel). There was a range of cytolytic scores across the CMS subtypes, with potentially higher cytolytic scores in tumors characterized as CMS1 and CMS4 (CMS4 versus CMS1; P = 0.0861) (Supplementary Fig. 4, bottom panel).
Acquired resistance alterations with Enco+Cetux±Bini
We analyzed paired cycle 1 day 1 (C1D1) and EoT samples using ctDNA genomic profiling to assess the landscape of acquired mutations by characterizing alterations as maintained, lost or acquired. Mutations in BRAF-V600E were predominantly maintained across all treatment arms (Fig. 5a).

a, Status of selected gene alterations (analyzed by ctDNA genomic profiling) at EoT versus baseline and survival outcomes by treatment arm. b, Mutational landscape of KRAS, NRAS and MAP2K1 variants in the Enco+Cetux and Enco+Bini+Cetux arms.
The most frequently (that is, top) acquired putative resistance alterations were mutations in KRAS, NRAS and MAP2K1 and amplification of MET (Fig. 5a). KRAS, BRAF and IGF1R amplifications were acquired at frequencies of 8.9%, 5.4% and 4.5%, respectively, in the Enco+Bini+Cetux arm and 4.5%, 1.8% and 0.9% in the Enco+Cetux arm. One patient in the control arm developed BRAF amplification (Supplementary Table 6).
Of the 318 patients with paired samples, acquired MET fusions were found in 13 (4.1%), BRAF fusions in 11 (3.5%) and ALK fusions in 7 (2.2%). Acquired fusions were also found in RET, NTRK1 and MERTK at very low frequencies. Acquired EGFR mutations were observed at a low frequency in the Enco+Bini+Cetux and Enco+Cetux arms (eight and four patients, respectively) (Supplementary Table 6).
Retrospective analyses in patients who did not have a mutation in KRAS, NRAS or MAP2K1 or MET amplification at baseline showed that patients who did not acquire any of these alterations at EoT had longer survival than those who did (OS: HR, 1.64 (95% CI, 1.134–2.365); PFS: HR, 1.75 (95% CI, 1.258–2.435)) (Extended Data Fig. 9, top panel and Supplementary Fig. 5a,b (adjusted analyses)). Patients whose tumors acquired MET amplification at EoT had shorter survival (OS: HR, 2.43 (95% CI, 1.616–3.650); PFS: HR, 2.28 (95% CI, 1.560–3.321)) than those whose tumors did not (Extended Data Fig. 9 (bottom panel) and Supplementary Fig. 5a,b (adjusted analyses)).
Among patients who did not have baseline alterations in the top acquired genes, the frequency of any alterations in these genes at EoT was 58.3%, 61.8% and 6.8% in the Enco+Bini+Cetux, Enco+Cetux and control arms, respectively (Supplementary Table 7). Of the patients with paired ctDNA data, the frequency for two or more acquired alterations was 34.8%, 41.1% and 0%, respectively (Supplementary Fig. 6). KRAS mutations were acquired in 42.5% and 45.0% of Enco+Bini+Cetux- and Enco+Cetux-treated patients, respectively, and NRAS mutations in 25.5% and 37.3%; these mutations were found predominantly in hotspot regions of the respective genes (Fig. 5b and Supplementary Table 7). Acquired MAP2K1 mutations were observed in 16.1% of patients treated with Enco+Cetux but in only 3.7% of patients treated with Enco+Bini+Cetux (P = 0.0028 for between-group difference) and were primarily RAF dependent, including F53L, K57E/N, C121S and E203K (Fig. 5b and Supplementary Tables 7 and 8). MET amplification was acquired in 22 of 112 (19.6%) and 19 of 112 (17.0%) Enco+Bini+Cetux- and Enco+Cetux-treated patients, respectively (Supplementary Table 7).
Characterization of acquired resistance alterations
To assess the clonality of acquired resistance alterations, the VAF of the alteration versus the maxVAF for any somatic alteration in the sample at EoT was determined. Acquired resistance alterations were predominately subclonal (≤ 0.3 EoT VAF/maxVAF) compared with alterations that were detected at baseline and maintained following treatment (P < 0.0001) (Extended Data Fig. 10). The rate of alterations acquired on treatment (number of acquired mutations per month) can be used to estimate subsequent rates of new alterations. Among patients in the Enco+Bini+Cetux and Enco+Cetux arms, the rate was approximately 1.7 and 1.5 times that in patients in the control arm, respectively (Fig. 6a). The rate at which alterations were acquired was found to represent a continuum across patients, with a subset showing >20 acquired mutations per month (Fig. 6a).

a, Subgroups based on rate of acquiring resistance. Poisson mixture model was used to identify the low-, medium- and high-rate subgroups. b, Number of mutations grouped by SBS mutational signature at C1D1 and EoT. c, GSEA against hallmark reactome signatures based on the association between baseline gene expression and acquiring one or more top alteration at EoT. The dashed red line corresponds to an adjusted P value of 0.05 from GSEA. d, Association of top acquired alterations with baseline TP53 mutation status and GSEA hallmark cell-cycle signature (split on median) (OR = 4.3; P = 0.006); Benjamini–Hochberg-adjusted P values (two-sided) are based on permutation tests (two-sided). aThe value 0 was coded as half of the smallest nonzero value, 0.03, for display.
To understand the nature of these acquired mutations, we evaluated the single-base substitution (SBS) mutational signatures (Catalogue of Somatic Mutations in Cancer)17 from ctDNA genomic profiling by pooling genomic data by treatment arm and timepoint (C1D1 and EoT). We observed that SBS1 and SBS5—potentially caused by spontaneous or clock-like mutation processes—and SBS15—associated with DNA mismatch repair18—were identified at both C1D1 and EoT across all treatment groups. However, SBS17, which is associated with reactive oxygen species (ROS)19, was observed only at EoT; SBS17b (mainly T>G mutations) was identified in all treatment arms and at higher frequencies in the Enco+Bini+Cetux and Enco+Cetux arms, while SBS17a (mainly T>C mutations) was observed in Enco+Cetux-treated patients (Fig. 6b).
To determine the baseline biological tumor characteristics associated with acquired resistance alterations following treatment with Enco+Cetux±Bini, we compared the genomic and transcriptomic profiles of baseline tumor tissues between patients who did and did not subsequently acquire a presumptive resistance alteration (that is, top acquired resistance alteration). GSEA analyses showed prominent enrichment of the hallmark cell-cycle signature in tumors of patients who acquired alterations compared with those who did not following treatment with Enco+Cetux±Bini, with CCNB1 and CDK1 in the leading edge and more highly expressed (Fig. 6c and Supplementary Figs. 7 and 8). A higher proportion of patients with the BM2 subtype acquired resistance than those with the BM1 subtype (P = 0.029) (Supplementary Fig. 9). Analysis of baseline tumor mutations showed that a higher proportion of patients with TP53 mutation acquired MET amplification following treatment with Enco+Cetux±Bini than those with wild-type TP53 (odds ratio = 4.3; P = 0.006) (Fig. 6d); this difference between the TP53 mutant and wild-type subgroups was consistent when considering the acquisition of any putative resistance amplification (that is, MET, KRAS, BRAF, IGF1R; odds ratio = 4.2; P = 0.0006) (Supplementary Fig. 10).
Discussion
The primary analysis of BEACON CRC showed that Enco+Cetux, with or without Bini, significantly improved OS compared with control (Cetux+irinotecan, or Cetux+folinic acid, fluorouracil and irinotecan (FOLFIRI)) in patients with BRAF-V600-mutant mCRC. Here, we report a prespecified, retrospective, exploratory biomarker analysis of this large, phase 3 dataset.
Overall, the clinical benefit observed with treatment with Enco+Bini+Cetux and Enco+Cetux compared with control was maintained across CMS and BM subtypes, regardless of specific gene mutations, MSI or TMB. CMS and BM subtypes have come to define molecular paradigms for CRC, and their analyses were included here to contextualize our findings. Nonetheless, results in this study were supported by unbiased differential expression and GSEA, which served to further highlight immunological features of the TME as potential determinants of treatment benefit. While most mCRCs have microsatellite stability (MSS) and hence are considered immune cold, those that have MSS and are BRAF-V600E mutant have been reported to show some degree of immune activation2,20,21,22. Immune signatures may be enriched in BM1 and CMS1/CMS4 CRC subtypes, which have been shown to have a stronger immune profile and greater inflammatory response compared with other CRC subtypes5,23.
In the present study, patients with tumors that had a stronger immune signature (for example, increased T cells or cytolytic score) showed a trend towards decreased benefit from treatment with Enco+Cetux and increased benefit from treatment with Enco+Bini+Cetux. The observed trend in treatment outcomes with Enco+Bini+Cetux compared with Enco+Cetux could be explained by immune or inflammatory signals within the tumor microenvironment. It was previously reported that BRAF inhibitors alone showed stimulatory effects on T cells, potentially through paradoxical activation of ERK24. In contrast, MEK inhibitors led to partial and transient inhibition of T cell proliferation and potentially induced regenerative stem cell-like memory CD8+ T cells25,26, which might confer protection against T cell exhaustion and support antitumor responses. Therefore, patients with BRAF-V600E-mutant CRC, who had higher levels of immune activity, might derive further benefit from the addition of a MEK inhibitor or immunotherapy in combination with BRAF/EGFR inhibition, possibly accounting for the aforementioned findings in the present study. However, these trends should be interpreted with caution due to the retrospective nature of the analyses and small sample sizes, warranting further evaluation in future prospective trials. Nevertheless, our findings also support ongoing trials, including SEAMARK (NCT05217446) in MSI-H CRC and other studies in MSS CRC (NCT04017650 and NCT05308446 (SWOG2107)), which are evaluating whether the combination of Enco+Cetux with checkpoint inhibitors will increase clinical efficacy27,28,29.
Although a potential association between clinical outcomes and immune gene signature was observed in the present study, this was not seen with specific gene mutations, including RNF43. However, a study previously reported that RNF43 mutations predicted response to BRAF inhibitor in combination with an anti-EGFR monoclonal antibody in BRAF-V600E-mutant mCRC30. The observed disparity in the predictive value of RNF43 mutations might be attributed to differences in patient population, sample size or comparator arm (chemotherapy ± antiangiogenic therapy). The predictive value of specific RNF43 mutations in patients who receive anti-BRAF/EGFR therapy requires further investigation in adequately powered prospective studies.
We observed a highly specific trend of acquired MAPK pathway alterations (KRAS and NRAS mutations) and MET amplifications that were similarly represented in the Enco+Bini+Cetux and Enco+Cetux arms but nearly absent in the control group. Our findings align with reports from previous smaller studies8,9,10,31, suggesting that targeting downstream MAPK activity or MET blockade could potentially help delay and overcome acquired resistance to Enco+Cetux, with or without Bini8,9,10. Activation of other receptor tyrosine kinases as part of a compensatory mechanism can also lead to the feedback activation of MEK, and the addition of SHP2 inhibitor has shown the potential to overcome resistance to MEK inhibitors32,33,34. Acquired MAP2K1 mutations observed following treatment in this study were also previously identified in patients with BRAF-V600E-mutant CRC treated with EGFR/BRAF inhibitors8,10. Most of the detected MAP2K1 mutations were primarily RAF dependent and therefore sensitive to MEK inhibition. Notably, acquired MAP2K1 mutations were around five times more common in Enco+Cetux-treated patients compared with Enco+Bini+Cetux-treated patients, which is consistent with the ability of Bini to inhibit common MAP2K1-activating mutations. Nevertheless, the frequency of RAF-dependent MEK resistance mutations was relatively low compared with that of alterations in KRAS, NRAS and MET, which were observed in both treatment arms. Acquired KRAS, BRAF and IGF1R amplifications were observed at slightly higher frequencies in the Enco+Bini+Cetux arm (8.9%, 5.4% and 4.5%, respectively) than in the Enco+Cetux arm (4.5%, 1.8% and 0.9%). Known EGFR resistance mutations in the EGFR extracellular domain, which may be associated with resistance to Cetux35, were rarely acquired. In the present study, patients who had acquired top putative resistance alterations (that is, in RAS, MAP2K1 and MET) had shorter PFS and OS, with similar HRs for both survival variables. These data suggest that patients who developed these alterations might experience faster progression on treatment, leading to shorter survival. Nevertheless, this analysis was only based on EoT samples, thus limiting interpretation; we do not have longitudinal sampling during treatment before progression, which would allow for more appropriate statistical evaluation of the association between acquired resistance alterations and survival.
Adaptive mutability refers to an increased mutation rate enabling cancer cells to evade therapeutic pressure36. This was reported in CRC in ref. 36; EGFR/BRAF inhibition downregulated DNA repair pathways and increased expression of low-fidelity DNA polymerases to promote DNA damage, mutability and microsatellite instability36. In a recent study of EGFR inhibition in RAS/BRAF/EGFR wild-type mCRC, disease progression on treatment was observed to be driven by the acquisition of newly acquired alterations rather than the expansion of preexisting resistance subclones, supporting the concept of adaptive mutability37. Results from our analysis suggest the presence of adaptive mutability in a cohort of patients with BRAF-V600E CRC treated with BRAF inhibitor in combination with an anti-EGFR monoclonal antibody, with or without a MEK inhibitor. Our results demonstrate a high number of emergent alterations in samples from patients on treatment. In particular, the rate of acquiring new alterations was higher with Enco+Cetux±Bini treatment, with a greater number of patients who had >20 acquired alterations per month in these treatment arms than in the control arm.
Acquired resistance alterations with Enco+Cetux±Bini showed a potential association with SBS17a/b mutational signatures, characterized mainly by T>C and T>G mutations17,38. SBS17b mutational signature was reported to be the main contributor of specific KRAS/NRAS and EGFR mutations that were enriched at acquired resistance to Cetux39. BRAF inhibitor-resistant melanoma has been reported to show elevated ROS levels40, consistent with the association of SBS17 signature with mutability and ROS19. Increased ROS levels may, in turn, induce DNA damage and affect the DNA damage response, potentially contributing to adaptive mutability36. The present analysis reports a potential association between SBS17a/b mutational signatures and acquired resistance alterations observed via ctDNA analysis in a large registrational study. Although limited by the size of the sequencing panel used, the potential to detect the SBS17b mutational signature in ctDNA samples provides an advantage by allowing easier sample collection for evaluation of resistance mechanisms during progression and warrants further exploration. Of note, SBS17a/b signatures were not observed in baseline tumor samples; however, paired progression biopsies were not available for longitudinal comparison.
The enrichment of cell-cycle gene expression at baseline in patients who acquired these top resistance alterations in the present study might also support the theory of adaptive mutability36 in that reduced DNA repair coupled with high-cycling tumors allows a greater chance for DNA errors to accumulate and mutations to emerge from treatment pressure, contributing to the acquired resistance phenotype and observed worse survival outcomes. In contrast, low-cycling tumors may take a longer time to accumulate putative resistance alterations, delaying the development of acquired resistance. Of note, we observed a potential association between the BM2 subtype and acquired resistance, consistent with the reported dysregulation of the cell cycle in the BM2 CRC subtype5. Additionally, consistent with TP53 mutations being associated with genomic instability41, we found that acquired MET amplification in the Enco+Cetux±Bini arms occurred in a higher proportion of patients with TP53 mutations than in those without. The roles of upregulated cell-cycle gene expression and TP53 mutations should be further evaluated in BRAF-V600E-mutant mCRC to determine whether they could be potential prognostic biomarkers in this patient population.
This study has several strengths. First, it is based on a large dataset for a BRAF-V600E-mutation-driven subset of CRC, from a phase 3 clinical trial with integrated tumor and ctDNA analyses via full WES/WTS sequencing and ctDNA genomic profiling. Second, the patients included in this analysis were representative of those randomly assigned to both targeted treatment arms and the control arm. Third, our detailed investigations provided insights into the potential effect of immune status within the tumor microenvironment on treatment outcomes in mCRC and explored characteristics of alterations acquired following treatment with BRAF inhibitor plus an anti-EGFR monoclonal antibody, with or without a MEK inhibitor, versus chemotherapy plus anti-EGFR therapy. A key limitation of this biomarker analysis is its retrospective nature; the biomarkers predictive of response and resistance to Enco+Cetux±Bini, in BRAF-V600E-mutant CRC need to be further evaluated prospectively. In addition, as acquired mutations were assessed using EoT samples, the exact timing of the occurrence of these mutations was unknown, which might limit interpretation of the association between acquired alterations and OS outcomes.
Results from this prespecified, exploratory biomarker analysis of BEACON CRC support the use of Enco+Cetux±Bini, for the treatment of patients with BRAF-V600E-mutant mCRC and provide important insights into the biology of response and resistance to MAPK-pathway-targeted therapy.
Methods
Ethics statement
The trial was approved by the institutional review board or independent ethics committee at each center (Supplementary Information) and was conducted in accordance with the requirements of the regulatory authorities of each country and with the provisions of the Declaration of Helsinki and the Good Clinical Practice guidelines of the International Council on Harmonisation. All patients provided written informed consent.
Study design and participants
The BEACON CRC trial was a global, multicenter, randomized, open-label phase 3 trial. The study design has been reported previously3. The latest version of the protocol is available as Supplementary Information.
Patients with histologically or cytologically confirmed, mCRC with the BRAF-V600E mutation who had disease progression after one or two previous treatment regimens were enrolled. From May 2017 through January 2019, patients were assigned randomly 1:1:1 to receive encorafenib, binimetinib and cetuximab (Enco+Bini+Cetux); encorafenib and cetuximab (Enco+Cetux); or investigators’ choice of either cetuximab with irinotecan or FOLFIRI (control). Encorafenib was administered at a dose of 300 mg daily; binimetinib at 45 mg twice daily; cetuximab at 400 mg m−2 as an initial dose, then 250 mg m−2 weekly; irinotecan at 180 mg m−2 on days 1 and 15; folinic acid at 180 mg m−2 on days 1 and 15; and fluorouracil at 400 mg m−2 as an initial dose, then 1,200 mg m−2 per day for 2 days, initiated on days 1 and 15. Treatment was administered in 28-day cycles until disease progression, unacceptable toxic effects, withdrawal of consent, initiation of subsequent anticancer therapy or death.
This prespecified, retrospective, exploratory biomarker analysis of the BEACON CRC study was performed in enrolled patients who received one or more dose of study drug (safety set) and had tumor and/or plasma specimens available for WES, WTS or ctDNA analyses, hereafter referred to as the biomarker analysis set. The provision and analysis of tumor samples at EoT were not mandatory.
Endpoints
This exploratory biomarker analysis evaluated genomic and transcriptomic correlates of survival outcomes. Patients were grouped by CRC subtype, individual gene alterations and gene signatures, on the basis of analyses of their blood and tissue samples at baseline and EoT, where available. PFS, assessed by blinded independent central review and investigators, was defined as the time from randomization to the earliest documented disease progression or death due to any cause. OS was defined as the time from randomization to death due to any cause. The association between acquired gene alterations in ctDNA and survival outcomes was analyzed to explore the potential mechanisms of resistance to encorafenib plus cetuximab, with or without binimetinib.
WES and analysis
WES of formalin-fixed paraffin-embedded (FFPE) samples collected at patient screening was performed by Personalis Inc. using their Accuracy and Content Enhanced (ACE) Cancer Exome panel v.3 (sequenced on Illumina NovaSeq)42. Variant calls were generated using BWA, GATK, MuTect, Vardict and Picard and further processed using Personalis proxy normal and custom filters to remove germline variants. Matched normal controls were not available, but variants were filtered for presumptive somatic status based on Personalis’ internal control pipeline. MSI status and TMB were also determined by Personalis. Somatic mutations were reported if above a 5% VAF threshold. Nonsynonymous variants with at least five unique supporting reads in a sample were retained for further analyses. Putative mutations in TET2 were among the most prevalent reported (in 37% of cases); however, as we were unable to verify that these were not germline and/or clonal hematopoiesis of indeterminant potential mutations, TET2 was excluded from further WES analyses.
WTS and analysis
RNA sequencing (RNA-seq) of FFPE tumor samples was conducted at Personalis Inc. DNA and RNA were dual isolated from FFPE samples with the AllPrep DNA/RNA FFPE Kit (Qiagen). Exome capture was performed using Agilent SureSelect Clinical Research Exome v.2 (Agilent Technologies) according to the manufacturer’s recommendations. Additional supplementation with Personalis ACE proprietary target probes was performed to enhance coverage in difficult-to-sequence regions within sets of biomedically and medically relevant genes. Details regarding the Personalis ACE assay design are further described in ref. 43. In brief, manufacturer protocols were modified such that the average library insert length was adjusted to around 250 bp, and the Stranded RNA-seq Kit (Kapa Biosystems) was used for RNA-seq. Sequencing was performed on NovaSeq 6000 sequencers (Illumina) with paired-end, 2 × 150-bp read lengths and using Illumina’s proprietary reversible terminator-based method. For RNA-seq, tumor specimens were sequenced to an average output of 100 million paired-end reads (total of 200 million reads) across the 74.8 Mb NeXT assay genomic footprint, and FASTQ files were generated. The assay provided normalized gene-level expression values as TPM, which were then log-transformed (as log2(TPM + 1)) for downstream analyses. We observed a potential modest batch effect in sequencing associated with sample type, where samples from patients with and without successful resections were separated in an exploratory principal component analysis (Supplementary Fig. 11). We thus confirmed key results on gene expression data batch-corrected for resection status using ComBat in the SVA R package v.3.50.0 (ref. 44).
Molecular subtyping
CMSs were assigned to each sample using the CMSclassifier package v.1.0.0 (ref. 45) and the single-sample predictor method after mapping gene symbols to Entrez IDs using AnnotationDbi (Bioconductor) v.1.62.2 (ref. 46). Not all patients received an unambiguous subtyping call; thus, the nearest subtype prediction was used to estimate a subtype for all cases. BM subtypes were assigned by summing the weighted z-score expression of each of 44 subtyping genes reported in ref. 5, using the gene weights provided therein. Samples with a score of >1.5 were categorized as BM1; those with a score of ≤1.5 were categorized as BM2.
Associations with outcome
Genes in the top quartile of variable expression across samples (by IQR) were included in analyses. Univariable expression (scaled as z-score) associations with OS were evaluated in each arm separately using Cox proportional hazards models. Genes were ranked according to model z-scores for GSEA tests using the fgsea package v.1.28.0 (Bioconductor) against hallmark genesets accessed via the MSigDB Rr package v.6.2.1 (refs. 47,48). To identify additional signals that might be predictive of therapeutic benefit in specific arms, Cox models were fitted for the interaction between gene and arm for analysis sets, including the control and Enco+Cetux arms and the Enco+Cetux and Enco+Bini+Cetux arms separately, with GSEA performed after ranking of genes based on interaction z-scores.
Signature estimation
Signatures of immune infiltration were estimated using several approaches. Briefly, the cytolytic score was estimated as the (geometric) mean of PRF1 and GZMA expression13; the immunological constant of rejection as the (geometric) mean of the expression of genes in the 20-gene signature49; and the CD8 T cell signature from ref. 50 as the (geometric) mean of CD8A and CD8B. xCell v.1.1.0 and MCPcounter v.1.2.0 were run with default parameters16,51. LM22 signature scores were calculated via ssGSEA of LM22 signature genes52.
Genomic ctDNA analysis
Plasma samples were collected for ctDNA analysis before first therapy exposure at C1D1 and at EoT and sequenced using the GuardantOMNI assay (Guardant Health, Inc.)—a targeted next-generation sequencing panel of around 500 genes—to evaluate somatic alterations, including single-nucleotide mutations; insertions and deletions; copy number alteration and gene fusion events. Variants were detected and reported based on molecule support, which corresponded to variants with ≥0.01% VAF.
All types of somatic alterations were called by Guardant Health using its proprietary bioinformatics pipeline53. Aneuploidy, silent mutations and mutations probably arising from clonal hematopoiesis of indeterminate potential were excluded from downstream analysis. For analyses of paired ctDNA data, the resulting alterations were further evaluated for each patient with both C1D1 and EoT samples and characterized at an alteration level as acquired (that is, found in EoT sample only), maintained (found in both C1D1 and EoT samples), or lost (found in C1D1 samples only) for all treatment arms. For gene-level data, if there was more than one alteration for a gene, maintained alteration are shown rather than acquired or lost alterations, and acquired alterations are shown rather than lost alterations. For ctDNA clonality analyses, clonality was a measurement of VAF/maxVAF at EoT on a sample level.
The association of baseline ctDNA BRAF-V600E status with baseline tumor size and baseline ctDNA maxVAF was examined. Baseline tumor size was defined as the sum of target lesions assessed before randomization; the latest measurement was used if a lesion was assessed several times.
Mutational signature analysis
For treatment group comparisons, we pooled the somatic protein-altering variants detected in ctDNA samples in a group-wise manner and then applied SigProfilerExtractor v.1.1 (ref. 54) to quantify the mutational signatures active in each group, based on a set of predefined SBS mutational signatures (Catalogue of Somatic Mutations in Cancer Mutational Signatures v.3.3, June 2022)17 obtained from the Wellcome Sanger Institute. Baseline alterations were defined as C1D1 VAF > 1%, and acquired alterations were defined as C1D1 VAF = 0 and EoT VAF > 0.
As with survival analyses, identification of gene expression correlates of mutation acquisition was performed using variance-filtered and z-scaled gene expression. GSEA analyses were based on genes ordered by t-statistics from univariable linear models.
Statistical analysis
Calculation of the overall sample size required for the primary endpoints, statistical testing schemes and preplanned interim analysis was described previously3. All data reported here are from prespecified exploratory biomarker analyses performed using data (cutoff 15 August 2019 (ref. 4)) from patients who received at least one dose of study drug; had at least one post-treatment assessment, which may include death; and had at least one valid biomarker measurement at the timepoint(s) of interest (baseline versus EoT). For analyses involving treatment, patients were analyzed according to the actual treatment received.
For biomarkers without a predefined cut point, subgroup analysis was performed based on a median split to maintain balance between subgroup size, and testing of treatment-by-biomarker interactions was performed using continuous measurements. Identification of optimal biomarker thresholds was not the objective of the analyses. Exploratory biomarker analyses (for example, subgroup analysis) are subject to both type I error (false-positive) due to multiple comparisons and type II error (false-negative) due to lack of statistical power (for example, test of interaction in subgroup analysis)55. The objective of the biomarker analysis is hypothesis generating; therefore, to reduce potential type II error56,57, the results of statistical analyses are presented without correction for multiple comparisons. To assess the effect of potential confounders, results based on models with adjustment of key baseline covariates (Eastern Cooperative Oncology Group performance status, C-reactive protein, number of organs, tumor status, cetuximab source and previous use of irinotecan at randomization) are provided.
Kaplan–Meier estimates of OS and PFS were presented by treatment arm and biomarker subgroup. The median PFS and OS (in months) were summarized, along with 95% CIs. Cox proportional hazards models were used to calculate HRs and 95% CIs in each treatment arm or biomarker subgroup, either with or without adjustment of baseline measurements of known prognostic factors. The interaction term was evaluated using the Cox proportional hazards model, including treatment, biomarker and the interaction term, with the P value determined by the two-sided Wald test. The Fisher exact test was used to compare proportions between independent samples with small sample size. The McNemar’s test was used to compare proportions between paired samples (for example baseline versus EoT). Depending on the data distribution, either the Wilcoxon rank-sum test or one-way analysis of variance was used to compare biomarker levels between independent samples. The Poisson mixture model was used to identify patient subgroups that acquired mutations at different rates. For visual display of patient subgroups, a rate was calculated using the number of acquired mutations and time on treatment for each patient, which allowed comparison of the number of mutations emerging that was adjusted for time on treatment. The negative binomial model was used to compare the rates of alteration acquisition across treatment arms. For all analyses involving rate of acquired mutations, a constant rate of acquisition was assumed. Statistical analyses were done using SAS v.9.4 or R v.4.0 and above.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The analyses in this paper were based on a data cutoff of 15 August 2019. Upon request, and subject to review, Pfizer will provide the data that support the findings of this study. Subject to certain criteria, conditions and exceptions, Pfizer may also provide access to the related individual deidentified participant data. Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. See https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information. Data may be requested from Pfizer trials 24 months after study completion. The BEACON CRC study was completed in November 2022. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
References
De Roock, W. et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 11, 753–762 (2010).
Tabernero, J., Ros, J. & Élez, E. The evolving treatment landscape in BRAF-V600E-mutated metastatic colorectal cancer. Am. Soc. Clin. Oncol. Educ. Book 42, 1–10 (2022).
Kopetz, S. et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N. Engl. J. Med. 381, 1632–1643 (2019).
Tabernero, J. et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J. Clin. Oncol. 39, 273–284 (2021).
Barras, D. et al. BRAF V600E mutant colorectal cancer subtypes based on gene expression. Clin. Cancer Res. 23, 104–115 (2017).
Guinney, J. et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 21, 1350–1356 (2015).
Ten Hoorn, S., de Back, T. R., Sommeijer, D. W. & Vermeulen, L. Clinical value of consensus molecular subtypes in colorectal cancer: a systematic review and meta-analysis. J. Natl Cancer Inst. 114, 503–516 (2022).
Xu, T. et al. Molecular mechanisms underlying the resistance of BRAF V600E-mutant metastatic colorectal cancer to EGFR/BRAF inhibitors. Ther. Adv. Med. Oncol. 14, 17588359221105022 (2022).
Ahronian, L. G. et al. Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discov. 5, 358–367 (2015).
Tan, L. et al. A phase Ib/II trial of combined BRAF and EGFR inhibition in BRAF V600E positive metastatic colorectal cancer and other cancers: the EVICT (Erlotinib and Vemurafenib In Combination Trial) study. Clin. Cancer Res. 29, 1017–1030 (2023).
Aandahl, E. M. et al. CD7 is a differentiation marker that identifies multiple CD8 T cell effector subsets. J. Immunol. 170, 2349–2355 (2003).
Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. & Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004).
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015).
Szabo, S. J. et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000).
Roelands, J. et al. An integrated tumor, immune and microbiome atlas of colon cancer. Nat. Med. 29, 1273–1286 (2023).
Aran, D., Hu, Z. & Butte, A. J. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220 (2017).
Catalogue of Somatic Mutations in Cancer (COSMIC) mutational signatures, v.3.3. Wellcome Sanger Institute https://cancer.sanger.ac.uk/signatures/# (2022).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Secrier, M. et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 48, 1131–1141 (2016).
Cen, S. et al. BRAF mutation as a potential therapeutic target for checkpoint inhibitors: a comprehensive analysis of immune microenvironment in BRAF mutated colon cancer. Front. Cell Dev. Biol. 9, 705060 (2021).
Clarke, C. N. & Kopetz, E. S. BRAF mutant colorectal cancer as a distinct subset of colorectal cancer: clinical characteristics, clinical behavior, and response to targeted therapies. J. Gastrointest. Oncol. 6, 660–667 (2015).
Sahin, I. H., Ciombor, K. K., Diaz, L. A., Yu, J. & Kim, R. Immunotherapy for microsatellite stable colorectal cancers: challenges and novel therapeutic avenues. Am. Soc. Clin. Oncol. Educ. Book 42, 1–12 (2022).
Middleton, G. et al. BRAF-mutant transcriptional subtypes predict outcome of combined BRAF, MEK, and EGFR blockade with dabrafenib, trametinib, and panitumumab in patients with colorectal cancer. Clin. Cancer Res. 26, 2466–2476 (2020).
Callahan, M. K. et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol. Res 2, 70–79 (2014).
Liu, L. et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 21, 1639–1651 (2015).
Verma, V. et al. MEK inhibition reprograms CD8(+) T lymphocytes into memory stem cells with potent antitumor effects. Nat. Immunol. 22, 53–66 (2021).
Elez, E. et al. SEAMARK: phase II study of first-line encorafenib and cetuximab plus pembrolizumab for MSI-H/dMMR BRAF V600E-mutant mCRC. Future Oncol. 20, 653–663 (2024).
Morris, V. K. et al. Randomized phase II trial of encorafenib and cetuximab with or without nivolumab for patients with previously treated, microsatellite stable, BRAFV600E metastatic and/or unresectable colorectal cancer: SWOG S2107. J. Clin. Oncol. 41, TPS265–TPS265 (2023).
Morris, V. K. et al. Phase I/II trial of encorafenib, cetuximab, and nivolumab in patients with microsatellite stable, BRAFV600E metastatic colorectal cancer. J. Clin. Oncol. 40, 12–12 (2022).
Elez, E. et al. RNF43 mutations predict response to anti-BRAF/EGFR combinatory therapies in BRAF(V600E) metastatic colorectal cancer. Nat. Med. 28, 2162–2170 (2022).
Yaeger, R. et al. Mechanisms of acquired resistance to BRAF V600E inhibition in colon cancers converge on RAF dimerization and are sensitive to its inhibition. Cancer Res. 77, 6513–6523 (2017).
Lu, H. et al. SHP2 inhibition overcomes RTK-mediated pathway reactivation in KRAS-mutant tumors treated with MEK inhibitors. Mol. Cancer Ther. 18, 1323–1334 (2019).
Ahmed, T. A. et al. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Rep. 26, 65–78.e5 (2019).
Kun, E., Tsang, Y. T. M., Ng, C. W., Gershenson, D. M. & Wong, K. K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat. Rev. 92, 102137 (2021).
Zhao, B. et al. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 8, 3980–4000 (2017).
Russo, M. et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 366, 1473–1480 (2019).
Parseghian, C. M. et al. Resistance mechanisms to anti-epidermal growth factor receptor therapy in RAS/RAF wild-type colorectal cancer vary by regimen and line of therapy. J. Clin. Oncol. 41, 460–471 (2023).
Christensen, S. et al. 5-Fluorouracil treatment induces characteristic T>G mutations in human cancer. Nat. Commun. 10, 4571 (2019).
Woolston, A. et al. Mutational signatures impact the evolution of anti-EGFR antibody resistance in colorectal cancer. Nat. Ecol. Evol. 5, 1024–1032 (2021).
Wang, L. et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell 173, 1413–1425.e14 (2018).
Donehower, L. A. et al. Integrated analysis of TP53 gene and pathway alterations in The Cancer Genome Atlas. Cell Rep. 28, 1370–1384.e5 (2019).
Powles, T. et al. Avelumab maintenance in advanced urothelial carcinoma: biomarker analysis of the phase 3 JAVELIN Bladder 100 trial. Nat. Med. 27, 2200–2211 (2021).
Patwardhan, A. et al. Achieving high-sensitivity for clinical applications using augmented exome sequencing. Genome Med. 7, 71 (2015).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Eide, P. W., Bruun, J., Lothe, R. A. & Sveen, A. CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci. Rep. 7, 16618 (2017).
Pagès, H., Carlson, M., Falcon S. & Li, N. AnnotationDbi: manipulation of SQLite-based annotations in Bioconductor. R package version 1.62.2. Bioconductor https://bioconductor.org/packages/AnnotationDbi (2023).
Korotkevich, G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv https://doi.org/10.1101/060012 (2021).
Dolgalev, I. MSigDB Gene sets for multiple organisms in a tidy data format, v.6.2.1. GitHub https://github.com/igordot/msigdbr (2018).
Wang, E., Worschech, A. & Marincola, F. M. The immunologic constant of rejection. Trends Immunol. 29, 256–262 (2008).
Danaher, P. et al. Gene expression markers of tumor infiltrating leukocytes. J. Immunother. Cancer 5, 18 (2017).
Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218 (2016).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Odegaard, J. I. et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin. Cancer Res. 24, 3539–3549 (2018).
Islam, S. M. A. et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genom. 2, 100179 (2022).
Guideline on the Investigation of Subgroups in Confirmatory Clinical Trials (European Medicines Agency, 2019); https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-subgroups-confirmatory-clinical-trials_en.pdf
Gao, Y. et al. Allele-specific mechanisms of activation of MEK1 mutants determine their properties. Cancer Discov. 8, 648–661 (2018).
Hanrahan, A. J. et al. Leveraging systematic functional analysis to benchmark an in silico framework distinguishes driver from passenger MEK mutants in cancer. Cancer Res. 80, 4233–4243 (2020).
Acknowledgements
We thank the participating patients and their families, as well as the staff at the participating sites. Additionally, we acknowledge E. Elez from the Vall d’Hebron Institute of Oncology and Vall d’Hebron University Hospital, for her contributions to patient recruitment and trial conduct. The BEACON CRC trial was sponsored by Pfizer and was conducted with support from Merck KGaA, Darmstadt, Germany (for sites outside of North America), ONO Pharmaceutical and Pierre Fabre. This study was also supported by a Cancer Center Core Grant P30 CA008748 to Memorial Sloan Kettering Cancer Center. Medical writing support was provided by Q.Y. Chong and A. Erden of Nucleus Global, an Inizio Company, and was funded by Pfizer.
Author information
Authors and Affiliations
Contributions
S.K., D.A.M., R.Y., F.C., J.D., H.S.W., T.Y. and J.T. conceptualized the study. S.K., F.C., J.D., E.V.C., H.S.W., T.Y., R.Y. and J.T. undertook the data acquisition. S.K., D.A.M., J.P., P.H., T.X., R.Y. and J.T. carried out the data analysis and interpretation. J.P. performed the statistical analysis. S.K., D.A.M., J.P. and P.H. drafted the manuscript. All authors reviewed the manuscript and gave approval for submission. All authors were accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
S.K.: Stock and Other Ownership Interests: Iylon, Lutris, MolecularMatch, Navire. Consulting or Advisory Role: AbbVie, Amal Therapeutics, AstraZeneca/MedImmune, Bayer Health, Bicara Therapeutics, Boehringer Ingelheim, Boston Biomedical, Carina Biotech, Daiichi Sankyo, EMD Serono, Endeavor BioMedicines, Flame Biosciences, Genentech, Gilead Sciences, GSK, HalioDx, Holy Stone Healthcare, Inivata, Ipsen, Iylon, Jacobio, Jazz Pharmaceuticals, Lilly, Lutris, Merck, Mirati Therapeutics, Natera, Novartis, Numab, Pfizer, Pierre Fabre, Redx Pharma, Repare Therapeutics, Servier, Xilis. Research Funding: Amgen (Inst), Array BioPharma (Inst), Biocartis (Inst), Daiichi Sankyo (Inst), EMD Serono (Inst), Genentech/Roche (Inst), Guardant Health (Inst), Lilly (Inst), MedImmune (Inst), Novartis (Inst), Sanofi (Inst). D.A.M.: Employment: Formerly Pfizer. Stock and Other Ownership Interests: Pfizer. J.P.: Employment: Formerly Pfizer. Stock and Other Ownership Interests: Pfizer. F.C.: Consulting or Advisory Role: Amgen, Bayer, Merck KGaA, Pfizer, Roche/Genentech. Research Funding: Amgen (Inst), Bayer (Inst), Bristol Myers Squibb (Inst), Ipsen (Inst), Merck KGaA (Inst), MSD (Inst), Roche/Genentech (Inst), Servier (Inst), Symphogen (Inst). J.D.: Consulting or Advisory Role: Amgen (Inst), Bayer, BeiGene, Daiichi Sankyo, Eisai, GSK, Merck KGaA, Pierre Fabre. Research Funding: AstraZeneca/MedImmune (Inst), BeiGene (Inst), Bionomics (Inst), Bristol Myers Squibb (Inst), GSK (Inst), Lilly (Inst), Novartis (Inst), Roche (Inst). E.V.C.: Consulting or Advisory Role: AbbVie, Agenus, ALX, Amgen, Arcus Biosciences, Astellas Pharma, AstraZeneca, Bayer, BeiGene, BioNTech, Boehringer Ingelheim, Bristol Myers Squibb, Daiichi Sankyo, Debiopharm, ElmediX, Eisai, GSK, Hookipa Biotech, Incyte, Ipsen, Lilly, Merck Sharp & Dohme, Merck KGaA, Mirati, Novartis, Nordic, Pierre Fabre, Pfizer, Roche, Seagen, Servier, Simcere, Takeda, Taiho Pharmaceutical, Terumo. H.S.W.: Consulting or Advisory Role: Amgen, Bayer, Bristol Myers Squibb (Celgene), Boehringer Ingelheim (DMC), BTG, EXACT Therapeutics, Erytech Pharma, Incyte, Merck KGaA, Oaktree Life Sciences, OncoSil, Pfizer, Pierre Fabre, Roche/Genentech, Seagen, Servier, Shire, Sirtex Medical, Takeda (Hutchinson Med), Zymeworks. Research Funding: Merck KGaA (Inst), MSD (Inst), Pfizer (Inst), Sirtex Medical (Inst). T.Y.: Honoraria: Bayer Yakuhin, Chugai Pharma, Merck KGaA, MSD, Ono Pharmaceutical, Sumitomo Corp., Takeda. Research Funding: Amgen (Inst), Boehringer Ingelheim (Inst), Chugai Pharma (Inst), Daiichi Sankyo Co., Ltd. (Inst), Eisai, FALCO Biosystems, Genomedia (Inst), Molecular Health, MSD (Inst), Nippon Boehringer Ingelheim, Ono Pharmaceutical (Inst), Pfizer (Inst), Roche Diagnostics, Sanofi (Inst), Sumitomo Dainippon (Inst), Sysmex (Inst), Taiho Pharmaceutical (Inst). H.S.: Employment: Pfizer. Stock and Other Ownership Interests: Pfizer. X.Z.: Employment: Pfizer. Stock and Other Ownership Interests: Pfizer. Patents, Royalties, Other Intellectual Property: Johns Hopkins University. P.H.: Employment: Pfizer. Stock and Other Ownership Interests: Pfizer. T.X.: Employment: Pfizer. Stock and Other Ownership Interests: Pfizer. R.Y.: Consulting or Advisory Role: Array BioPharma/Pfizer, Mirati Therapeutics, Zai Lab, Amgen. Research Funding: Array BioPharma (Inst), Boehringer Ingelheim (Inst), Mirati Therapeutics (Inst), Pfizer (Inst), Daiichi Sankyo (Inst). J.T.: Consulting or Advisory Role: Array BioPharma, AstraZeneca, Bayer, Boehringer Ingelheim, Chugai Pharma, Daiichi Sankyo, F. Hoffmann-La Roche, Genentech, HalioDx, Hutchison MediPharma, Ikena Oncology, Inspirna Inc., IQVIA, Lilly, Menarini, Merck Serono, Merus, Mirati Therapeutics, MSD, NeoPhore, Novartis, Ona Therapeutics, Orion Biotechnology, Peptomyc, Pfizer, Pierre Fabre, Samsung Bioepis, Sanofi, Scandion Oncology, Scorpion Therapeutics, Seagen Inc., Servier, Taiho Pharmaceutical, Tessa Therapeutics, TheraMyc. Other Relationship: Amgen, Array Biopharma, BeiGene, Boehringer Ingelheim, BMS, Cancer Research UK, Celgene, Debiopharm, F. Hoffman-La Roche, Fundación Científica de la Asociación Española Contra el Cáncer, Genentech, HalioDX, Hutchinson Medipharma, Imedex, Janssen-Cilag, MedImmune, Medscape, Menarini, Merck Health KGaA, MJH Life Sciences, MSD, Merus, Mirati, Novartis, Oniria Therapeutics, PeerView Institute for Medical Education, Pfizer, PharmaMar, Physicans’ Education Resource, Sanofi-Aventis, Servier, Taiho Pharmaceutical.
Peer review
Peer review information
Nature Medicine thanks Pashtoon Kasi, Noelia Tarazona and Jeanne Tie for their contribution to the peer review of this work. Primary Handling Editor: Ulrike Harjes, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Genomic ctDNA profiling of baseline plasma samples.
*Multiple mutations were defined as genes with ≥ 2 alterations of different categories (for example missense mutation + amplification). ctDNA, circulating tumor DNA; MSI-H, microsatellite instability-high; mut; mutation; TMB, tumor mutational burden.
Extended Data Fig. 2 Association between BRAF V600E VAF and ctDNA maxVAF at baseline.
a, Percentages of baseline BRAF V600E positivity across ctDNA maxVAF subgroups. The low maxVAF subgroup included patients with ctDNA maxVAF below the first quartile (2.01%); the high maxVAF subgroup included patients with ctDNA maxVAF in the third quartile (33.77%) and above; the medium maxVAF subgroup included the remaining patients. P-value was calculated based on logistic regression analysis (two-sided Wald test). b, Pearson correlation and the associated two-sided P value of the association between BRAF V600E VAF and ctDNA maxVAF at baseline. Only patients with BRAF V600E ctDNA detected were included in the analysis involving VAF. Silent mutations, clonal hematopoiesis of indeterminate potential, and alterations from genes with copy number alterations were excluded. ctDNA, circulating tumor DNA; max, maximum; VAF, variant allele frequency.
Extended Data Fig. 3 Kaplan–Meier plots of OS by ctDNA maxVAF status in each treatment arm.
ctDNA maxVAF was categorized as ≤ or > the median of 11.005. Cox proportional hazard models with or without adjustment of baseline covariates (ECOG performance status, C-reactive protein, number of organs, tumor status, cetuximab source, and prior use of irinotecan at randomization) were used. A two-sided Wald test was used for P value of interaction terms, where ctDNA maxVAF was regarded as a continuous variable. Bini, binimetinib; Cetux, cetuximab; CI, confidence interval; ctDNA, circulating tumor DNA; ECOG, Eastern Cooperative Oncology Group; Enco, encorafenib; HR, hazard ratio; max, maximum; OS, overall survival; VAF, variant allele frequency.
Extended Data Fig. 4 Association between OS and CRC subtypes.
Forest plots of HRs for OS with Enco+Cetux (top panel) or Enco+Bini+Cetux (bottom panel) vs control in subgroups defined by BM subtypes and CMS. The diamonds show HRs and the lines show 95% CI. P values for interaction (two-sided Wald test) were based on the Cox model without adjustment for baseline covariates. *The CMS2 subgroup was excluded due to small sample size.
Extended Data Fig. 5 Immune gene associations with OS benefit in each treatment arm.
a, Associations between OS and immune gene expression (scaled) in each treatment arm were determined by Cox proportional hazards models (two-sided Wald test). Genes in the top quartile of variance in expression (based on IQR and measured in log2[TPM]) were tested. Genes showing an interaction (unadjusted P value < 0.001) for Enco+Cetux vs Control are shown with blue circles and for Enco+Cetux vs Enco+Bini+Cetux (top panel) are shown as red dots across panels. b, Enrichment plots from GSEA against the hallmark IFN-γ gene signature in each treatment arm.
Extended Data Fig. 6 Correlation between tumor immune status and cytolytic score.
a, Correlation between cytolytic score and ssGSEA scores for the IFN-γ response (top panel), allograft rejection (middle panel) and inflammatory response (bottom panel) hallmark signatures; r denotes Pearson correlation. Data points are color coded based on CMS classification. b, Pearson correlation between cytolytic score and sample scores for multiple metrics of CD8+ T-cell infiltration and gene signatures of immune infiltration. CMS, consensus molecular subtypes; CYT, cytolytic; ssGSEA, single-sample gene set enrichment analysis; ICR, immune constant of rejection.
Extended Data Fig. 7 Kaplan–Meier plots of PFS by cytolytic score between and within each treatment arm.
a, Kaplan–Meier plots of PFS by cytolytic score between treatment arms (inter-arm comparison). b, Kaplan–Meier plots of PFS by cytolytic score within each treatment arm (intra-arm comparison). Tumors were categorized as cytolytic high or low, defined as score > and ≤ the median value of 3.11, respectively. Cox proportional hazard models with or without adjustment of baseline covariates (ECOG performance status, C-reactive protein, number of organs, tumor status, cetuximab source, and prior use of irinotecan at randomization) were used. A two-sided Wald test was used for P value of interaction terms, where cytolytic score was regarded as a continuous variable. Bini, binimetinib; Cetux, cetuximab; CYT, cytolytic; ECOG, Eastern Cooperative Oncology Group; Enco, encorafenib; HR, hazard ratio; PFS, progression-free survival.
Extended Data Fig. 8 Abundance of immune cell types and fibroblasts (estimated using xCell) by CYT score and CRC subtype.
Bini, binimetinib; BM, BRAF mutant; Cetux, cetuximab; CMS, consensus molecular subtypes; CRC, colorectal cancer; CYT, cytolytic; Enco, encorafenib; HR, hazard ratio; IQR, interquartile range; NES, normalized enrichment score; NK, natural killer; OS, overall survival; TPM, transcripts per million.
Extended Data Fig. 9 Kaplan-Meier plots of OS by presence of top acquired alterations (top panel) or MET amplification (bottom panel) following treatment with Enco+Cetux±Bini.
Cox proportional hazard models with and without adjustment of baseline covariates (ECOG performance status, C-reactive protein, number of organs, tumor status, cetuximab source, and prior use of irinotecan at randomization) were used. Bini, binimetinib; Cetux, cetuximab; CI, confidence interval; Enco, encorafenib; HR hazard ratio; OS, overall survival.
Extended Data Fig. 10 Clonality of acquired alterations determined by ctDNA genomic profiling analysis.
Violin plots showing clonality (EoT VAF/EoT max VAF) of all acquired putative resistance alterations (left); acquired KRAS, NRAS, and MAP2K1 mutations (middle); and all maintained putative resistance alterations (right). *P value for comparison between all acquired vs maintained putative resistance alterations was based on the Wilcoxon rank-sum test (two-sided). The resulting p-value from the analysis is < 0.0001. †Only mutations were evaluated in the KRAS, NRAS, and MAP2K1 group. ‡Silent mutations, clonal hematopoiesis of indeterminate potential, and alterations from genes that also had copy number alterations were excluded from the analysis.
Supplementary information
Supplementary Information
List of IRBs and IECs, Protocol, and Supplementary Tables 1–8 and Figs. 1–11.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kopetz, S., Murphy, D.A., Pu, J. et al. Molecular profiling of BRAF-V600E-mutant metastatic colorectal cancer in the phase 3 BEACON CRC trial. Nat Med (2024). https://doi.org/10.1038/s41591-024-03235-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-024-03235-9
- Springer Nature America, Inc.