

Abstract
Trastuzumab deruxtecan (T-DXd) demonstrated significantly improved efficacy over trastuzumab emtansine (T-DM1) in DESTINY-Breast03 (median follow-up, 28 months). We report updated efficacy and safety analyses, including secondary and exploratory efficacy endpoints (median follow-up, 41 months) of DESTINY-Breast03. Patients with advanced HER2-positive metastatic breast cancer previously treated with taxane and trastuzumab were randomized to T-DXd (5.4 mg per kg (261 patients)) or T-DM1 (3.6 mg per kg (263 patients)). The primary endpoint was progression-free survival (PFS) by blinded independent central review and was previously reported. The key secondary endpoint was overall survival (OS). Other secondary endpoints included objective response rate, duration of response and PFS (all by investigator assessment) and safety. At data cutoff, 20 November 2023, median PFS by investigator assessment was 29.0 versus 7.2 months (hazard ratio (HR), 0.30; 95% confidence interval (CI), 0.24–0.38), the 36-month PFS rate was 45.7% versus 12.4% and median OS was 52.6 versus 42.7 months (HR, 0.73; 95% CI, 0.56–0.94) with T-DXd versus T-DM1, respectively. Treatment-emergent adverse events were consistent with the previous analyses. No new instances of grade ≥3 interstitial lung disease or pneumonitis occurred (all grade rate, 16.7% (T-DXd) versus 3.4% (T-DM1)). With longer follow-up, T-DXd continued to demonstrate superior efficacy over T-DM1 with a manageable safety profile. ClinicalTrials.gov registration: NCT03529110.
Similar content being viewed by others

Main
Human epidermal growth factor receptor 2 (HER2)-positive breast cancer is characterized by amplification of the HER2 (ERBB2) gene and/or overexpression of the HER2 protein, which stimulates cell proliferation, survival, differentiation, angiogenesis and invasion1,2,3,4,5. High levels of HER2 expression have been reported in approximately 20% of all breast cancer tumors, resulting in a more aggressive subtype that metastasizes at a faster rate than breast tumors that do not overexpress HER2 (refs. 1,2,5,6,7,8). The discovery of HER2 alterations led to the development of treatments that specifically target HER2, resulting in improved prognosis for patients with this subtype of breast cancer1,2,7,9,10,11,12.
T-DXd is approved in several regions across the globe, including the United States, the European Union and Japan, for patients with HER2-positive metastatic breast cancer after disease progression on taxane and trastuzumab or in patients who have developed disease recurrence during or within 6 months of completing neoadjuvant and/or adjuvant therapy; T-DXd is now a guideline-recommended treatment11,13,14,15,16. The approval of T-DXd in this setting was based on the results of DESTINY-Breast03 (NCT03529110), a multicenter phase 3 trial conducted to investigate the efficacy and safety of T-DXd versus T-DM1 (ref. 13). Before approval of T-DXd, T-DM1 was primarily used in this setting.
T-DXd and T-DM1 are both antibody–drug conjugates composed of humanized monoclonal antibodies targeting HER2, linked to a cytotoxic payload17,18,19. T-DM1 incorporates a microtubule-disrupting agent, which is tethered by a durable thioether bond, whereas T-DXd employs a topoisomerase I inhibitor connected through a tetrapeptide-based cleavable linker, which enables greater specificity in targeting cancer cells, thereby diminishing unintended toxicity17,19,20. T-DXd has a high, homogeneous drug-to-antibody ratio of approximately 8, while T-DM1 has a drug-to-antibody ratio of approximately 3.5 (refs. 17,19,21).
The primary endpoint of DESTINY-Breast03 was PFS, as determined by blinded independent central review (BICR), and the key secondary endpoint was OS. In the primary (first interim) analysis (data cutoff, 21 May 2021) of DESTINY-Breast03, the primary endpoint was met, with median PFS not reached for T-DXd compared with 6.8 months for T-DM1 (HR, 0.28; 95% CI, 0.22–0.37; P < 0.001)22. In the second OS interim analysis (data cutoff, 25 July 2022), T-DXd demonstrated a statistically significant and clinically meaningful OS improvement versus T-DM1, with a reduction in the risk for death of approximately 36% (HR, 0.64; 95% CI, 0.47–0.87; P = 0.0037)23. However, median OS was not reached in either treatment group at the primary analysis or the second OS interim analysis22,23.
After the demonstrated statistically significant improvement of PFS with T-DXd versus T-DM1 in the first interim analysis22 and updated analysis of PFS at the time of the second OS interim analysis23, further assessment of tumor response by BICR was discontinued. We report on an exploratory analysis of DESTINY-Breast03 (data cutoff, 20 November 2023), with updated efficacy, including median OS, and safety data with longer follow-up.
Results
Patients
From 20 July 2018 to 23 June 2020, 699 patients were screened for eligibility to enroll in the trial. Five hundred twenty-four patients with HER2-positive, unresectable or metastatic breast cancer were enrolled and randomly assigned 1:1 to receive either T-DXd at 5.4 mg per kg (n = 261) or T-DM1 at 3.6 mg per kg (n = 263) intravenously once every 3 weeks (Fig. 1). Demographic and baseline characteristics were similar between the two treatment groups (Table 1). The median age was 54.3 years (range, 27.9–83.1 years) in the T-DXd group and 54.2 years (range, 20.2–83.0 years) in the T-DM1 group. An Eastern Cooperative Oncology Group performance score (ECOG PS) of 0 at baseline was reported for 154 patients (59.0%) in the T-DXd group and for 175 patients (66.5%) in the T-DM1 group, whereas 106 patients (40.6%) and 87 patients (33.1%), respectively, had an ECOG PS of 1. In both groups, the majority of patients had a HER2 immunohistochemistry (IHC) score of 3+ (T-DXd, 234 patients (89.7%); T-DM1, 232 patients (88.2%)). Baseline central nervous system (CNS) metastases were reported in 43 patients (16.5%) in the T-DXd group and in 39 patients (14.8%) in the T-DM1 group.

Efficacy analysis was conducted in the full analysis set (all patients who were randomly assigned to a treatment group), and safety analysis was conducted in the safety analysis set (all patients who were randomly assigned and received at least one dose of T-DXd or T-DM1).
In both the T-DXd and T-DM1 groups, patients had received a median of two prior lines of therapy in the metastatic setting. As of 20 November 2023, 50 patients (19.5%) in the T-DXd group and ten patients (3.8%) in the T-DM1 group remained on treatment (Fig. 1). The most common reasons patients discontinued study treatment were progressive disease or clinical progression (T-DXd, 107 patients (41.6%) and five patients (1.9%); T-DM1, 183 patients (70.1%) and 16 patients (6.1%)), adverse events (T-DXd, 61 patients (23.7%); T-DM1, 24 patients (9.2%)) and withdrawal by patient (T-DXd, 22 patients (8.6%); T-DM1, 14 patients (5.4%)). Median duration of follow-up was 43.0 months (range, 0.0–62.9 months) for T-DXd and 35.4 months (range, 0.0–60.9 months) for T-DM1.
Efficacy
The confirmed objective response rate (ORR) by investigator assessment was 78.9% (206 patients; 95% CI, 73.5–83.7%) with T-DXd and 36.9% (97 patients; 95% CI, 31.0–43.0%) with T-DM1 (Table 2). In the T-DXd and T-DM1 groups, respectively, 33 patients (12.6%) and 11 patients (4.2%) experienced a complete response and 173 patients (66.3%) and 86 patients (32.7%) experienced a partial response. The median duration of response (DoR) by investigator assessment was 30.5 months (95% CI, 23.0 months to not estimable (NE)) with T-DXd and 17.0 months (95% CI, 14.1–23.7 months) with T-DM1 (Extended Data Fig. 1).
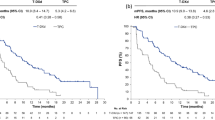
Median PFS by investigator assessment was 29.0 months (95% CI, 23.7–40.0 months) with T-DXd and 7.2 months (95% CI, 6.8–8.3 months) with T-DM1 (HR, 0.30; 95% CI, 0.24–0.38) (Fig. 2a). The PFS rate at 36 months was 45.7% (95% CI, 38.9–52.2%) with T-DXd and 12.4% (95% CI, 8.1–17.7%) with T-DM1.

a, PFS. b, PFS2. c, OS. Crosses indicate where data were censored; numbers of patients censored are not stated.
In the T-DXd and T-DM1 groups, of the patients who discontinued treatment, 144 patients (69.6%) and 198 patients (78.9%), respectively, received anticancer systemic therapy after the trial (Extended Data Table 1). In the T-DXd group, 75 patients (52.1%) received T-DM1 and 12 patients (8.3%) received T-DXd; in the T-DM1 group, 64 patients (32.3%) received T-DXd and 26 patients (13.1%) received T-DM1 after the trial. Median PFS2 (PFS from the time of randomization to progression on the next line of therapy or death) by investigator assessment was 45.2 months (95% CI, 39.3 months to NE) with T-DXd and 23.1 months (95% CI, 17.8–29.7 months) with T-DM1 (HR, 0.53; 95% CI, 0.41–0.68) (Fig. 2b). The PFS2 rate at 36 months was 62.1% (95% CI, 55.5–68.0%) with T-DXd and 40.3% (95% CI, 33.3–47.2%) with T-DM1.
Two hundred thirty-six OS events were observed up to the data cutoff of 20 November 2023: 110 (42.1%) in the T-DXd group and 126 (47.9%) in the T-DM1 group. Median OS was 52.6 months (95% CI, 48.7 months to NE) with T-DXd and 42.7 months (95% CI, 35.4 months to NE) with T-DM1; the risk of death was reduced by 27% (HR, 0.73; 95% CI, 0.56–0.94) (Fig. 2c and Table 2). The OS rate at 24 months was 77.5% (95% CI, 71.8–82.2%) with T-DXd versus 70.1% (95% CI, 64.0–75.4%) with T-DM1, and the OS rate at 36 months was 67.6% (95% CI, 61.3–73.0%) versus 55.7% (95% CI, 49.2–61.7%), respectively.
In the sensitivity analysis (Extended Data Fig. 2), using a median rank-preserving structural failure time model (RPSFTM), the adjusted median OS for the T-DM1 group was 39.8 months (95% CI, 32.4 months to NE). The HR for OS between the T-DXd group and the RPSFTM-adjusted T-DM1 group was 0.66 (95% CI, 0.51–0.87%).
Safety
Median treatment duration was 18.2 months (range, 0.7–56.6 months) with T-DXd and 6.9 months (range, 0.7–55.2 months) with T-DM1 at the data cutoff. Similar rates of any-grade treatment-emergent adverse events (TEAEs) were observed in both treatment groups (Table 3; 99.6% (256 patients) with T-DXd versus 95.4% (249 patients) with T-DM1). Grade ≥3 TEAEs occurred in 149 T-DXd–treated patients (58.0%) and 136 T-DM1–treated patients (52.1%), of which 48.6% and 42.5%, respectively, were drug-related. In the T-DXd and T-DM1 groups, 58 patients (22.6%) and 19 patients (7.3%), respectively, discontinued treatment due to drug-related TEAEs. In the T-DXd group, the most common drug-related TEAEs associated with discontinuation were pneumonitis (6.6% (17 of 257)) and interstitial lung disease (ILD) (5.4% (14 of 257)). In the T-DM1 group, the most common drug-related TEAEs associated with discontinuation were pneumonitis (1.5% (four of 261)) and platelet count decrease (1.5% (four of 261)) (Extended Data Table 2). Drug-related TEAEs associated with dose reduction occurred in 72 patients (28.0%) with T-DXd and in 40 patients (15.3%) with T-DM1, and drug-related TEAEs leading to drug interruption occurred in 113 patients (44.0%) and 48 patients (18.4%), respectively (Table 3).
Exposure-adjusted incidence rates (EAIRs) were measured to account for differences in treatment duration between the T-DXd and T-DM1 groups. EAIRs for any-grade TEAEs per patient-year were 0.53 with T-DXd and 1.10 with T-DM1 (Extended Data Table 3). EAIRs for grade ≥3 TEAEs were 0.31 and 0.60, and EAIRs for serious TEAEs were 0.15 and 0.26 with T-DXd and T-DM1, respectively. The most common TEAEs (reported in ≥20% of patients) were similar between the current and previous data cutoff analyses (Extended Data Table 4)23.
Adjudicated drug-related ILD and/or pneumonitis occurred in 43 patients (16.7%) in the T-DXd group and in nine patients (3.4%) in the T-DM1 group during the entire study period through the 20 November 2023 data cutoff (Table 4). In the T-DXd group, 11 patients (4.3%) had a grade 1 event, 30 patients (11.7%) had a grade 2 event and two patients (0.8%) had a grade 3 event. Since the previous data cutoff (25 July 2022), four new adjudicated drug-related ILD events (all grade 2) were reported with T-DXd. In the T-DM1 group, five patients (1.9%) had a grade 1 event, three patients (1.1%) had a grade 2 event and one patient (0.4%) had a grade 3 event. No grade 4 or 5 events of ILD or pneumonitis were reported in either treatment group. In the T-DXd group, any-grade adjudicated drug-related ILD was reported in 14 patients (5.4%) within 6 months of the first dose, in 12 patients (4.6%) between 6 and 12 months, in 11 patients (4.2%) between 12 and 24 months and in six patients (2.3%) after 24 months (Extended Data Table 5). EAIRs for ILD and pneumonitis were 0.09 with T-DXd and 0.04 with T-DM1.
Left ventricular dysfunction or left ventricular ejection fraction (LVEF) decrease occurred in 11 patients (4.3%) in the T-DXd group and in four patients (1.5%) in the T-DM1 group. Since the previous data cutoff, there were two events of left ventricular dysfunction or LVEF decrease in each treatment group (one grade 1 event in the T-DXd group and one grade 2 event in the T-DM1 group). EAIRs were 0.02 for both the T-DXd and T-DM1 groups.
Discussion
In this updated analysis of the DESTINY-Breast03 phase 3 clinical trial in patients with previously treated HER2-positive metastatic breast cancer, T-DXd continued to demonstrate clinically meaningful improvement in efficacy compared with T-DM1 and a manageable safety profile that was consistent with previous results23. The median PFS and ORR by investigator assessment reinforced the clinical benefit of T-DXd over T-DM1 and were consistent with the analysis at the previous data cutoff23. Median OS was reached in both treatment groups in this updated analysis, with an approximate 10-month improvement over T-DM1 observed with T-DXd and a reduction in the risk of death by approximately 27%, which has not been previously observed in this setting.
The ORR by investigator assessment reported in the T-DXd group in the current analysis was consistent with the ORR by BICR and by the investigator with T-DXd reported in the previous analysis23. However, there were differences in the number of complete responses reported in the T-DXd group by the investigator in this analysis (12.6%, n = 33) and in the previous analysis (11%, n = 30) compared with that reported by BICR in the previous data cutoff analysis (21%, n = 55), possibly indicating that the investigators were more conservative in declaring a complete response23. Responses appeared to be more durable with T-DXd treatment, with a median DoR by investigator assessment of 30.5 months (median follow-up, 43.0 months) in the T-DXd group compared with 17.0 months (median follow-up, 35.4 months) in the T-DM1 group.
The clinical benefit of T-DXd over T-DM1 in this updated data cutoff is evidenced by the improved median PFS, which was approximately four times longer with T-DXd at 29.0 months than with T-DM1 at 7.2 months, consistent with the previous analysis23. Furthermore, almost half of the patients (45.7%) in the T-DXd group were progression free at 3 years and more than 40% (41.5%) of patients were progression free at 4 years (however, several patients were censored at that time point). The median PFS in the T-DXd group was longer than the median PFS reported with first-line pertuzumab, trastuzumab and docetaxel combination therapy at the end-of-study analysis of the CLEOPATRA trial (18.7 months), and the PFS rate was below 40% at 4 years in that trial9; however, these cross-trial comparisons should be interpreted cautiously given the continuously changing treatment landscape of HER2-positive metastatic breast cancer. The median PFS2 by investigator assessment with T-DXd was approximately twice as long as that with T-DM1 in this updated analysis, which suggests that patients may derive a better clinical benefit when treated with T-DXd before T-DM1.
To our knowledge, the median OS with T-DXd in DESTINY-Breast03 is the longest reported OS in this disease setting (median OS, 52.6 months; median follow-up, 43 months). This is in the range of the CLEOPATRA trial in the first-line setting, which demonstrated a median OS of 57.1 months (median follow-up, 99.9 months) at the end-of-study analysis in patients with HER2-positive metastatic breast cancer treated with pertuzumab, trastuzumab and docetaxel combination therapy9. The median OS observed in the T-DM1 group in DESTINY-Breast03 was 42.7 months, which is longer than that reported in the EMILIA trial (29.9 months)24. However, cross-trial comparisons should be interpreted with caution, as the differences observed in median OS between the trials may be due to variations in study design and post-trial therapies used. Since the EMILIA trial was completed, several therapies have been approved for the treatment of HER2-positive metastatic breast cancer25. Treatment crossover was not part of the study design of DESTINY-Breast03; however, patients received a range of systemic therapies after the trial and after progression (Table 3). These therapies included other anti-HER2 agents (beyond trastuzumab, T-DM1, T-DXd and pertuzumab), such as HER2-directed tyrosine kinase inhibitors (48.0% for T-DM1) and new HER2-targeted agents (11.6% for T-DM1), which may have impacted OS in the T-DM1 group.
In the post-trial (clinical) setting, 64 patients (32.3%) in the T-DM1 group subsequently received T-DXd. Notably, when adjusting the OS of these patients in the T-DM1 group who received T-DXd after the trial in a sensitivity analysis, the approximate OS improvement with T-DXd versus T-DM1 was >1 year (adjusted median OS of 39.8 months with T-DM1). The efficacy of T-DXd following progression on T-DM1 was previously demonstrated in the DESTINY-Breast02 (NCT03523585) trial (median OS of 39.2 months with T-DXd versus 26.5 months with treatment of physician’s choice)26. When taking the data from these two studies together, the better outcomes demonstrated by T-DXd in the present study, including ORR, DoR, PFS and OS, support the potential benefit of T-DXd when used in earlier treatment settings.
Overall, drug-related TEAEs associated with drug discontinuation, dose reduction and drug interruption continued to be higher with T-DXd than with T-DM1, as observed in previous analyses22,23. Although more patients in the T-DXd group discontinued treatment due to drug-related TEAEs than in the T-DM1 group, more patients remained on T-DXd treatment than T-DM1 at this updated data cutoff. The safety profile of T-DXd continued to be manageable in this longer-term follow-up of DESTINY-Breast03. Incidence rates of any-grade, grade ≥3 and serious TEAEs were slightly higher with T-DXd than with T-DM1, consistent with reports from previous analyses22,23. The median duration of treatment was more than 2.5 times longer with T-DXd than with T-DM1; however, EAIRs, which account for differences between treatment duration, were lower with T-DXd than with T-DM1 for any-grade TEAEs, grade ≥3 TEAEs and serious TEAEs. No new safety signals were observed with long-term treatment, supporting the favorable benefit–risk profile of T-DXd versus T-DM1 in previously treated HER2-positive metastatic breast cancer.
With the additional follow-up since the previous analysis23, four new ILD and/or pneumonitis events occurred in the T-DXd group (all grade 2). Most new events resolved or resolved with sequalae (75%) and occurred during the third year of treatment (time to onset between 832 and 961 d). As previously reported23, only two patients had grade 3 events in the T-DXd group (both events resolved); no grade 4 or 5 events were observed. Consistent with a previous study, most ILD and/or pneumonitis events occurred within the first year of T-DXd treatment (Extended Data Table 5)27. In the T-DM1 group, rates of ILD and pneumonitis increased from 3% to 3.4% at this updated data cutoff; there was only one additional grade 1 event compared with the previous data cutoff23. These results support continuous patient monitoring and prompt management of potential ILD and/or pneumonitis when symptoms are detected in patients treated with T-DXd.
Potential limitations of the DESTINY-Breast03 trial have been published22,23. In the current analysis, PFS, ORR and DoR were assessed by the investigators, not by BICR; consequently, no formal statistical comparisons were made. We report median OS for both the T-DXd and T-DM1 groups, with an HR supporting improved OS with T-DXd treatment; however, this was an exploratory analysis. Longer follow-up is needed to determine a more precise estimate of the median OS in the T-DXd group due to the number of patients censored; more mature data are expected at the next data cutoff as the study continues.
This long-term analysis reinforces the superiority of T-DXd over T-DM1 in patients with metastatic breast cancer previously treated with taxane and trastuzumab, with the longest median OS reported in this disease setting and more than two-thirds (67.6%) of patients still alive at 3 years. The clinically meaningful improvement in efficacy was consistent with the previous data cutoff. The safety profile of T-DXd continues to be manageable with no cumulative toxicities observed with longer follow-up. Analyses on the impact of T-DXd on long-term responders across studies and exploring the efficacy of T-DXd in the earlier metastatic breast cancer setting (DESTINY-Breast09, NCT04784715) are ongoing.
Methods
Trial design
Details of the DESTINY-Breast03 (NCT03529110) study design have been published22,23. In summary, this was an open-label, multicenter, phase 3 trial conducted to compare T-DXd with T-DM1 in patients with HER2-positive, unresectable or metastatic breast cancer who were previously treated with trastuzumab and taxane. Patients were randomly assigned 1:1 to receive either T-DXd at 5.4 mg per kg or T-DM1 at 3.6 mg per kg intravenously every 3 weeks. Patients were stratified based on hormone receptor status, prior pertuzumab treatment and history of visceral disease via a web-based system.
Eligible patients had received prior treatment with trastuzumab and taxane, either in an advanced or metastatic setting or with progression that occurred within 6 months of post-neoadjuvant or adjuvant therapy, and had confirmed HER2 positivity according to the American Society of Clinical Oncology–College of American Pathologists guidelines assessed by a central laboratory. Patients were considered to have HER2-positive disease if the tumor was IHC 3+ or IHC 2+ with a positive in situ hybridization result28. Documented evidence of radiologic progression either during or after recent treatment or within 6 months after adjuvant therapy was required. Patients were included only if they had adequate renal and hepatic function. Patients with notable or uncontrollable cardiovascular disease, such as recent myocardial infarction, symptomatic heart failure, abnormal troponin levels, prolonged QT intervals or an LVEF below 50% within 28 d of randomization were excluded from the study. Patients previously treated with any HER2-directed antibody–drug conjugate or patients with a history of (noninfectious) ILD and/or pneumonitis requiring steroids or current or unconfirmed ILD and/or pneumonitis were ineligible for the study. Patients with inactive brain metastases or asymptomatic brain metastases that did not require treatment with corticosteroids or anticonvulsants or who had recovered from the acute toxic effect of radiotherapy were eligible for inclusion. A minimum of 2 weeks must have elapsed between the end of whole-brain radiotherapy and study enrollment.
Randomization of patients involved balanced block randomization with a 1:1 allocation ratio for T-DXd and T-DM1. Due to distinct administration protocols and adverse event profiles of the treatments, blinding of patients and investigators was not possible. However, tumor assessments were performed by BICR, which were previously reported22,23.
Baseline study assessments preceded the first treatment, followed by assessments on day 1 of each 21-d cycle, including additional evaluations on days 8 and 15 of the first cycle. Tumor assessments occurred every 6 weeks from randomization, irrespective of the treatment cycle. End-of-treatment assessments were conducted within 7 d of discontinuation, with a follow-up at 40 d after treatment or before new anticancer treatment. Subsequent long-term visits were scheduled every 3 months until death, consent withdrawal, loss to follow-up or study closure.
Trial oversight
The trial was designed by Daiichi Sankyo. Before initiation of the study, the trial protocol was approved by the ethical bodies or institutional review boards at each site. The study was conducted in accordance with the standards set by the Declaration of Helsinki, the International Conference on Harmonization Guideline for Good Clinical Practice, any local regulations and the study protocol. All participating patients provided their informed consent in writing before enrollment. Patients did not receive any compensation for participating in the study.
Endpoints
The primary endpoint was PFS assessed by BICR22,23. The key secondary endpoint was OS. Other secondary and exploratory endpoints reported in this study included ORR, DoR, PFS, PFS2 by investigator assessment and safety. PFS2 was defined as the time from the date of randomization to the first documented progression on the next line of therapy or death due to any cause, whichever occurred first. The next line of therapy was defined as the first new systemic antineoplastic therapy initiated after discontinuation of study treatment regardless of the reason for end of treatment.
Safety
Adverse events were graded based on Common Terminology Criteria for Adverse Events version 5.0 and coded according to the Medical Dictionary for Regulatory Activities version 25.0. Suspected ILD and/or pneumonitis events were adjudicated by an external independent adjudication committee. Patients with suspected ILD and/or pneumonitis had treatment interrupted until further evaluation, and ILD and/or pneumonitis events were carefully monitored until complete resolution, including after drug discontinuation.
Sensitivity analysis
The RPSFTM with recensoring techniques was applied to the DESTINY-Breast03 median OS to calculate the estimated acceleration factor exp(ψ) for OS. A hypothetical OS was derived for patients in the T-DM1 group using the estimated acceleration factor exp(ψ) = 0.425; this represents the OS that would have been observed if T-DXd treatment had not been administered after the trial. The sensitivity analysis adhered to the stratified Cox proportional hazards model, which incorporates stratification factors such as hormone receptor status, prior pertuzumab treatment and history of visceral disease, as identified by the interactive response technology platform.
Statistical analysis
The study aimed to enroll approximately 500 patients, with random assignment determined using EAST software version 6.4. Efficacy analysis was conducted on the full analysis set and included all patients who were randomly assigned to a treatment group. The safety analysis was conducted on the safety analysis set and included all randomly assigned patients who received at least one dose of T-DXd or T-DM1. Analysis of PFS and OS between treatment groups employed a stratified log-rank test, considering randomization factors. This involved presenting Kaplan–Meier survival estimates and curves, including median event times and two-sided 95% CIs (Brookmeyer and Crowley method). Kaplan–Meier estimates at specified intervals with 95% CIs were also provided. HRs and 95% CIs were calculated using a stratified Cox proportional hazards model. The median follow-up duration for OS and its two-sided 95% CI were calculated for each treatment group using the Kaplan–Meier method by reversing the OS censoring and event indicators. Based on a prespecified hierarchical testing procedure, OS (the key secondary endpoint) was tested if PFS by BICR (the primary efficacy endpoint) was statistically significant22,23. The current updated OS analysis was exploratory because the prespecified threshold for statistical significance was reached at the second OS interim analysis, although the median OS was not reached previously.
Cochran–Mantel–Haenszel tests, stratified by randomization factors, were used to evaluate ORR. Estimates of ORR were presented with 95% CIs (Clopper–Pearson method). The DoR included median event times and 95% CIs (Brookmeyer and Crowley method), along with Kaplan–Meier estimates. Statistical analysis used SAS version 9.3 or later and R 4.2.0 for the RPSFTM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Anonymized individual participant data on completed studies and applicable supporting clinical study documents may be available upon request at https://vivli.org/. In cases where clinical study data and supporting documents are provided pursuant to our company policies and procedures, Daiichi Sankyo Companies will continue to protect the privacy of the company and our clinical study participants. Details on data sharing criteria and the procedure for requesting access can be found at this web address: https://vivli.org/ourmember/daiichi-sankyo/. Additional information can be found in the Supplementary Information.
References
Iqbal, N. & Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol. Biol. Int. 2014, 852748 (2014).
Gutierrez, C. & Schiff, R. HER2: biology, detection, and clinical implications. Arch. Pathol. Lab. Med. 135, 55–62 (2011).
Slamon, D. J. et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244, 707–712 (1989).
Slamon, D. J. et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 (1987).
National Cancer Institute. SEER Cancer Stat Facts: Female Breast Cancer Subtypes seer.cancer.gov/statfacts/html/breast-subtypes.html (2017).
Wolff, A. C. et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 31, 3997–4013 (2013).
Swain, S. M. et al. Multidisciplinary clinical guidance on trastuzumab deruxtecan (T-DXd)-related interstitial lung disease/pneumonitis—focus on proactive monitoring, diagnosis, and management. Cancer Treat. Rev. 106, 102378 (2022).
American Cancer Society. Breast Cancer HER2 Status www.cancer.org/cancer/types/breast-cancer/understanding-a-breast-cancer-diagnosis/breast-cancer-her2-status.html (2022).
Swain, S. M. et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 21, 519–530 (2020).
Grinda, T. et al. Evolution of overall survival and receipt of new therapies by subtype among 20 446 metastatic breast cancer patients in the 2008–2017 ESME cohort. ESMO Open 6, 100114 (2021).
Gennari, A. et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. 32, 1475–1495 (2021).
Slamon, D. J. et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792 (2001).
Daiichi Sankyo Co. Ltd. ENHERTU® Approved in the U.S. for Patients with HER2 Positive Metastatic Breast Cancer Treated with a Prior Anti-HER2-Based Regimen daiichisankyo.us/press-releases/-/article/enhertu-approved-in-the-u-s-for-patients-with-her2-positive-metastatic-breast-cancer-treated-with-a-prior-anti-her2-based-regimen (2022).
Giordano, S. H. et al. Systemic therapy for advanced human epidermal growth factor receptor 2-positive breast cancer: ASCO guideline update. J. Clin. Oncol. 40, 2612–2635 (2022).
Im, S. A. et al. Pan-Asian adapted ESMO Clinical Practice Guidelines for the diagnosis, staging and treatment of patients with metastatic breast cancer. ESMO Open 8, 101541 (2023).
Daiichi Sankyo Europe GmbH. Summary of Product Characteristics https://www.medicines.org.uk/emc/product/12135/smpc (2024).
Barok, M., Joensuu, H. & Isola, J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 16, 209 (2014).
Nicolò, E., Zagami, P. & Curigliano, G. Antibody–drug conjugates in breast cancer: the chemotherapy of the future? Curr. Opin. Oncol. 32, 494–502 (2020).
Ogitani, Y. et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 22, 5097–5108 (2016).
Ogitani, Y., Hagihara, K., Oitate, M., Naito, H. & Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody–drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 107, 1039–1046 (2016).
Nakada, T., Sugihara, K., Jikoh, T., Abe, Y. & Agatsuma, T. The latest research and development into the antibody–drug conjugate, [fam-] trastuzumab deruxtecan (DS-8201a), for HER2 cancer therapy. Chem. Pharm. Bull. 67, 173–185 (2019).
Cortés, J. et al. Trastuzumab deruxtecan versus trastuzumab emtansine for breast cancer. N. Engl. J. Med. 386, 1143–1154 (2022).
Hurvitz, S. A. et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: updated results from DESTINY-Breast03, a randomised, open-label, phase 3 trial. Lancet 401, 105–117 (2023).
Diéras, V. et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 18, 732–742 (2017).
Swain, S. M., Shastry, M. & Hamilton, E. Targeting HER2-positive breast cancer: advances and future directions. Nat. Rev. Drug Discov. 22, 101–126 (2023).
André, F. et al. Trastuzumab deruxtecan versus treatment of physician’s choice in patients with HER2-positive metastatic breast cancer (DESTINY-Breast02): a randomised, open-label, multicentre, phase 3 trial. Lancet 401, 1773–1785 (2023).
Powell, C. A. et al. Pooled analysis of drug-related interstitial lung disease and/or pneumonitis in nine trastuzumab deruxtecan monotherapy studies. ESMO Open 7, 100554 (2022).
Wolff, A.C. et al. Human epidermal growth factor receptor 2 testing in breast cancer. Arch. Pathol. Lab. Med. 147, 993–1000 (2023).
Acknowledgements
We thank the patients who participated in this study as well as their families and caregivers. We also thank staff and investigators at all study sites, the members of the independent data monitoring committee, the members of the ILD adjudication committee as well as R. Tang, the Study Team Lead, and J. Perritt, the Data Management Lead, for their valuable contribution to the study. I. Boltsis, E. Wolmarans, C.M. Rigby and S. Duggan of ApotheCom provided assistance in medical writing and editorial support, which was funded by Daiichi Sankyo. This study was funded by Daiichi Sankyo and AstraZeneca. In March 2019, AstraZeneca entered into a global development and commercialization collaboration agreement with Daiichi Sankyo for T-DXd (DS-8201).
Author information
Authors and Affiliations
Contributions
J.C., S.A.H., S.-A.I., H.I., G.C., S.N., S.A., Z.L., A.E. and E.H. contributed to the conception and/or design of the study and the development of the study protocol. J.C., S.A.H., S.-A.I., H.I., G.C., S.-B.K., J.W.Y.C., J.L.P., W.L., K.Y., G.B., S.L., G.S.B., X.W., T.B. and E.H. were involved in data collection and quality control. Z.L. performed the data analysis. All authors participated in the interpretation of data. All authors were involved in drafting and revision of the paper, and all authors approved the final version of the paper for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: J.C. has received research grants from Roche, ARIAD Pharmaceuticals, AstraZeneca, Baxalta/Servier Affaires, Bayer Healthcare, Eisai, F. Hoffmann-La Roche, Guardant Health, Merck Sharp & Dohme, Pfizer, Piqur Therapeutics, IQVIA and the Queen Mary University of London; has received honoraria from Roche, Novartis, Eisai, Pfizer, Lilly, Merck Sharp & Dohme, Daiichi Sankyo, AstraZeneca, Gilead and Stemline Therapeutics; has received stock from Leuko (relative) and MAJ3 Capital; has received support for attending meetings, accommodations and/or travel from Roche, Novartis, Eisai, Pfizer, Daiichi Sankyo, AstraZeneca, Gilead, Merck Sharp & Dohme and Stemline Therapeutics; has held consulting or advisory roles for Roche, AstraZeneca, Seattle Genetics, Daiichi Sankyo, Lilly, Merck Sharp & Dohme, Leuko, Bioasis, Clovis Oncology, Boehringer Ingelheim, Ellipses, HiberCell, BioInvent, GEMoaB, Gilead, Menarini, Zymeworks, Reveal Genomics, Scorpion Therapeutics, ExpreS2ion Biotechnologies, Jazz Pharmaceuticals, AbbVie, BridgeBio, BioNTech and Biocon; and has the following patents: pharmaceutical combinations of a PI3K inhibitor and a microtubule destabilizing agent (J. Cortés Castán, A. Piris Giménez, V. Serra Elizalde; WO 2014/199294A (issued)) and HER2 as a predictor of response to dual HER2 blockade in the absence of cytotoxic therapy (A. Prat, A. Llombart, J.C.; US 2019/0338368 A1 (licensed)). S.A.H. has received research grants from Genentech/Roche, Novartis, GlaxoSmithKline, Sanofi, Pfizer, Amgen, OBI Pharma, Puma Biotechnology, Dignitana, Bayer, BioMarin, Lilly, Merrimack, Cascadian Therapeutics, Seagen, Daiichi Sankyo, MacroGenics, Ambryx, Immunomedics, Pieris Pharmaceuticals, Radius Health, Arvinas, Zymeworks, Gilead Sciences, Phoenix Molecular Designs, CytomX Therapeutics, Samumed, Dantari, Orinove, Greenwich LifeSciences, AstraZeneca/Daiichi Sankyo, G1 Therapeutics and Orum Therapeutics; has received stock and other ownership interests from ROM Tech; and has received support for attending meetings and/or travel from Lilly. S.-A.I. has received research grants from AstraZeneca, Pfizer, Roche/Genentech, Daewoong Pharmaceutical, Eisai and Boryung Pharmaceuticals; and has held consulting or advisory roles at AstraZeneca, Novartis, Roche/Genentech, Eisai, Pfizer, Amgen, Hanmi, Lilly, MSD and Daiichi Sankyo. H.I. has received research grants from MSD, AstraZeneca, Kyowa Kirin, Daiichi Sankyo, Chugai Pharma, Nihokayaku, Lilly Japan, Novartis, Bayer, Pfizer, Boehringer Ingelheim, Sanofi and Amgen; has received honoraria from Chugai Pharma, AstraZeneca, Eisai, Pfizer, Daiichi Sankyo, Lilly Japan, Kyowa Kirin, Taiho Pharmaceutical and MSD; and has held consulting or advisory roles at Chugai Pharma, Daiichi Sankyo, Pfizer, AstraZeneca, Lilly Japan, Kyowa Kirin, Novartis, MSD and Sanofi. G.C. has received research grants from Merck; has received honoraria from Ellipses Pharma; has received support for attending meetings and/or travel from Roche/Genentech, Pfizer, Daiichi Sankyo and AstraZeneca; has a leadership role for the ESMO, the European Society of Breast Cancer Specialists and ESMO Open; is a speakers’ bureau member for Roche/Genentech, Novartis, Pfizer, Lilly, Foundation Medicine, Samsung, Daiichi Sankyo, Seagen, Menarini, Gilead Sciences, AstraZeneca and Exact Sciences; and has held consulting or advisory roles for Roche/Genentech, Pfizer, Novartis, Lilly, Foundation Medicine, Bristol Myers Squibb, Samsung, AstraZeneca, Daiichi Sankyo, Boehringer Ingelheim, GlaxoSmithKline, Seagen, Guardant Health, Veracyte, Celcuity, Hengrui Therapeutics, Menarini, Merck, Exact Sciences, Blueprint Medicines and Gilead Sciences. S.-B.K. has received research grants from Genzyme; has received honoraria from DAEHWA Pharmaceutical, LegoChem Biosciences and Kalbe Farma; has received stock and other ownership interests from Genopeaks; and has held consulting or advisory roles for Lilly, AstraZeneca, DAEHWA Pharmaceutical, ISU Abxis, BeiGene, Daiichi Sankyo/AstraZeneca, OBI Pharma and Ensol Biosciences. J.W.Y.C. has a leadership role for Precision Oncology; and has held consulting or advisory roles at Lilly. K.Y. has received research grants from Ono Pharmaceutical, MSD, Daiichi Sankyo/AstraZeneca, AstraZeneca/MedImmune, Taiho Pharmaceutical, Pfizer, Novartis, Takeda, Sanofi, Seagen, Eisai, Lilly, Genmab, Boehringer Ingelheim, Kyowa Kirin, Haihe Pharmaceutical and Nihokayaku; has received honoraria from Eisai, Pfizer, AstraZeneca, Novartis, Taiho Pharmaceutical, Lilly Japan, Daiichi Sankyo/AstraZeneca, Takeda, Fujifilm, Ono Pharmaceutical, Chugai Pharma and MSD Oncology; and has held consulting or advisory roles for Chugai Pharma, Ono Pharmaceutical, Novartis, Eisai, OncXerna Therapeutics and Henlius. G.B. has received research grants from Gilead Sciences; has received honoraria from Roche, Eisai, MSD, Lilly, Pfizer, Novartis, Exact Sciences, AstraZeneca/Daiichi Sankyo, Gilead Sciences and Seagen; has received support for attending meetings and/or travel from Novartis, Roche, Pfizer, AstraZeneca/Daiichi Sankyo and Gilead Sciences; and has held consulting or advisory roles for Roche, AstraZeneca/Daiichi Sankyo, Pfizer, Lilly, Novartis, Gilead Sciences, Exact Sciences, Eisai, MSD, Seagen and Agendia. S.L. has received research grants from Roche/Genentech, Novartis, Merck, Puma Technology, Bristol Myers Squibb, Seagen, AstraZeneca, Nektar and Lilly; has received honoraria from Roche/Genentech, Novartis, MSD Oncology and Mersana; has a leadership role for Bristol Myers Squibb; has received support for attending meetings and/or travel from Bristol Myers Squibb; and has held consulting or advisory roles for Roche/Genentech, Novartis, G1 Therapeutics, Puma Biotechnology, GlaxoSmithKline, AstraZeneca, Seagen, Bristol Myers Squibb, Silverback Therapeutics, Pfizer, Gilead Sciences, Daiichi Sankyo/Lilly and Tallac Therapeutics. T.B. has received research grants from Novartis, AstraZeneca, Seagen, Pfizer and Daiichi Sankyo/AstraZeneca; has received support for attending meetings and/or travel from Roche, Pfizer, AstraZeneca and Novartis; and has held consulting or advisory roles for Novartis, Pfizer, Seagen, Daiichi Sankyo/AstraZeneca and Lilly. S.N. is employed by Daiichi Sankyo/Arqule; and has received stock and other ownership interests from Daiichi Sankyo. S.A. is employed by Daiichi Sankyo. Z.L. is employed by Daiichi Sankyo. A.E. is employed by Daiichi Sankyo/AstraZeneca; and has received stock and other ownership interests from Daiichi Sankyo/AstraZeneca. E.H. has received research grants from AstraZeneca, Hutchison MediPharma, OncoMed, MedImmune, Stemcentrx, Genentech/Roche, Curis, Verastem, Zymeworks, Syndax, Lycera, Rgenix, Novartis, Mersana, Millennium, TapImmune, Lilly, Pfizer, Tesaro, Boehringer Ingelheim, H3 Biomedicine, Radius Health, Acerta Pharma, MacroGenics, AbbVie, Immunomedics, Fujifilm, eFFECTOR Therapeutics, Merus, NuCana, Regeneron, Leap Therapeutics, Taiho Pharmaceutical, EMD Serono, Daiichi Sankyo, ArQule, Syros Pharmaceuticals, Clovis Oncology, CytomX Therapeutics, InventisBio, Deciphera, Sermonix Pharmaceuticals, Sutro Biopharma, Zenith Epigenetics, Arvinas, Harpoon, Black Diamond Pharmaceuticals, Orinove, Molecular Templates, Seagen, Compugen, G1 Therapeutics, Karyopharm Therapeutics, the Dana Farber Cancer Hospital, Onconova Therapeutics, Shattuck Labs, PharmaMar, Olema Pharmaceuticals, ImmunoGen, Plexxikon, Amgen, Akeso Biopharma, ADC Therapeutics, AtlasMedx, Aravive, Ellipses Pharma, Incyte, MabSpace Biosciences, ORIC Pharmaceuticals, Pieris Pharmaceuticals, Pionyr, Repertoire Immune Medicines, Treadwell Therapeutics, Jacobio, Accutar Biotech, Artios, Bliss Biopharmaceutical, Cascadian Therapeutics, Dantari, Duality Biologics, Elucida Oncology, Infinity Pharmaceuticals, Relay Therapeutics, Tolmar, Torque, BeiGene, Context Therapeutics, K-Group Beta, Kind Pharmaceuticals, Loxo, Oncothyreon, Orum Therapeutics, Prelude Therapeutics, ProfoundBio, Cullinan Oncology, Bristol Myers Squibb, Eisai, Fochon Pharmaceuticals, Gilead Sciences, Inspirna, Myriad Genetics, Silverback Therapeutics and Stemline Therapeutics; and has held consulting or advisory roles for Pfizer, Genentech/Roche, Lilly, Daiichi Sankyo, Mersana, AstraZeneca, Novartis, Greenwich LifeSciences, Orum Therapeutics, Ellipses Pharma, Olema Pharmaceuticals, Stemline Therapeutics, Tubulis, Verascity Science, Theratechnology, Accutar Biotechnology, Entos, Fosun Pharma, Gilead Sciences, Jazz Pharmaceuticals, Medical Pharma Services and Zentalis. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Yu Shen, Melinda Telli and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ulrike Harjes, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Kaplan-Meier estimates of DoRa.
DoR, duration of response; NE, not estimable; T-DM1, trastuzumab emtansine; T-DXd, trastuzumab deruxtecan. aThe DoR rate at 36 months was 48.9% (95% CI, 41.3-56.1%) with T-DXd and 28.7% (95% CI, 18.9-39.2%) with T-DM1.
Extended Data Fig. 2 Sensitivity analysis for OS.
RPSFTM adjusted Kaplan-Meier curve of OS for the T-DM1 group plotted with the unadjusted OS curves for the T-DXd group and T-DM1 group. OS, overall survival; RPSFTM, rank-preserving structural failure time models; T-DM1, trastuzumab emtansine; T-DXd, trastuzumab deruxtecan.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 2
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cortés, J., Hurvitz, S.A., Im, SA. et al. Trastuzumab deruxtecan versus trastuzumab emtansine in HER2-positive metastatic breast cancer: long-term survival analysis of the DESTINY-Breast03 trial. Nat Med (2024). https://doi.org/10.1038/s41591-024-03021-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-024-03021-7
- Springer Nature America, Inc.