

Abstract
Organisms evolve mechanisms that regulate the properties of biogenic crystals to support a wide range of functions, from vision and camouflage to communication and thermal regulation. Yet, the mechanism underlying the formation of diverse intracellular crystals remains enigmatic. Here we unravel the biochemical control over crystal morphogenesis in zebrafish iridophores. We show that the chemical composition of the crystals determines their shape, particularly through the ratio between the nucleobases guanine and hypoxanthine. We reveal that these variations in composition are genetically controlled through tissue-specific expression of specialized paralogs, which exhibit remarkable substrate selectivity. This orchestrated combination grants the organism with the capacity to generate a broad spectrum of crystal morphologies. Overall, our findings suggest a mechanism for the morphological and functional diversity of biogenic crystals and may, thus, inspire the development of genetically designed biomaterials and medical therapeutics.

Similar content being viewed by others


Main
Across the animal kingdom, many organisms harness intracellular crystals made from organic materials, particularly guanine, for a wide variety of purposes1,2. These range from enhancing vision3,4, through creating the vibrant colors observed in fish5, copepods6 and chameleons7, to nitrogen storage in unicellular organisms8,9,10. Within these systems, guanine crystals are intricately tailored to serve a range of functions. Prism-shaped crystals contribute to light scattering11, arrays of elongated plates are used for light reflection12 and square and hexagonal crystals tile light-focusing surfaces3,13. Remarkably, multiple crystal morphologies coexist in different tissues of the same organism4,14,15,16. Despite the evident importance of crystal shape, the precise mechanisms controlling crystal morphogenesis and the associated cellular processes remain elusive.
Crystal formation relies on the synthesis of large quantities of guanine, an essential nucleobase in purine metabolism17. Crystal-forming cells execute these intricate processes with precision and control. Notably, each crystal is composed of billions of nucleobases and a single cell may contain hundreds of these crystals2,3,4,5,6. These specialized cells manage to produce vast quantities of crystal-forming nucleobases while maintaining cellular homeostasis, including DNA and RNA synthesis and overall cell function2.
It is generally believed that, in both unicellular and multicellular crystal-forming organisms, nucleobases are synthesized within the cytoplasm8,17. Then, they are transported by dedicated machinery into membrane-bound organelles, where the meticulously regulated process of crystal formation takes place12,13,18,19,20,21,22. This process results in the generation of crystals with precise morphologies that, in turn, determine their optical and mechanical properties and subsequent functions. The control of these biogenic crystals often exceeds the capabilities of state-of-the-art materials science and solid-state chemistry23,24,25,26.
Recent studies have shown that thin crystal plates are formed through templated nucleation on protein scaffolds12,13. However, the regulation of the morphogenesis of these intracellular crystals remains enigmatic. Additionally, these biogenic crystals rarely consist of a single nucleobase; rather, they are typically a blend of nucleobases, mainly guanine and hypoxanthine cocrystals1,5,27,28. In certain species, the guanine crystals integrate up to 25% hypoxanthine28,29. A recent study exploring the nucleobase composition of guanine crystals in various organisms further concluded that these structures are solid solutions primarily of guanine and hypoxanthine with a consistent distribution within the crystal28. The study did not find a clear correlation between crystal morphology and dopant content, suggesting that these dopants may not have a notable role in determining crystal shape but are rather used as cheap building blocks28. Prior research has unveiled multiple, often conflicting, biosynthetic pathways for guanine and hypoxanthine, leaving the precise route of nucleobase biosynthesis in crystal formation uncertain30,31,32,33,34,35.
To unravel the intricate molecular mechanisms enabling the control over crystal morphogenesis, we embarked on a multifaceted investigation encompassing proteomics and spatial metabolomics, in vitro enzymatic activity assays, cellular localization studies, genetic manipulations and electron microscopy. Our model system constituted the crystal-forming cells (termed iridophores) of zebrafish (Danio rerio) (Fig. 1a). Zebrafish harness the remarkable properties of guanine crystals for a multitude of purposes. These crystals, located across their skin and eyes12,15,16, have a pivotal role in color and pattern formation14,36,37,38, serve for camouflage4,37, act as light barriers4 and enhance visual sensitivity, especially in low-light conditions4,12. Despite a uniform, thin, plate-like shape, the guanine crystals in zebrafish exhibit notable morphological variations between tissues, influenced by their specific functions12,14. This variability within the same organism provides a unique opportunity to study the underlying genetic regulation of crystal morphogenesis.

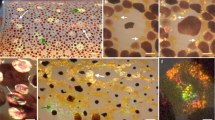
Left: optical microscope images of different crystal-containing tissues in adult zebrafish, showing crystals with varying morphologies. Middle: TEM images of biogenic guanine crystals isolated from WT zebrafish operculum, eye and body, showing increasingly more pronounced (012) facets and underexpressed (001) facets. Right: TEM images of in vitro formed (synthetic) crystals mirror the trend observed in the isolated biogenic crystals, with more pronounced (012) facets as hypoxanthine levels increase. We note that the difference in contrast between the synthetic and biogenic crystals is because of the synthetic crystals being larger and thicker than their biogenic counterparts. A schematic illustration of the in vitro and in vivo observations revealing differences in crystal morphology. Crystal isolation was performed on four independent biological replicates.
In this work, we combine proteomics and spatial metabolomics analyses with in vitro enzymatic assays, cellular localization studies, genetic manipulations and electron microscopy to unravel the molecular mechanisms underlying crystal morphogenesis in zebrafish iridophores. We demonstrate that the chemical composition of the crystals dictates their shape, particularly through the ratio of the nucleobases guanine and hypoxanthine. Furthermore, we uncover that these compositional variations are genetically regulated by the tissue-specific expression of specialized paralogs, which exhibit notable substrate selectivity. This coordinated mechanism enables the organism to produce a diverse array of crystal morphologies. Overall, our findings suggest a mechanism for the morphological and functional diversity of biogenic crystals.
Results
Variability in crystal composition and morphology in tissues
To investigate the origin of the observed variability in crystal morphologies in zebrafish, we isolated crystals from distinct tissues of adult fish. Our analysis focused on crystals obtained from the operculum (a bony structure protecting the gills), responsible for creating silvery iridescence for camouflage (Fig. 1, top), as well as shorter, wider crystals from the fish’s eye, designed as a light barrier and for low-light vision (Fig. 1, middle). Additionally, we examined even shorter crystals from the skin, which contribute to the formation of the alternating blue and yellow stripes (Fig. 1, bottom). Transmission electron microscopy (TEM), electron diffraction and micro-Raman analyses showed that the isolated crystals are all plate-like β-anhydrous guanine (Supplementary Figs. 1 and 2a,b). However, the skin crystals were observed to have a much smaller aspect ratio when compared with the crystals from the operculum and eye (Fig. 1 and Supplementary Fig. 2a,b). Specifically, the skin crystals displayed overdeveloped (012) facets and underdeveloped (010) facets, compared with the operculum and eyes, contributing to the distinct differences in crystal morphology observed across the various tissues (Fig. 1 and Supplementary Figs. 1 and 2a).
Given the reported variability in the composition of guanine crystals across different organisms, we posited that the observed differences in crystal morphology in zebrafish might be correlated with their compositions. To investigate this, we conducted a comparative analysis, focusing on two distinct crystal morphologies from adult zebrafish: the thin and elongated crystals isolated from the operculum and the shorter skin crystals with a much lower aspect ratio. Orbitrap secondary ion mass spectrometry (Orbitrap-SIMS) imaging (Methods) revealed substantial differences in crystal composition. Specifically, the hypoxanthine-to-guanine ratios in the shorter skin crystals surpassed those in the operculum by more than 30% (Supplementary Fig. 2e). To provide further validation using a quantifiable method, we turned to high-pressure liquid chromatography (HPLC) and observed a similar trend with 25% more hypoxanthine in the operculum versus in the skin (Supplementary Fig. 2g). This marked discrepancy strongly suggests that the incorporation of hypoxanthine within the crystal lattice has a pivotal role in determining its morphology.
To further validate the effect of the chemical composition on crystal morphology, we conducted a series of in vitro experiments producing crystals with increasing ratios of hypoxanthine to guanine (Fig. 1 and Supplementary Fig. 2c,d). While we observed variations in crystal morphologies during the crystallization process, a consistent pattern emerged. As the quantity of hypoxanthine increased, the crystals exhibited greater width and more prominent (012) facets (Supplementary Fig. 2c,d,f,h). Furthermore, micro-Raman analysis of synthetic guanine–hypoxanthine crystals and isolated biogenic skin crystals showed the characteristic modes of guanine–hypoxanthine mixed crystals28 (Supplementary Fig. 1j). This finding confirms the influence of hypoxanthine incorporation on crystal morphology in both in vivo and in vitro systems (Fig. 1 and Supplementary Figs. 1 and 2).
Enrichment in the de novo purine metabolism pathway
To explore the effect of the proteomic landscape on the variability in crystal composition, we isolated iridophores from zebrafish larvae at 5 days postfertilization (dpf), a stage at which intensive crystal formation takes place, by fluorescence-activated cell sorting (FACS) and performed proteomic analysis using liquid chromatography–mass spectrometry (LC–MS) (Fig. 2a–c and Supplementary Fig. 3a–d). We identified 190 significantly upregulated iridophore-specific proteins, including those encoded by the established iridophore marker genes pnp4a, slc2a15a, gmps, impdh1b, gpnmb and alx4a (refs. 15,30,33,38,39,40) (Fig. 2b,c). Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes pathway analyses unveiled key pathways enriched in iridophores, notably de novo inosine monophosphate (IMP) biosynthesis and purine nucleobase biosynthesis (Fig. 2d and Supplementary Fig. 3e).

a, Illustration of an iridophore packed with iridosomes (membrane-bound crystals); different organelles are color-coded. Insets in grayscale show high-resolution Cryo-electron tomography images of iridosomes. b, Volcano plot displaying differentially expressed proteins between iridophores (n = 3) and a control, heterogenous cell population (n = 3) in zebrafish larvae at 5 dpf (P = 0.01 and fold change (FC) > 2). Upregulated proteins are shown in blue and downregulated proteins are shown in pink. c, Bar graph depicting the elevated expression of key iridophore marker proteins. d, GO enrichment analysis highlighting upregulated (purple) and downregulated biological processes (pink) for identified proteins using a modified Fisher’s extract test from STRING41.
To delve into the functional role of these upregulated iridophore-specific proteins, we applied MetaScape and STRING analyses41,42. As expected, the purine metabolism pathway emerged as the most prominently enriched and interconnected network, underscoring its pivotal role in iridophore biology (Fig. 2b–d and Supplementary Fig. 3e–g). Specifically, proteins involved in the conversion of phosphoribosyl pyrophosphate to IMP in the early steps of purine metabolism, such as Ppat, Gart, Adsl, Atic and Pfas, were significantly upregulated43 (Fig. 2b and Supplementary Fig. 3e). Interestingly, our analysis also identified downregulated proteins, such as Gda and Uox, which are involved in the breakdown of guanine and its derivatives44,45 (Fig. 2b,d).
Unique biosynthetic enzymes are upregulated in iridophores
Subsequently, we extended our proteomic analysis to comprehensively map the intricate purine metabolic network within iridophores, starting from IMP. Strikingly, we observed a comprehensive upregulation of the entire guanine metabolic network. This encompassed the enzymatic conversions of guanosine monophosphate (GMP), guanosine diphosphate (GDP) and deoxyguanosine diphosphate (dGDP), ultimately leading to the formation of guanine (Fig. 3 and Supplementary Figs. 4 and 5). These results indicate that, in addition to guanosine, deoxyguanosine is also converted into guanine. This finding underscores the importance of coordinated expression and activity of multiple proteins and enzymes in the intricate guanine biosynthesis pathway within iridophores (Fig. 2b,d and Supplementary Figs. 4 and 5).

Schematic representation of the guanine biosynthetic pathway, highlighting differential protein regulation. The selective upregulation of specific enzymes within iridophores emphasizes their distinct roles in guanine biosynthesis and crystal formation. XMP, xanthine monophosphate; GTP, guanine triphosphate.
To validate our findings and gain insight into the metabolic regulation of these processes, we performed spatial metabolomics on iridophores in zebrafish larva eyes at various developmental stages (2, 3 and 10 dpf) using matrix-assisted laser desorption–ionization (MALDI) imaging46 (Methods and Supplementary Figs. 4 and 5). We observed a consistent and steady rise in guanine, coupled with a consistent decrease in guanosine and deoxyguanosine levels throughout eye development (Supplementary Figs. 4 and 5). These results further validate the notion that guanine is produced by the de novo purine metabolism pathway and that both guanosine and deoxyguanosine are converted into guanine (Fig. 3).
In-depth examination of the enzymes within the guanine biosynthetic pathway unveiled the selective upregulation of specific paralogs in iridophores (Figs. 2b,c and 3 and Supplementary Fig. 6). These encompassed IMP dehydrogenase 1b (Impdh1b), but not Impdh1a or Impdh2, nucleotidase 5C1bb (Nt5C1bb), but not Nt5c1ba, Nt5c1aa or Nt5c1ab, and guanylate kinase 1a (Guk1a), rather than Guk1b (Figs. 2b,c and 3 and Supplementary Fig. 6). The targeted enhancement of a subset of paralogs highlights their specialized role in the guanine biosynthesis pathway and the subsequent crystal formation process. This raises the intriguing possibility that these enzymes may have evolved specialized catalytic activities and increased specificity for this purpose.
Distinct substrate selectivity within purine nucleoside phosphorylase enzyme family
Of particular importance was the substantial upregulation of members of the purine nucleoside phosphorylase (PNPase) family, notably Pnp4a, a recognized marker for iridophores (Fig. 2b,c). To elucidate the evolutionary relationships and functional distinctions among the PNPase enzymes involved in guanine biosynthesis, we first performed phylogenetic analyses. Phylogenetic tree analysis revealed the distinct nature of Pnp4a compared with other PNPase members in zebrafish, suggesting its potential evolution toward acquiring a unique catalytic activity for the specific formation of guanine crystals within iridophores (Fig. 4a). To explore this hypothesis, we conducted a comprehensive series of in vitro enzymatic activity assays and structural predictions. For this purpose, we expressed and purified all zebrafish PNPase variants, including Pnp4a, from Escherichia coli and exposed them to a diverse range of nucleosides as substrates (Fig. 4b). We found that Pnp4a displayed heightened enzymatic activity when using guanosine and deoxyguanosine as substrates, in contrast to some of its paralogs such as Pnp4b and Pnp6 (Fig. 4b). Conversely, for nucleosides such as inosine, the precursor of hypoxanthine, and cytidine, the precursor of cytosine, Pnp4a activity was markedly lower compared with its highly active paralogs, namely Pnp5a or Pnp5b (Fig. 4b). This trend was also evident in substrate competition experiments (Fig. 4c). Collectively, these results offer compelling evidence for the selective and distinct functional role of Pnp4a in guanine crystal biosynthesis.

a, Phylogenetic analysis of the vertebrate PNPase protein family. Clades are highlighted in the background, with their phylogenetic classes color-coded: blue for fish, green for amphibians, light pink for birds, yellow for reptiles and pink for mammals. Species abbreviations: salmon, Atlantic salmon; cod, Atlantic cod; lizard, green anole; turtle, painted turtle; snake, eastern brown snake; frog, African clawed frog; medaka, Japanese rice fish. The MTAP protein served as an outgroup. b, Enzymatic activity assays of the zebrafish PNPase family using different nucleosides as substrates. The mean ± s.e.m. are shown for at least duplicates, where each data point represents an independent biological repeat. c, Substrate competition experiments, in which residual nucleoside concentrations were determined for each enzyme using LC–MS following a 30-min incubation with the specified nucleoside mixture. The mean ± s.e.m. are shown for independent biological duplicates. d, An overlay of the structures of PNPases 4a, 4b, 5a, 5b and 6 predicted by AlphaFold2 (refs. 50,52). pLDDT, predicted local distance difference test.
To elucidate the observed differences in catalytic activities of D. rerio PNPases, we compared their sequences and computationally predicted structures using several algorithms47,48,49,50,51, including AlphaFold2 (refs. 50,52). Although different prediction tools can yield different structural models, the algorithms we used produced almost identical structures for each variant, and their AlphaFold2 models were assigned high confidence values (Fig. 4d and Supplementary Fig. 7). Additionally, when overlaid, the structures of all PNPases were highly similar, with differences localized primarily at their N and C termini (Fig. 4d). On the basis of their specificity toward 6-oxopurine nucleosides (Fig. 4b) and sequence similarities, they belong to the trimeric PNPase family53. A comparison of the predicted structure of Pnp4a and the crystal structure of bovine PNPase (Protein Data Bank (PDB) 1B8O) revealed notable sequence and structural similarities between them (60% sequence identity, 0.55 Å root-mean-square deviation (r.m.s.d.)). We then compared bovine PNPase active-site residues to their corresponding positions in D. rerio PNPases and found that most were highly conserved (Supplementary Fig. 7). However, the divergence of some of these conserved residues in Pnp4b (for example, Glu63Val, Ala122Ser and Asn150His) and in Pnp6 (for example, Asp56Lys, Glu62His and Phe204Tyr) may account for the variations in substrate specificities and reduced enzymatic activities we observed relative to Pnp4a, 5a and 5b (Fig. 4b–d and Supplementary Fig. 8).
Given the upregulation of the purine metabolic network and the role of Pnp4a in directly catalyzing guanosine and deoxyguanosine, we next investigated the subcellular localization of Pnp4a. To achieve this, we cloned and engineered a transgenic zebrafish expressing a fluorescent tag fused to Pnp4a under the conditional expression of a heat-shock promoter (hsp70:Pnp4a-mKate2m) (Methods). We then examined the fish following heat shock using confocal microscopy (Supplementary Fig. 9a–d). Live imaging revealed limited colocalization between Pnp4a-mKate2 and the crystal-forming iridosomes. This observation suggests a predominant localization of Pnp4a in the iridophore cytoplasm (Supplementary Fig. 9a–d). To validate this, we examined the catalytic activity rate of the different purified zebrafish PNPases across a pH range, using guanosine as a substrate. For most PNPases, peak activity occurred at pH 7, with Pnp4a performing optimally at pH 7.5 (Supplementary Fig. 9e). This pH value is consistent with cytoplasmic conditions, rather than within the presumably acidic iridosome20.
Reduced crystal production and altered morphology in pnp4a −/−
Given the notable substrate selectivity exhibited by Pnp4a, we hypothesized that inhibiting this enzyme would have a notable impact on nucleobase biosynthesis by iridophores, potentially altering crystal composition and subsequent morphology. To test this hypothesis, we used clustered regularly interspaced short palindromic repeats (CRISPR)–Cas9 technology to generate pnp4a−/− mutants (Fig. 5a–c). We successfully generated two pnp4a−/− mutant variants: one with a 5-nt deletion, referred to as pnp4acbg20, and the other with a 10-nt deletion, designated as pnp4awz19 (Fig. 5c and Methods). Both homozygous pnp4a−/− mutants exhibited a marked reduction in crystal quantity compared with the wild type (WT) (Fig. 5a, b). A similar phenotype was observed in knockdown experiments using morpholinos targeting the AUG region of pnp4a (Supplementary Fig. 10), thus confirming the specificity of the pnp4a mutations. Intriguingly, crystals produced by both pnp4a−/− mutants and morpholino-injected fish displayed a distinct square-like morphology, compared with the typical elongated semihexagonal shape observed in the eye of WT fish (Fig. 5 and Supplementary Fig. 10).

a,b, WT (a) and pnp4acbg20 mutant (b) larvae iridescence phenotypes. Larval gross morphology at 72 hpf; eyes with iridophores labeled (green, transgenic TDL358:GFP) and crystal reflection (gray); zoomed-in view of crystal morphology in WT and mutant conditions. Note the reduced crystal formation and altered morphology compared with the WT control (insets in a‴ and b‴). Scale bars: 500 μm (a,b), 50 μm (a′,b′,a″,b″), 10 μm (a‴,b‴) and 5 μm (insets). c, Schematic representation of the CRISPR–Cas9-mediated knockout strategy targeting the pnp4a gene. Illustration created with BioRender.com. In detail, the pnp4acbg20 mutant results in a 6-nt deletion and 1-nt insertion, leading to a 5-nt frameshift and truncated protein resulting in 150 aa instead of 291 aa. The pnp4awz19 mutant results in a 10-nt deletion and truncated protein resulting in 142 aa. d, Comparison of the average iridophore reflection per 50 μm in different WT, mutant and rescue conditions. au, arbitrary units. WT control animals were subjected to the same treatment as the pnp4acbg20 rescue condition (Methods). The mean ± s.e.m. are shown for independent biological replicates; WT, n = 7; WT control, n = 12; pnp4acbg20, n = 16; pnp4acbg20 rescue, n = 20 (two-tailed, unpaired, non-parametric Mann–Whitney test; ****P < 0.0001, **P < 0.01 and *P < 0.05). e, RT–qPCR analysis of pnp5a and pnp5b mRNA expression levels from iridophores of WT (pnp5a, n = 5; pnp5b, n = 5) or pnp4awz19 mutants (pnp5a, n = 4; pnp5b, n = 5). RNA was extracted at 6 dpf. The mean ± s.d. are shown for independent biological replicates (two-tailed, unpaired Student’s t-test; ****P < 0.0001 and ***P < 0.001).
To better understand the effect of Pnp4a on crystal morphogenesis, we conducted structural analyses on crystals from mutant and WT fish. Despite the differences in pnp4a−/− mutant crystal morphology, micro-Raman spectroscopy and electron diffraction confirmed that these crystals were still β-anhydrous guanine (Supplementary Fig. 11a–e). However, TEM imaging unveiled distinct characteristics in the pnp4a−/− mutant crystals, displaying strongly overdeveloped (012) facets alongside minimal, barely visible (010) facets (Fig. 5a and Supplementary Fig. 11a–d). TEM imaging of the mutant larvae revealed multiple small, underdeveloped iridosomes and cryo-scanning electron microscopy imaging of these larvae showed thin, plate-like crystals similar to those found in the WT (Fig. 5a and Supplementary Fig. 11f,h).
To gain deeper insights into the specific contribution of Pnp4a to crystal formation, we conducted rescue experiments and restored pnp4a expression in the null mutants. The rescued embryos exhibited elevated crystal quantities compared with the control group, confirming that the observed phenotype was specific to the perturbation of Pnp4a (Fig. 5d). Interestingly, the square-like crystal morphology remained unchanged in the rescued mutant larvae (Fig. 5a–c). To determine whether other members of the PNPase family contribute to crystal morphology, we performed morpholino knockdown experiments targeting each enzyme (Supplementary Fig. 10). Interestingly, the knockdown of pnp4b, pnp5a, pnp5b and pnp6 did not notably affect the crystal quantities or morphology (Supplementary Fig. 10).
Given the selective substrate affinity of Pnp4a, we hypothesized that the distinctive morphological alterations observed in pnp4a−/− mutant crystals might be linked to changes in their guanine-to-hypoxanthine ratio. Thus, we conducted a direct comparison of the composition between WT and pnp4a−/− mutant crystals in situ, using Orbitrap-SIMS imaging (Methods). The results showed that hypoxanthine-to-guanine ratios in the mutant crystals were almost two times higher than in the WT. This notable shift in composition mirrors our findings in both naturally occurring and in vitro formed crystals, further establishing the correlation between crystal composition and morphology (Supplementary Fig. 12).
PNPase expression regulates crystal morphogenesis in tissues
A surprising result of these experiments was that, although Pnp4a is recognized as the primary enzyme in guanine biosynthesis, crystal formation occurred in its absence. Furthermore, adult pnp4a−/− mutant fish eventually displayed nearly normal stripe patterns and eye iridescence (Supplementary Fig. 13). To delve deeper into the intricacies of guanine crystal formation and compensatory mechanisms, we conducted reverse transcription (RT)–qPCR to evaluate mRNA levels of other PNPase family members in both pnp4a−/− mutants and WT fish at 6 dpf (Fig. 5e and Supplementary Fig. 14). While pnp4a expression levels were reduced compared with the WT control, the levels of its paralogs, pnp5a and pnp5b, showed a significant increase in the mutants (Fig. 5e and Supplementary Fig. 14). This suggests a cellular adaptive response or compensatory mechanism at play, where the absence of Pnp4a triggers the upregulation of these closely related family members. Notably, the efficiency of Pnp5a and Pnp5b in converting inosine to hypoxanthine (Fig. 4b,c) offers a plausible explanation for the considerable elevation in intracrystalline levels of hypoxanthine. This intricate interplay between PNPase paralogs not only sheds light on the compensatory processes triggered by the absence of Pnp4a but also underscores the multifaceted regulatory role of PNPases in determining crystal composition and morphology.
The observed variations in the expression pattern of the PNPase family in mutant fish prompted us to hypothesize that similar fluctuations in expression levels might underlie the distinct composition of naturally occurring crystals in WT fish. Our investigation indeed substantiated this hypothesis, revealing that, while pnp4a expression levels in the operculum and skin were comparable, the expression levels of pnp5a and pnp5b were significantly higher in the skin. This coincided with the heightened intracrystalline levels of hypoxanthine (Fig. 6 and Supplementary Fig. 15). This important finding suggests that organisms intricately regulate crystal morphogenesis by fine-tuning the expression levels of biosynthetic enzymes, thereby modulating crystal composition.

A schematic illustration showing the coupling of the expression levels and selectivity of PNPases to crystal composition and subsequent morphology. Morphogenesis relies on the development of paralogous enzymes with exceptional substrate selectivity. The tissue-specific expression patterns of these enzymes yield varied substrate compositions that are instrumental in forming crystals with distinct compositions, directly influencing their morphology and, consequently, their optical characteristics.
Lastly, to determine the onset of tissue-specific expression differences among paralogs, we used RT–qPCR to analyze the expression levels of pnp4a, pnp5a and pnp5b at two developmental stages, 3 and 8 dpf, within the head and trunk regions. At 3 dpf, we observed similar expression patterns in the trunk and head, with a notable upregulation of pnp4a relative to pnp5a and pnp5b (Supplementary Fig. 16a,b). Contrastingly, at 8 dpf, we detected a significant upregulation of pnp5a compared with pnp4a exclusively in the trunk (Supplementary Fig. 16c,d). These results indicate that the differentiation in expression among these paralogs emerges as early as 8 dpf.
Discussion
To foster a range of functions critical for survival such as vision, camouflage, communication and thermoregulation, organisms have had to adapt and evolve mechanisms to control and fine-tune the properties of biogenic crystals. Here, we show that the complex biochemical process of crystal morphogenesis depends on a series of sequential regulatory steps, starting with the development of paralogous enzymes characterized by high substrate selectivity. Thus, the tissue-specific expression of these enzymes results in a variety of substrate combinations within different iridophores. This in turn gives rise to crystals with distinct compositions, which is instrumental in shaping crystal morphology and, consequently, in enabling tissue-specific optical characteristics.
Contrary to the prevailing notion that hypoxanthine is merely an economical building block28, our work demonstrates that the guanine-to-hypoxanthine ratio has a pivotal role in defining crystal properties. We first show that zebrafish crystals differ morphologically, depending on their tissue of origin and hypoxanthine content. Crystals from skin, which align into arrays for coloration, display lower aspect ratios and a higher hypoxanthine content than those from the operculum, whose disorganized structure contributes to a silvery appearance. This implies that zebrafish finely tune crystal composition and morphology for optimized functionality. In vitro crystallization assays corroborated the pivotal role of guanine-to-hypoxanthine ratio in defining crystal properties. We further demonstrated that the incorporation of hypoxanthine resulted in the overdevelopment of (012) facets and the underdevelopment of (010) facets both in vitro and in vivo. Considering that the expression of a crystallographic facet is inversely proportional to its growth rate54, this suggests that hypoxanthine selectively hinders growth along the (012) plane. A plausible mechanism for this could be the preferential adsorption of hypoxanthine molecules onto the (012) facets. Such an adsorption process could hinder further growth in the perpendicular direction by reducing the attachment energy of the next guanine molecules because of the lower number of hydrogen bond donors in the hypoxanthine molecules compared with guanine55,56.
This study also addresses the mechanism behind tissue-specific regulation of crystal composition. By applying a multidisciplinary approach integrating proteomics, metabolomics, electron microscopy, biochemical assays, cellular studies and genetical manipulations, we identified the biosynthetic pathways in iridophores, revealing a sophisticated regulatory network. This network modulates enzyme activity to manage elevated guanine levels while avoiding the accumulation of toxic metabolites. Additionally, our research contributes to the understanding of the evolutionary occurrence of guanine–hypoxanthine cocrystals in various species. Because the enzymes responsible for the synthesis of guanine and hypoxanthine from IMP (that is, nucleotidase and PNPase) are shared, the upregulation of guanine production also leads to an increase in hypoxanthine levels, possibly leading to the formation of these cocrystals.
Particularly noteworthy is the elevation of a distinct subset of paralogous enzymes uniquely expressed in crystal-forming cells. Our study highlights the pivotal role of the PNPase family and, particularly, Pnp4a in nucleobase biosynthesis and crystal formation within iridophores. Furthermore, although the differentiation and development of iridophores appeared minimally affected in pnp4a−/− mutant larvae, they produced fewer crystals in their eyes and skin. The residual crystal formation and normal patterning in adult mutants suggest compensatory mechanisms by other PNPase family members, as evidenced by the increased expression of pnp5a and pnp5b in these mutants.
Lastly, we found that differential substrate selectivity among PNPase family members leads to nucleobase diversity within iridophores. Comparing the sequence of these enzymes showed that several amino acid substitutions, some of them within the active site, may account for the differences in substrate selectivity we observed. The precise, tissue-specific expression of PNPase variants fosters a diverse array of substrate compositions. This diversity can be seen in the different guanine-to-hypoxanthine ratios observed in crystals from different WT tissues and, even more pronounced, in the unique square and hypoxanthine-enriched crystals of the pnp4a−/− mutants.
In conclusion, our findings propose a mechanism governing the emergence of diverse biogenic crystal morphologies, each uniquely adapted to fulfill specific biological functions. By elucidating these intricate processes, our work not only contributes to the understanding of fundamental biological phenomena but also paves the way for potential applications in diverse fields, from genetically designed biomaterials to medical therapeutics.
Methods
Ethics and safety statement
This study followed the Weizmann Institute of Science Animal Care and Use Committee (protocol number 02000223-1) and was conducted according to the guidelines of the European Union directives (2010/63/EU) and German law, with project license 25-5131/522/54 (TVV52/2021). Genetic engineering work was carried out in an S1 area, following regulations from the German Genetic Engineering Act and German Genetic Engineering Safety Ordinance (license no. Az.: 54-8451/103).
Zebrafish husbandry and handling
Zebrafish (D. rerio) were housed at ∼28 °C in 14-h light, 10-h dark conditions and fed with Artemia and flake food. Fish were maintained and fed following the standard protocols. Crosses were performed with 3–12-month old WT AB strain adults. Embryos were kept in E3 zebrafish embryo medium at 28.5 °C until reaching the desired developmental stage. Published lines used were as follows: Et(Ola.Edar:GAL4,14xUAS:GFP)TDL358Et (RRID: ZDB-FISH-150901-2380)57, denominated TDL358 throughout the paper for simplicity, and Tg(pnp4a:PALM-mCherry)wprt10Tg (RRID: ZDB-FISH-210414-18)58.
Cell dissociation, isolation and FACS
Following the protocol adapted from ref. 12, fish were anesthetized with Tricaine and immersed in TrypLE Express (Invitrogen, 12604039) with 200 μg ml−1 Liberase TL Research Grade (Roche, 05401020001). Fish were incubated at 37 °C and with gentle shaking at 200 r.p.m. for 40 min, followed by mechanical disruption to further dissociate the cells. The resulting cell suspension was then strained through a 40-µm cell strainer with HL-15 buffer (Hank’s balanced salt solution 40% (Sigma, H8264) and 60% Leibovitz’s L-15 medium (Gibco, 21083-027)) and centrifuged. The dissociated cells were then centrifuged at 1,000 r.c.f. for 10 min at 4 °C and then resuspended in 5 ml of fresh HL-15 for FACS. Cells were isolated from Tg(pnp4a:PALM-mCherry)-positive fish as in ref. 12. Following resuspension in 5 ml of cold HL-15, the isolated cells were incubated with Hoechst to mark the nuclei for 30 min before FACS. Cells were analyzed and sorted using a BD FACSAria III Cell Sorter with a 100-µm nozzle. Cells were gated on the basis of size and fluorescence attributes to exclude debris and non-target cells. Forward scatter and side scatter, along with Hoechst signals, were used to filter out particles smaller than 1 mm. The target cells expressing the pnp4a marker were enriched using the 561-nm laser. Sorted cells were collected in ice-cold PBS and kept at 4 °C. After sorting, the cells were centrifuged at 1,000 r.c.f. for 5 min; the pellet was flash-frozen in liquid nitrogen and stored at −80 °C until further proteomics analysis.
Proteomics sample preparation
Cell pellets were lysed with 5% SDS in 50 mM Tris-HCl. Lysates were incubated at 96 °C for 5 min, followed by six cycles of 30-s sonication (Bioruptor Pico, Diagenode). Protein concentration was measured using the BCA assay (Thermo Fisher Scientific) and 7.3 μg of protein was reduced with 5 mM dithiothreitol and alkylated with 10 mM iodoacetamide in the dark. Each sample was loaded onto S-Trap microcolumns (Protifi) according to the manufacturer’s instructions. In brief, after loading, samples were washed with a 9:1 ratio of methanol and 50 mM ammonium bicarbonate. Samples were then digested with trypsin (1:50 trypsin to protein) for 1.5 h at 47 °C. The digested peptides were eluted using 50 mM ammonium bicarbonate; trypsin was added to this fraction and incubated overnight at 37 °C. Two more elutions were made using 0.2% formic acid and 0.2% formic acid in 50% acetonitrile. The three elutions were pooled together and vacuum-centrifuged to dry. Samples were kept at −20 °C until analysis59.
LC
Ultraperformance (UP)LC–MS-grade solvents were used for all chromatographic steps. Each sample was loaded using splitless nanoUPLC (10 kpsi nanoAcquity, Waters). Mobile phase A was H2O + 0.1% formic acid and mobile phase B was acetonitrile + 0.1% formic acid. Desalting of the samples was performed online using a reversed-phase Symmetry C18 trapping column (180 µm internal diameter, 20 mm length and 5 µm particle size; Waters). The peptides were then separated using a T3 HSS nanocolumn (75 µm internal diameter, 250 mm length and 1.8 µm particle size; Waters) at 0.35 µl min−1. Peptides were eluted from the column into the MS instrument using the following gradient: 4% to 30% B in 105 min, 30% to 90% B in 10 min, maintained at 90% for 7 min, and then back to initial conditions.
MS
The nanoUPLC was coupled online through a nanoESI emitter (10-μm tip; New Objective) to a quadrupole orbitrap MS instrument (Exploris 480, Thermo Fisher Scientific) using a FlexIon nanospray apparatus (Proxeon). Data were acquired in data-dependent acquisition mode using a 2-s cycle time. The MS1 resolution was set to 120,000 (at 200 m/z), with a mass range of 380–1,500 m/z, 200% automatic gain control (AGC) and a maximum injection time of 50 ms. The MS2 resolution was set to 15,000, with quadrupole isolation at 1.4 m/z, 75% AGC, a dynamic exclusion of 30 s and maximum injection time set to auto.
Data processing for MS
Raw data were processed with MetaMorpheus (version 0.0.320). The data were searched against the D. rerio UniProt proteome database (appended with common lab protein contaminants and the following modifications: carbamidomethylation of C as a fixed modification and oxidation of M and protein N-terminal acetylation as variable modifications. Quantification was performed using the embedded FlashLFQ60 and protein inference61 algorithms. The label-free quantitation (LFQ) intensities were imported into Perseus (version 1.6.2.3)62. Decoy hits were removed and the LFQ intensities were log2-transformed. The data were filtered to include only proteins that had at least two valid values in at least one experimental group. The remaining missing values were randomly imputed and a Student’s t-test was used to detect proteins that were significantly differentially expressed.
Phylogenetic analysis of the vertebrates PNPase protein family
The orthologous genes were identified and their associated protein sequences was extracted Ensembl, using the canonical transcript for each gene. Multiple alignment was performed with the Clustal method in MEGA11. The tree was constructed using the maximum likelihood method and Jones–Taylor–Thornton matrix-based model with MEGA11 (ref. 63). The numbers indicate the percentage of replicate trees in which the associated clades clustered together in a bootstrap test (100 replicates). The phylogenetic clades are highlighted in the background, with each clade’s phylogenetic class indicated by a specific color code: orange for fishes, green for amphibians, blue for birds, gray for reptiles and pink for mammals. Species used were as follows: Atlantic Salmon, Atlantic cod, green anole, painted turtle, eastern brown snake, African clawed frog and medaka. The methylthioadenosine phosphorylase (MTAP) protein served as an outgroup and was scaled outside of the figure.
pnp4a cloning and transgenic generation
The pnp4a (ID: ENSDARG00000057575) complementary DNA (cDNA) was amplified from 5-dpf zebrafish total cDNA together with primers containing AscI and FseI digestion sites at their 5′ and 3′ ends, respectively (forward, 5′-ATGGCGCGCCCATGCATAGTAAAGACC-3′; reverse, 5′ ATATGGCCGGCCTAATGCCGTGTTGTTGTTGATGTC-3′). The 876-bp amplified fragment was purified using a gel extraction kit (Qiagen, 28706), digested with restriction enzymes AscI and FseI (New England Biolabs) and cloned into pCS2+8CmCherry (Addgene, 34935), previously digested with the same restriction enzymes and treated with calf intestinal alkaline phosphatase (New England Biolabs, M0290S). For Tg(hsp70:Pnp4a-mkate2)cbg19Tg generation, Gateway technology was used. A pnp4a middle entry clone was generated by amplifying pnp4a from the pCS2+pnp4a-mCherry construct with primers containing attB1 and attB2 sites. (forward, 5′GGGGACAAGTTTGTACAAAAAAGCAGGCTTAATGCATAGTAAAGACCAAATCTGCCAT-3′; reverse, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTTAATGCCGTGTTG TTGTTGATGTCCATGC-3′).
The 937-bp amplified fragment was cleaned using a PCR clean up kit (Promega, A9281) and recombined with pDONOR221 (plasmid 208 from the Tol2 Kit) in a BP reaction (BP Clonase II enzyme mix; Invitrogen, 11789020). The generated middle entry clone pME-pnp4a was then recombined in a LR reaction using pDEST-Tol2pA (plasmid 426 from the Tol2 Kit), p5E- hsp70 (plasmid 222 from the Tol2 kit) and p3E-mKate2 (gift from the Oates Lab) (LR Clonase II Plus enzyme; Invitrogen, 12538120). One-cell-stage TDL358 transgenic embryos were injected with 25 pg of transposase mRNA and 25 pg of the generated hsp70:Pnp4a-mKate2 Tol2 construct. The embryos were then grown for 3 months. Afterward, F0 fish were crossed with WT AB. The obtained embryos were heat-shocked at 24 hpf for 1 h at 37 °C and screened for red fluorescence with an Olympus SZX16 fluorescence microscope. Positive F1 fish in the TDL358 background were selected and raised.
pnp4a cbg20 and pnp4a wz19 CRISPR mutant and rescue
The pnp4acbg20 and pnp4awz19 CRISPR mutants were generated as described previously64. Briefly, trans-activating crispr RNAs (crRNAs) were designed for specific loci of the pnp4a gene using the predesigned crRNAs dataset from Integrated DNA Technologies (IDT). Several crRNAs were tested for ribonucleoprotein (RNP) mutagenesis and were chosen considering where the start codon is located, as well as high on-target and low off-target scores. The most efficient crRNA tested was located on exon 4 of the pnp4a canonical transcript, with target sequence 5′-CGTGACCACATTAACTTCCC-3′ (Dr.Cas9.PNP4A.1.AB, IDT). Each tested crRNA was separately annealed with an equal molar amount of tracrRNA (1072533, IDT) and diluted to 57 μM in Duplex buffer (11-01-03-01, IDT), generating the single guide RNA (sgRNA). The RNP mixes were assembled using Cas9 protein (Alt-R S.p. Cas9 Nuclease V3; 1081058, IDT; 61 μM stock) and an sgRNA, in equimolar amounts, generating a 28.5 μM RNP solution. To improve mutagenesis efficiency, the mixes were kept overnight at −20 °C before injections the following day. One-cell-stage WT AB or TDL358;hsp70:Pnp4a-mKate2 double transgenic embryos were injected with 1 nl of each of the RNP mixes. To screen for the efficiency of RNP mutagenesis and potential genotype-to-phenotype relationships, ten embryos of each RNP-injected condition were individually genotyped and reflection phenotypes were assessed using incident light from a stereoscope (Leica M165C) at 3 dpf, with fish partially grown to adulthood. Founder fish containing frameshift mutations were identified by genotyping the resulting F1 progeny, resulting from outcrosses with WT AB fish. Two mutant lines were generated: (1) the pnp4acbg20 resulting in a 6-nt deletion and 1-nt insertion, leading to an overall 5-nt frameshift and truncated protein, resulting in 150 aa instead of 291 aa, on the background of TDL358;hsp70:Pnp4a-mKate2 and (2) the pnp4awz19 resulting in a 10-nt frameshift and truncated protein, resulting in 142 aa on the background of WT AB. For rescue experiments, 400 pg of full-length pnp4a-mCherry mRNA was injected at the one-cell stage into in-crossed heterozygous pnp4acbg20;TDL358;hsp70:Pnp4a-mKate2 mutant embryos. Then, injected embryos were heat-activated at 36, 48 and 56 hpf in a water bath at 37 °C for 1 h (petri dishes containing 30 ml of embryo medium), subsequently kept at 28.5 °C, selected for fluorescence and finally imaged at 72 hpf. After imaging, genotyping was performed in individual larvae, as described below. The mRNA synthesis details are also described below.
Genotyping
Genomic DNA from individual larvae was extracted using the Kapa Express Extract kit (Kapa Biosystems) according to the manufacturer’s protocol. This was followed by performing PCR with KAPA2G Robust HotStart ReadyMix (Kapa Biosystems) with primers surrounding the pnp4a mutation region (forward, 5′-CAGAATTTGTGCTTGTGTTC3′; reverse, 5′-CCTTGTACTGGTGATTGTAATG-3′). As these are located in intron regions, amplicons were specific to the mutation and not the transgenes present. Then, PCR products were sent for sequencing. Sequences were analyzed using Snapgene software.
Microinjection of embryos with mRNAs and morpholinos
One-cell-stage WT AB or TDL358;hsp70:Pnp4a-mkate2 embryos were injected using standard procedures with different pnp4a mRNA concentrations. This was generated by linearization of the pCS2+Pnp4a-mCherry vector with NotI (New England Biolabs) and transcription using the SP6 mMESSAGE mMACHINE high-yield capped RNA transcription kit (AM1340, Ambion), following the manufacturer’s protocol. The same procedure was applied for Tol2 transposase mRNA synthesis, generated from a pCS2FA-transposase plasmid65. mRNAs were aliquoted and stored at −80 °C until use. Embryos were left to develop at 28.5 °C until the desired stage and then live-imaged or grown to adulthood. A PV-820 Pico-injector (World Precision Instruments) and a Narashige micromanipulator were used for microinjection.
Gene knockdown experiments were performed using the following morpholino-modified antisense oligonucleotides (MO, Gene Tools):
pnp4a, 5′ GGCAGATTTGGTCTTTACTATGCAT 3′;
pnp4b, 5′ CATGATCTCGGGTAATCACAGGCTG 3′;
pnp5a, 5′ GGGAAACATGATTCAAGTCGCTGCT 3′;
pnp5b, 5′ ATAGAGGTGAAACCTGCCCTGCATG 3′;
pnp6, 5′GCTGGTCGAATTACTGAACGTGATA 3′;
control morpholino, 5′ CCTCTTACCTCAGTTACAATTTATA 3′ (Gene Tools stock control).
Each oligonucleotide was diluted with ultrapure (Milli-Q) water to 1 mM and 1–2 pmol of MO was injected into one-cell-stage embryos.
Live imaging and mounting
Confocal live imaging was performed using an upright Zeiss LSM880 confocal microscope, with a C-Apochromat ×40 water-dipping objective. Briefly, at 72 hpf, larvae were anesthetized with 0.1% MS-222 (Sigma) diluted in embryo medium and then mounted on a concave slide (Sigma-Aldrich, BR475505) with 0.5% low-melting-point agarose (Sigma A9414) in E3 fish medium.
Iridophore area and reflection quantification
After image acquisition, maximum intensity z-projections (MIPs) of all acquired signals were obtained using Fiji66. The obtained images from the green fluorescent protein channel, corresponding to cytoplasmic signal from the TDL358-positive cells (iridophores), were segmented using Ilastik67 using the pixel classification method. This provided a binary mask that labeled the contour of all iridophores in the samples. The mask was used to define the regions of interest (ROIs) within the MIPs of other obtained channels, allowing measurements of the corresponding reflectance within iridophores and the iridophore area for each sample using a custom-made Fiji macro. Individual sample reflection was normalized by dividing the reflectance signal per corresponding iridophore area, allowing statistical comparisons between samples.
RT–qPCR
The relative mRNA levels of pnp4a (NM_001002102.1), pnp4b (NM_205643.1), pnp5a (NM_213311.2), pnp5b (NM_001004628.1) and pnp6 (NM_205655.2) were determined using RT–qPCR.
Total mRNA was extracted using the NucleoSpin RNA II Mini Spin Kit (740955, Macherey-Nagel) according to the manufacturer’s instructions from the following samples:
-
1.
Larvae at 6 dpf (15 larvae per sample) from three groups of WT and three groups of pnp4awz19 mutant.
-
2.
Dissected tissue from the skin and operculum of an adult zebrafish.
-
3.
Head and trunk from 3-dpf and 8-dpf larvae.
A 0.5-μg sample of mRNA was reverse-transcribed using the qScript cDNA synthesis kit (95047-100, Quanta BioSciences). Relative transcript levels were determined by the QuantStudio 3 real-time PCR system (Thermo Fisher Scientific). Triplicates of each cDNA sample were PCR-amplified using the Fast SYBR green master mix (4385612, Thermo Fisher Scientific) and the following specific primers were used: pnp4a, 5′-TCATTTGTGGCTCTGGACTG-3′ and 5′-TTTTCCCTTTGAGCTCCCC-3′; pnp4b, 5′-TGAAGGGTGTCTGGTATTTGG-3′ and 5′-TGTCTCCCACTTTGAAATCCTG-3′; pnp5a, 5′-ACATTCCTAACTTTCCCCAGAG-3′ and 5′-CCCAGCATCTTGAAAATCCG-3′; pnp5b, 5′-CTAGAGATTGAGGCATGTCAGG-3′ and 5′-GCCATTTCACTCTTCATTGTCC-3′; pnp6, 5′-CAAAATCCCACGCTTTCCAC-3′ and 5′-GGTCACAATCAAAGTCTCAATTCC-3′. The relative quantification of gene expression levels was normalized against rpl13 (NM_198143.1) in all assays with the rpl13 primers 5′-AGCTCAAGATGGCAACAG-3′ and 5′- AAGTTCTTCTCGTCCTCC-3′ and ΔΔCt analysis68. The rpl13 gene was selected because its relative expression did not significantly change between pnp4awz19 mutants and WT during the various developmental stages69. Gene levels were normalized by dividing the absolute levels of each sample by the average of all WT samples.
MALDI imaging
MALDI imaging analysis and data processing were performed as described previously46 using PICA (pixel intensity correlation analysis for deconvolution and metabolite identification in MS imaging)70. In brief, zebrafish at different developmental stages were embedded with M1 embedding matrix (Thermo Fisher Scientific) in Peel-A-Way disposable embedding molds (Peel-A-Way Scientific). The embedded tissues were transferred to a cryostat (Leica CM3050) and allowed to thermally equilibrate at −18 °C for at least 2 h. The frozen tissues were cut into 70–100-μm-thick sections. The sections were then thaw-mounted onto Superfrost Plus slides (Thermo Fisher Scientific) and vacuum-dried in a desiccator. TM sprayer (HTX Technologies) was used to coat the slide with DHB matrix (40 mg ml−1 in 30:70 water–methanol (v/v) containing 0.2% trifluoroacetic acid). The nozzle temperature was set at 70 °C and the DHB matrix solution was sprayed in 16 passes over the tissue sections at a linear velocity of 120 cm min−1 with a flow rate of 50 μl min−1. MALDI imaging measurements were performed using a 7-T Solarix FT-ICR (Fourier-transform ion cyclotron resonance) mass spectrometer (Bruker Daltonics). MSI datasets were collected with 10-μm spatial resolution in positive ion mode using lock mass calibration (DHB matrix peak: [3DHB + H-3H2O]+, m/z 409.0554) at a frequency of 1 kHz and a laser power of 50%, with 100 laser shots per pixel. Unless stated otherwise, each mass spectrum was recorded in the range of 118–1,000 m/z in broadband mode with a time domain for acquisition of 1 m, providing an estimated resolving power of 115,000 at 400 m/z. Raw data were converted into the imzML format using FlexImaging software (version 5.0; Bruker Daltonics). MALDI images were plotted with the R package Cardinal71 with a mass bin width of ±0.001 Da. Images were optimized with Gaussian smoothing and contrast enhancement as previously described.
Orbitrap-SIMS imaging
Cryosections, prepared as described above were analyzed using the M6 Hybrid SIMS instrument (IONTOF) using a top-mount sample holder. To detect, assign and determine the distribution of guanine and hypoxanthine, sample areas (field of view, 400 × 400 µm; raster size, 200 × 200 pixels) were analyzed with a 20-keV AR1610 gas cluster ion beam in positive ionization mode (three replicates of WT at 10 dpf and three replicates of pnp4awz19 mutants at 10 dpf). Ion transfer was performed with a He collision cell pressure of 5 × 10−2 bar and radiofrequency (RF) amplitude of 70%. The primary ion current was 27 pA. For detection, the Orbitrap HF high-resolution mass analyzer (mass range, 50–750 m/z; mass resolution, 120.000 at 200 m/z; injection time, 261 ms) was used. Because the sample setup on glass slides resembles an insulator, all measurements were performed with charge compensation and Ar gas flooding of the main chamber (surface potential, −40 V; floodgun, on; Pmain = 9 × 10−7 mbar). Data analysis was performed with Surfacelab 7.3 software (IONTOF). To determine the ratios of guanine and hypoxanthine, five ROIs per imaging run were selected on the basis of the highest relative intensity of guanine [M + H+]. Mass peaks of guanine ([M + H+], m/z 152.056; [M + Na+], m/z 174.038) and hypoxanthine ([M + H+], m/z 137.046; no sodium adduct observed) were integrated and intensity values were used to calculate the ratio of the two molecules ((Iguanine + H+ + Iguanine + Na+)/Ihypoxanthine + H+) for each individual ROI.
Orbitrap-SIMS spectrometry
Isolated crystals from operculum and skin tissues were fixed onto glass slides using double-sided tape and analyzed using the M6 Hybrid SIMS instrument (IONTOF) using a top-mount sample holder. Five random areas per sample (480 × 480 µm) were analyzed with a 20-keV AR1610 gas cluster ion beam in positive ionization mode with a primary ion current of 26.5 pA and a spot size of 2 µm. Ion transfer was performed with a He collision cell pressure of 5 × 10−2 bar and RF amplitude of 70%. For detection, the Orbitrap HF high-resolution mass analyzer (mass range, 50–750 m/z; mass resolution, 240.000 at 200 m/z; injection time, 512 ms) was used. For each measurement, 600 scans were summed up. The floodgun was on, the surface potential was set to −40 V and the main chamber was flooded with Ar gas and regulated to Pmain = 9 × 10−7 mbar. Mass signals of guanine ([M + H+], m/z 152.056; [M + Na+], m/z 174.038) and hypoxanthine ([M + H+], m/z 137.046; no sodium adduct observed) were integrated and intensity values were used to calculate the ratio of the two molecules ((Iguanine + H+ + Iguanine + Na+)/Ihypoxanthine + H+) for each individual measurement.
Cryo-electron tomography
Sorted iridophore cells (3.5 μl) with 15-nm gold beads (1 μl) were applied to glow-discharged holey carbon R2/2 Cu 200 SiO2 mesh grids (Quantifoil) coated with collagen, Type I, rat tail (EMD Millipore, 08-115) for cell adherence. The grids were blotted and vitrified by plunging into liquid ethane using a Leica EM GP automatic plunger at 4 °C and 90% humidity. Frozen grids were kept in liquid nitrogen until used. Data were collected on a Titan Krios TEM G3i (Thermo Fisher Scientific) equipped with a BioQuantum energy filter and a K3 direct electron detector (Gatan). Datasets were collected at 300 kV with the K3 camera (counting mode) using SerialEM software72. The TEM magnification corresponded to a camera pixel size of 1.6 Å and the target defocus was set to 3 μm. The total dose for a full tilt series was 120 electrons per Å2. Tomogram tilt series were collected using the dose-symmetric scheme, ±60° at 2° steps. The tilt series images alignment and reconstruction were performed in IMOD73.
In vitro crystallization
Crystals were produced by customizing a protocol described previously74. Briefly, the following solutions were prepared: (1) 1 mg of PVP-co-VA (Mw 50,000; 190845-250G, Sigma-Aldrich) in 10 ml of formamide (F7503, Sigma-Aldrich); (2) 4.5 mg of guanine powder (G6779, Sigma-Aldrich) in 1 ml of 0.4 M NaOH solution (CAS 1310-73-2, BioLab); (3) 1 mg of adenine powder (A8626, Sigma-Aldrich) in 1.5 ml of distilled water; and (4) different percentages of hypoxanthine powder (0%–30% (w/w); H9377, Sigma-Aldrich) dissolved in 1.5 ml of distilled water. Guanine solution (2) was then filtered using a PVDF filter (0.22 µm; SLGV033RS, Merck Millipore) and adenine (3) and hypoxanthine (4) solutions were filtered using Whatman filters (0.2 µm; 10462200, Cytiva). Guanine crystals were produced by adding solutions (3) and (4) into solution (1) and mixing, followed by the addition of solution (2). The obtained suspensions were collected and filtered after 24 h.
Chemical fixation and conventional TEM
Samples (whole larvae) were fixed with 4% paraformaldehyde and 2% glutaraldehyde in 0.1 M cacodylate buffer containing 5 mM CaCl2 (pH 7.4) for 1 h, postfixed in 1% osmium tetroxide supplemented with 0.5% potassium hexacyanoferrate tryhidrate and potassium dichromate in 0.1 M cacodylate for 1 h, stained with 2% uranyl acetate in double-distilled water for 1 h, dehydrated in graded ethanol solutions and embedded in epoxy resin. Ultrathin sections (70–90 nm) were obtained with a Leica EMUC7 ultramicrotome and transferred to Formvar support film slot grids (EMS). Grids were stained with lead citrate and examined with a Tecnai SPIRIT TEM instrument (Thermo Fisher Scientific). Digital electron micrographs were acquired with a bottom-mounted Gatan OneView camera.
TEM imaging and electron diffractions of in vitro formed and isolated crystals
Crystals were placed on top of a copper-carbon coated TEM grid, allowing the crystals to dry. Samples were observed on a Tecnai T12 TEM instrument (Thermo Fisher Scientific) operated at 120 kV. Images and diffraction patterns were recorded on a bottom-mounted TVIPS TemCam-XF416 4k × 4k complementary metal–oxide–semiconductor using imaging and diffraction modes.
HPLC
HPLC analysis was carried out by customizing a protocol described previously28. Ammonium formate buffer (10 mM, pH 3.7) was prepared by dissolving 630 mg of ammonium formate (70221-25G-F, Sigma-Aldrich) in 1 L of double-distilled water and used as mobile phase A. Next, the pH was adjusted using formic acid (~200 µl; 27001-1L-R, Sigma-Aldrich). Acid hydrolysis of the in vitro, biogenic and standard samples was carried out before the analysis. For in vitro samples, 1 mg of crystals were vortexed with 500 µl of perchloric acid (244252-1L, Sigma-Aldrich) at room temperature. For the biogenic crystals, the pellet was dissolved in 120 µl of perchloric acid. For standard samples, 90 mg of guanine, 4.5 mg of hypoxanthine and 1 mg of adenine were dissolved in 90 ml of perchloric acid. Ultrasonication for 15 min at room temperature was applied to all samples. All sample dilutions were performed with ammonium formate buffer. Standard samples of guanine, hypoxanthine and adenine were diluted 5–700 times for the calibration curves. Biogenic and in vitro samples were diluted 20:1. All the samples were filtered with a PVDF filter (0.22 µm; SLGV033RS, Merck Millipore) before injection into the HPLC system. All analytical HPLC measurements were carried out on an Agilent 1260 Infinity HPLC system (Agilent Technologies) using an Agilent Zorbax Eclipse plus column (250 mm × 4.5 mm, 5 μm). The optimum separation was obtained under gradient elution with phase A (ammonium formate buffer, 10 mM, pH 3.7) and phase B (methanol; 1368350100, BioLab). The following gradient elution was carried out: 0–10 min, 1% B; 10–13 min, linear gradient 80% B; 13–19 min, hold at 80% B; 19–21 min, linear gradient 1% B; 21–30 min, hold at 1% B. The total run time was 30 min. The flow rate was 1 ml min−1, the injected volume was 10 μl and the column temperature was maintained at 30 °C. The ultraviolet detection wavelength used for analysis was 260 nm. The retention time was 7.22 ± 0.06 min for guanine, 7.73 ± 0.07 min for hypoxanthine and 9.29 ± 0.09 min for adenine. The hypoxanthine levels within the crystals were quantified by peak integration using standard calibration curves: guanine, y = 27,299x (R2 = 0.9995); hypoxanthine, y = 32,318x (R2 = 0.9987); adenine, y = 53,529x (R2 = 0.9991).
Micro-Raman spectroscopy
Micro-Raman spectroscopy (λ = 532 nm, excitation) was collected using a Horiba LabRAM HR Evolution spectrometer (Horiba) equipped with four laser lines (325 nm, 532 nm, 633 nm and 785 nm). The system had an 800-mm focal length spectrograph for high resolution and low stray light, with several interchangeable gratings, and it was mounted with an open-electrode, front-illuminated, cooled charge-coupled device detector. The sample was placed under a modular microscope (Olympus BX-FM) with a suitable objective. For this study, LUMPlanFL N ×60 (1.0 numerical aperture (NA)) WI and MPlanFL N ×150 (0.9 NA) BD (Olympus Japan) objectives with a spatial resolution better than 1 µm were used. The light from the Raman-scattered 532-nm laser was dispersed on gratings of 600 or 1,800 grooves per mm with pixel resolution better than 2 cm−1 or 0.5 cm−1, respectively. Spectra were normally collected between 100 and 1,800 cm−1, with a power of up to 2 mW and an exposure of 20–60 s using 2–5 averages. The data underwent polynomial baseline subtraction and spike removal. The spectra were smoothed using a Savitzky Golay algorithm (third order, 11 points), as is commonly done.
Cloning, expression and purification of PNPases
The sequences of PNPase variants 4a, 4b, 5a, 5b and 6 were codon-optimized for expression in E. coli, synthesized and cloned into a pET28-His-bdSUMO expression vector (Twist Bioscience). Plasmids were transformed into electrocompetent BL21/DE3 cells harboring a chaperone-expressing plasmid pGJKE8 (Takara) and plated on Luria–Bertani agar plates with 35 µg ml−1 chloramphenicol, 50 µg ml−1 kanamycin and 1% (w/v) glucose. Following overnight incubation at 37 °C, individual colonies from each transformation were randomly picked into 5-ml culture tubes (Falcon) containing growth medium (2YT + Cap 35 µg ml−1 + kanamycin 50 µg ml−1; 1 ml) and grown overnight at 37 °C with shaking (250 r.p.m.). The resulting cultures were used to inoculate 100-ml growth medium cultures (1:100 dilution) and grown at 37 °C to an optical density at 600 nm of ~0.5. Protein expression was induced by IPTG (1 mM) and cultures were grown for 48 h at 16 °C with shaking (250 r.p.m.). Subsequently, the cells were pelleted (4,000 r.p.m., 4 °C, 20 min), frozen at −80 °C, thawed and resuspended in 3 ml of cell lysis buffer (20 mM Tris pH 7.5, 50 mM NaCl, 0.6 mg ml−1 lysozyme, 10 U per ml benzonase nuclease and bacterial protease inhibitor cocktail without EDTA; 1:50). Cells were lysed by sonication and the lysate was clarified by centrifugation (30 min, 4 °C, 20,000g). The clarified lysates were loaded onto columns (BioRad) containing Ni-NTA-resin (Merck), washed with wash buffer (50 mM Tris pH 7.5, 100 mM NaCl and 35 mM imidazole) and eluted using elution buffer (50 mM Tris pH 7.5, 100 mM NaCl and 300 mM imidazole). Eluted proteins were concentrated following exchange to storage buffer (100 mM Tris pH 7.5, 200 mM NaCl and 10% glycerol). The His-SUMO tag was cleaved by incubation (18–20 h, 4 °C) with 1 mM DTT and 0.1 mg ml−1 bdSumo protease and the untagged proteins were then purified by size-exclusion chromatography using a Superdex 75-Increase 10/300 GL (Cytiva) equilibrated with PBS buffer + 100 mM NaCl. Purified PNPase variants were eluted as a single peak corresponding to a ~90-kDa trimer and were pooled, concentrated, buffer-exchanged to storage buffer and then frozen in aliquots at −80 °C.
Kinetic analyses of PNPases
Freshly made stock solutions of guanosine or deoxyguanosine were diluted to varying final concentrations (0–500 μM) in potassium phosphate buffer (50 mM, pH 7.5) containing purified PNPases (0.1 μM). The absorbance of the reaction mixtures was monitored at 258 nm in a quartz 96-well plate using an ELISA plate reader (BioTek) for 10 min. Initial velocities of purified PNPases with other nucleoside substrates were similarly obtained using their respective maximal absorbance wavelengths (inosine, 249 nm; adenosine, deoxyadenosine or xanthosine, 258 nm; cytidine, deoxycytidine or thymidine, 270 nm). For substrate competition experiments, four nucleosides (guanosine, deoxyguanosine, inosine and adenosine) were mixed in equimolar concentrations (200 μM). The nucleoside mix was added to a potassium phosphate buffer solution (50 mM, pH 7.5) containing a purified PNPase (0.25 μM) and incubated for 30 min at room temperature. Following incubation, the reaction mixtures were inactivated (65 °C, 10 min) and frozen at −80 °C. Residual nucleoside concentrations were determined using LC–MS. The pH rate profiles of individual PNPases were determined by mixing dilute enzyme solutions (20 μl, 2.5 μM) with potassium phosphate-buffered guanosine solutions (430 μM, 180 μM) at different pH values (2–9) in a 96-well quartz ELISA plate, while monitoring solution absorbance at 258 nm for 10 min at room temperature. The enzyme solutions were obtained by titrating a stock solution of guanosine in potassium phosphate buffer (100 mM) using K2HPO4 (100 mM, pH 4.2) or KH2PO4 (100 mM, pH 9) to the appropriate pH values.
Structural predictions of PNPases
The three-dimensional (3D) structures of PNPase variants 4a, 4b, 5a, 5b and 6 were predicted using AlphaFold2, locally installed on May 29, 2023 (https://github.com/deepmind/alphafold), a ColabFold version of AlphaFold2 (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb, with and without template -pdb 100)49 with relaxation in all cases, a ColabFold version of OmegaFold (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/beta/omegafold.ipynb)50, an API version of ESM (https://esmatlas.com/resources?action=fold)51, the Uni-Fold Colab Notebook (https://colab.research.google.com/github/dptech-corp/Uni-Fold/blob/main/notebooks/unifold.ipynb)48 and a Rosetta server implementation of RoseTTAFold (https://robetta.bakerlab.org/submit.php)47.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software by carrying out non-parametric, unpaired, two-tailed Mann–Whitney and Student’s t-tests between the different conditions. Statistical experimental details can be found in the relevant figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The MS proteomics data were deposited to the ProteomeXchange Consortium through the PRIDE75 partner repository with the dataset identifier PXD052468 (https://doi.org/10.6019/PXD052468). The 3D structures of PNPase variants 4a, 4b, 5a, 5b and 6 were predicted using AlphaFold2, locally installed on May 29, 2023 (https://github.com/deepmind/alphafold), a ColabFold version of AlphaFold2 (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb, with and without template -pdb 100)49 with relaxation in all cases, a ColabFold version of OmegaFold (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/beta/omegafold.ipynb)50, an API version of ESM (https://esmatlas.com/resources?action=fold)51, the Uni-Fold Colab Notebook (https://colab.research.google.com/github/dptech-corp/Uni-Fold/blob/main/notebooks/unifold.ipynb)48 and a Rosetta server implementation of RoseTTAFold (https://robetta.bakerlab.org/submit.php)47. The structural coordinates for the protein of interest discussed in this study (PDB 1B8O) can be accessed freely. The relative mRNA levels of pnp4a (NM_001002102.1), pnp4b (NM_205643.1), pnp5a (NM_213311.2), pnp5b (NM_001004628.1) and pnp6 (NM_205655.2) were determined using RT–qPCR. Data are available from the corresponding author upon request.
References
Land, M. F. The physics and biology of animal reflectors. Prog. Biophys. Mol. Biol. 24, 75–106 (1972).
Gur, D., Palmer, B. A., Weiner, S. & Addadi, L. Light manipulation by guanine crystals in organisms: biogenic scatterers, mirrors, multilayer reflectors and photonic crystals. Adv. Funct. Mater. 27, 1603514 (2017).
Palmer, B. A. et al. The image-forming mirror in the eye of the scallop. Science 358, 1172–1175 (2017).
Gur, D. et al. The dual functional reflecting iris of the zebrafish. Adv. Sci. 5, 1800338 (2018).
Denton, E. J. Review lecture: on the organization of reflecting surfaces in some marine animals. Philos. Trans. R. Soc. Lond. B 258, 285–313 (1970).
Gur, D. et al. Structural basis for the brilliant colors of the sapphirinid copepods. J. Am. Chem. Soc. 137, 8408–8411 (2015).
Teyssier, J., Saenko, S. V., van der Marel, D. & Milinkovitch, M. C. Photonic crystals cause active colour change in chameleons. Nat. Commun. 6, 6368 (2015).
Pilátová, J., Pánek, T., Oborník, M., Čepička, I. & Mojzeš, P. Revisiting biocrystallization: purine crystalline inclusions are widespread in eukaryotes. ISME J. 16, 2290–2294 (2022).
Pavan, M. E. et al. Guanine crystal formation by bacteria. BMC Biol. 21, 66 (2023).
Mojzeš, P. et al. Guanine, a high-capacity and rapid-turnover nitrogen reserve in microalgal cells. Proc. Natl Acad. Sci. USA 117, 32722–32730 (2020).
Levy-Lior, A. et al. Guanine-based biogenic photonic-crystal arrays in fish and spiders. Adv. Funct. Mater. 20, 320–329 (2010).
Eyal, Z. et al. Plate-like guanine biocrystals form via templated nucleation of crystal leaflets on preassembled scaffolds. J. Am. Chem. Soc. 144, 22440–22445 (2022).
Wagner, A. et al. Macromolecular sheets direct the morphology and orientation of plate-like biogenic guanine crystals. Nat. Commun. 14, 589 (2023).
Gur, D. et al. In situ differentiation of iridophore crystallotypes underlies zebrafish stripe patterning. Nat. Commun. 11, 6391 (2020).
Hirata, M., Nakamura, K. & Kondo, S. Pigment cell distributions in different tissues of the zebrafish, with special reference to the striped pigment pattern. Dev. Dyn. 234, 293–300 (2005).
Hirata, M., Nakamura, K., Kanemaru, T., Shibata, Y. & Kondo, S. Pigment cell organization in the hypodermis of zebrafish. Dev. Dyn. 227, 497–503 (2003).
Pedley, A. M. & Benkovic, S. J.Anew view into the regulation of purine metabolism: the purinosome. Trends Biochem. Sci. 42, 141–154 (2017).
Wagner, A. et al. The non-classical crystallization mechanism of a composite biogenic guanine crystal. Adv. Mater. 34, e2202242 (2022).
Gur, D. et al. Guanine-based photonic crystals in fish scales form from an amorphous precursor. Angew. Chem. 52, 388–391 (2013).
Ullate-Agote, A. et al. Genome mapping of a LYST mutation in corn snakes indicates that vertebrate chromatophore vesicles are lysosome-related organelles. Proc. Natl Acad. Sci. USA 117, 26307–26317 (2020).
Jantschke, A. et al. Anhydrous β-guanine crystals in a marine dinoflagellate: structure and suggested function. J. Struct. Biol. 207, 12–20 (2019).
Bagnara, J. T. et al. Common origin of pigment cells. Science 203, 410–415 (1979).
Palmer, B. A., Gur, D., Weiner, S., Addadi, L. & Oron, D. The organic crystalline materials of vision: structure–function considerations from the nanometer to the millimeter scale. Adv. Mater. 30, e1800006 (2018).
Gur, D. et al. Guanine crystallization in aqueous solutions enables control over crystal size and polymorphism. Cryst. Growth Des. 16, 4975–4980 (2016).
Hirsch, A. et al. Biologically controlled morphology and twinning in guanine crystals. Angew. Chem. 56, 9420–9424 (2017).
Guo, D. et al. Formation mechanism of twinned β-form anhydrous guanine platelets in scallop eyes. CrystEngComm 25, 4521–4530 (2023).
Greenstein, L. Nacreous pigments and their properties. Proc. Sci. Sect. Toilet Goods Assoc. 26, 20–26 (1966).
Pinsk, N. et al. Biogenic guanine crystals are solid solutions of guanine and other purine metabolites. J. Am. Chem. Soc. 144, 5180–5189 (2022).
Department of Health and Human Services, Food and Drug Administration Guanine. Code of Federal Regulations https://www.ecfr.gov/current/title-21/chapter-I/subchapter-A/part-73/subpart-C/section-73.2329 (1977).
Higdon, C. W., Mitra, R. D. & Johnson, S. L. Gene expression analysis of zebrafish melanocytes, iridophores, and retinal pigmented epithelium reveals indicators of biological function and developmental origin. PLoS ONE 8, e67801 (2013).
Kimura, T. Pigments in teleosts and their biosynthesis. In Pigments, Pigment Cells and Pigment Patterns (eds Hashimoto, H., Goda, M., Futahashi, R., Kelsh, R. & Akiyama, T.) (Springer, 2021).
Jang, H. S. et al. Epigenetic dynamics shaping melanophore and iridophore cell fate in zebrafish. Genome Biol. 22, 282 (2021).
Li, Y. et al. Integrative analysis of circadian transcriptome and metabolic network reveals the role of de novo purine synthesis in circadian control of cell cycle. PLoS Comput. Biol. 11, e1004086 (2015).
Kimura, T., Takehana, Y. & Naruse, K. pnp4a is the causal gene of the medaka iridophore mutant guanineless. G3 (Bethesda) 7, 1357–1363 (2017).
Ide, H. & Hama, T. Guanine formation in isolated iridophores from bullfrog tadpoles. Biochim. Biophys. Acta 286, 269–271 (1972).
Owen, J., Yates, C. & Kelsh, R. N. Pigment patterning in teleosts. In Pigments, Pigment Cells and Pigment Patterns (eds Hashimoto, H. et al.) (Springer, 2021).
Irion, U., Singh, A. P. & Nüsslein-Volhard, C. The developmental genetics of vertebrate color pattern formation: lessons from zebrafish. Curr. Top. Dev. Biol. 117, 141–169 (2016).
Subkhankulova, T. et al. Zebrafish pigment cells develop directly from persistent highly multipotent progenitors. Nat. Commun. 14, 1258 (2023).
Lister, J. A., Lane, B. M., Nguyen, A. & Lunney, K. Embryonic expression of zebrafish MiT family genes tfe3b, tfeb, and tfec. Dev. Dyn. 240, 2529–2538 (2011).
Saunders, L. M. et al. Thyroid hormone regulates distinct paths to maturation in pigment cell lineages. eLife 8, e45181 (2019).
Szklarczyk, D. et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Ng, A., Uribe, R. A., Yieh, L., Nuckels, R. & Gross, J. M. Zebrafish mutations in gart and paics identify crucial roles for de novo purine synthesis in vertebrate pigmentation and ocular development. Development 136, 2601–2611 (2009).
Fernández, J. R., Byrne, B. & Firestein, B. L. Phylogenetic analysis and molecular evolution of guanine deaminases: from guanine to dendrites. J. Mol. Evol. 68, 227–235 (2009).
Wu, X. W., Lee, C. C., Muzny, D. M. & Caskey, C. T. Urate oxidase: primary structure and evolutionary implications. Proc. Natl Acad. Sci. USA 86, 9412–9416 (1989).
Dong, Y. et al. High mass resolution, spatial metabolite mapping enhances the current plant gene and pathway discovery toolbox. New Phytol. 228, 1986–2002 (2020).
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876 (2021).
Li, Z. et al. Uni-Fold: an open-source platform for developing protein folding models beyond AlphaFold. Preprint at bioRxiv https://doi.org/10.1101/2022.08.04.502811 (2022).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Wu, R. et al. High-resolution de novo structure prediction from primary sequence. Preprint at bioRxiv https://doi.org/10.1101/2022.07.21.500999 (2022).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Zhang, Y. & Ealick, S. E. Purine nucleoside phosphorylase. In Computational and Structural Approaches to Drug Discovery: Ligand–Protein Interactions (eds Stroud, R. & Finer-Moore, J.) (RSC, 2007).
Hartman, P. & Perdok, W. G. On the relations between structure and morphology of crystals. I. Acta Crystallogr. 8, 49–52 (1955).
Addadi, L. et al. Growth and dissolution of organic crystals with `tailor-made' inhibitors—implications in stereochemistry and materials science. Angew. Chem. 24, 466–485 (1985).
Berkovitch-Yellin, Y. et al. Crystal morphology engineering by ‘tailor-made’ inhibitors; a new probe to fine intermolecular interactions. J. Am. Chem. Soc. 107, 3111–3122 (1985).
Levesque, M. P., Krauss, J., Koehler, C., Boden, C. & Harris, M. P. New tools for the identification of developmentally regulated enhancer regions in embryonic and adult zebrafish. Zebrafish 10, 21–29 (2013).
Lewis, V. M. et al. Fate plasticity and reprogramming in genetically distinct populations of Danio leucophores. Proc. Natl Acad. Sci. USA 116, 11806–11811 (2019).
Elinger, D., Gabashvili, A. & Levin, Y. Suspension trapping (S-Trap) is compatible with typical protein extraction buffers and detergents for bottom-up. J. Proteome Res. 18, 1441–1445 (2019).
Millikin, R. J., Solntsev, S. K., Shortreed, M. R. & Smith, L. M. Ultrafast peptide label-free quantification with FlashLFQ. Proteome Res. 17, 386–391 (2018).
Miller, R. M. et al. Improved protein inference from multiple protease bottom-up mass spectrometry data. Proteome Res. 18, 3429–3438 (2019).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Kroll, F. et al. A simple and effective F0 knockout method for rapid screening of behaviour and other complex phenotypes. eLife 10, e59683 (2021).
Kwan, K. M. et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev. Dyn. 236, 3088–3099 (2007).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Haubold, C. et al. Segmenting and tracking multiple dividing targets using ilastik. Adv. Anat. Embryol. Cell Biol. 219, 199–229 (2016).
Schmittgen, T. & Livak, K. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108 (2008).
Tang, R. et al. Validation of zebrafish (Danio rerio) reference genes for quantitative real-time RT–PCR normalization. Acta Biochim. Biophys. Sin. 39, 384–390 (2007).
Dong, Y. et al. PICA: pixel intensity correlation analysis for deconvolution and metabolite identification in mass spectrometry imaging. Anal. Chem. 95, 1652–1662 (2023).
Bemis, K. D. et al. Cardinal: an R package for statistical analysis of mass spectrometry-based imaging experiments. Bioinformatics 31, 2418–2420 (2015).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Mastronarde, D. N. & Held, S. R. Automated tilt series alignment and tomographic reconstruction in IMOD. J. Struct. Biol. 197, 102–113 (2017).
Chen, F., Liu, Y., Li, L., Qi, L. & Ma, Y. Synthesis of bio-inspired guanine microplatelets: morphological and crystallographic control. Chemistry 26, 16228–16235 (2020).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
We thank A. Baram for his help with the HPLC measurements and method development. We thank L. Leiserowitz and L. Addadi for helpful discussions on this project. This research was cofunded by the European Union (ERC, CRYSTALCELL, 101077470). Views and opinions expressed are, however, ours and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. This research was also supported by a research grant from the Harmstieg New Scientist Fund, the Thomas A. and Georgina T. Russo New Scientist Fund, the Alisa and Peter Savitz Foundation, R. Cohen, M. Levine, A. Schatz, the Lancovici-Fallmann Memorial Fund, established by Ruth and Henry Yancovich, the Ullmann Family Foundation, Mr. and Mrs. Alan Franco, the Horwitz Research Fund and the Barton Award for Young Scientists. S.K. and R.M. acknowledge funding from the Max Planck Society and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy (EXC-2068-390729961, Cluster of Excellence Physics of Life of TU Dresden). R.D. is a fellow of the Ariane de Rothschild Women Doctoral Program.
Author information
Authors and Affiliations
Contributions
Conceptualization, R.D. and D.G. Investigation, R.D., T.L.G., O.B., Z.E., D.B.B., M.G., S.K., U.H., Y.D., N.V., M.K., Z.P., S.M.L.Z., I.P., R.M. and D.G. Formal analysis, R.D., Z.E., M.G., S.H., U.H., Y.D., S.L., T.O. and M.K. Visualization, R.D., T.L.G., Z.E., M.G., S.K., U.H., Y.D. and N.V. Funding acquisition, R.M. and D.G. Supervision, R.M. and D.G. Writing—original draft, R.D., T.L.G. and D.G. Writing—review and editing, R.D., T.L.G., Z.E., M.G., S.K., R.M. and D.G.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks Vern Schramm, Peter Vekilov, Monika Warzecha, Julie Watts and the other, anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–16.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deis, R., Lerer-Goldshtein, T., Baiko, O. et al. Genetic control over biogenic crystal morphogenesis in zebrafish. Nat Chem Biol (2024). https://doi.org/10.1038/s41589-024-01722-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41589-024-01722-1
- Springer Nature America, Inc.