

Abstract
Evolutionary theory suggests that lifespan-reducing alleles should be purged from the gene pool, and yet decades of genome-wide association and model organism studies have shown that they persist. One potential explanation is that alleles that regulate lifespan do so only in certain environmental contexts. We exposed outbred Drosophila to control and high-sugar diets and genotyped more than 10,000 adult flies to track allele frequency changes over the course of a single adult lifespan. We identified thousands of lifespan-associated alleles associated with early versus late-life trade-offs, late-onset effects and genotype-by-environment interactions. Remarkably, a third of lifespan-associated genetic variation had environmentally dependent effects on lifespan. We find that lifespan-reducing alleles are often recently derived, have stronger effects on a high-sugar diet and show signatures of selection in wild Drosophila populations, consistent with the evolutionary mismatch hypothesis. Our results provide insight into the highly polygenic and context-dependent genetic architecture of lifespan variation and the evolutionary processes that shape this key trait.





Similar content being viewed by others
Data availability
Sequencing data have been deposited on SRA with the accession data under the under BioProject PRJNA725602. Additional data used to estimate population genetic parameters (Fig. 5,b–d) and validation experiments (Fig. 3b) are available on Dryad under accession hhmgqnkgr. There are no restrictions on data availability.
Code availability
Custom code used for data analysis is available on GitHub at https://doi.org/10.5281/zenodo.7139896 and https://doi.org/10.5281/zenodo.7143559.
References
Ruby, J. G. et al. Estimates of the heritability of human longevity are substantially inflated due to assortative mating. Genetics 210, 1109–1124 (2018).
Kuningas, M. et al. Genes encoding longevity: from model organisms to humans. Aging Cell. 7, 270–280 (2008).
Joshi, P. K. et al. Variants near CHRNA3/5 and APOE have age- and sex-related effects on human lifespan. Nat. Commun. 7, 11174 (2016).
Sella, G. & Barton, N. H. Thinking about the evolution of complex traits in the era of genome-wide association studies. Annu. Rev. Genom. Hum. Genet. 20, 461–493 (2019).
Medawar, P. B. An Unsolved Problem of Biology (H.K. Lewis and Company, 1952).
Williams, G. C. Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411 (1957).
Gibson, G. Decanalization and the origin of complex disease. Nat. Rev. Genet. 10, 134–140 (2009).
Corbett, S., Courtiol, A., Lummaa, V., Moorad, J. & Stearns, S. The transition to modernity and chronic disease: mismatch and natural selection. Nat. Rev. Genet. 19, 419–430 (2018).
Gibson, G. & Dworkin, I. Uncovering cryptic genetic variation. Nat. Rev. Genet. 5, 681–690 (2004).
Di Rienzo, A. & Hudson, R. R. An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends Genet. 21, 596–601 (2005).
Quintana-Murci, L. Understanding rare and common diseases in the context of human evolution. Genome Biol. 17, 225 (2016).
Musselman, L. P. et al. A high-sugar diet produces obesity and insulin resistance in wild-type Drosophila. Dis. Models Mech. 4, 842–849 (2011).
Na, J. et al. Model of high sugar diet-induced cardiomyopathy. PLoS Genet. 9, e1003175 (2013).
Mostafavi, H. et al. Identifying genetic variants that affect viability in large cohorts. PLoS Biol. 15, e2002458 (2017).
Huang, W. et al. Context-dependent genetic architecture of Drosophila life span. PLoS Biol. 18, e3000645 (2020).
Burger, J. M. S. & Promislow, D. E. L. Sex-specific effects of interventions that extend fly life span. Sci. Aging Knowl. Environ. 2004, pe30 (2004).
Tacutu, R. et al. Human ageing genomic resources: new and updated databases. Nucleic Acids Res. 46, D1083–D1090 (2018).
Piper, M. D. W. & Partridge, L. Drosophila as a model for ageing. Biochim. Biophys. Acta 1864, 2707–2717 (2018).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Hoedjes, K. M. et al. Distinct genomic signals of lifespan and life history evolution in response to postponed reproduction and larval diet in Drosophila. Evol. Lett. 3, 598–609 (2019).
Fabian, D. K. et al. Evolution of longevity improves immunity in Drosophila. Evol. Lett. 2, 567–579 (2018).
Deelen, J. et al. A meta-analysis of genome-wide association studies identifies multiple longevity genes. Nat. Commun. 10, 3669 (2019).
Buszczak, M., Lu, X., Segraves, W. A., Chang, T. Y. & Cooley, L. Mutations in the midway gene disrupt a Drosophila acyl coenzyme A: diacylglycerol acyltransferase. Genetics 160, 1511–1518 (2002).
Xu, Y. & Wang, T. LOVIT is a putative vesicular histamine transporter required in Drosophila for vision. Cell Rep. 27, 1327–1333 (2019).
Streeper, R. S. et al. Deficiency of the lipid synthesis enzyme, DGAT1, extends longevity in mice. Aging 4, 13–27 (2012).
Johnson, A. A. & Stolzing, A. The role of lipid metabolism in aging, lifespan regulation, and age-related disease. Aging Cell 18, e13048 (2019).
Crow, J. F. & Kimura, M. An Introduction to Population Genetics Theory (Blackburn Press, 2009).
Durham, M. F., Magwire, M. M., Stone, E. A. & Leips, J. Genome-wide analysis in Drosophila reveals age-specific effects of SNPs on fitness traits. Nat. Commun. 5, 4338 (2014).
Musselman, L. P. & Kühnlein, R. P. Drosophila as a model to study obesity and metabolic disease. J. Exp. Biol. 221, jeb163881 (2018).
Rodríguez, J. A. et al. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat. Ecol. Evol. 1, 55 (2017).
Carnes, M. U. et al. The genomic basis of postponed senescence in Drosophila melanogaster. PLoS One 10, e0138569 (2015).
Remolina, S. C., Chang, P. L., Leips, J., Nuzhdin, S. V. & Hughes, K. A. Genomic basis of aging and life-history evolution in Drosophila melanogaster. Evolution 66, 3390–3403 (2012).
Thornton, K. & Andolfatto, P. Approximate Bayesian inference reveals evidence for a recent, severe Bottleneck in a Netherlands population of Drosophila melanogaster. Genetics 172, 1607–1619 (2006).
Picelli, S. et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040 (2014).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Lesnoff, M. & Lancelot, R. aod: analysis of overdispersed data. R package version 1.3.2. https://cran.r-project.org/web/packages/aod/index.html (2016).
Kofler, R., Pandey, R. V. & Schlötterer, C. PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 27, 3435–3436 (2011).
Kapun, M., Fabian, D. K., Goudet, J. & Flatt, T. Genomic evidence for adaptive inversion clines in Drosophila melanogaster. Mol. Biol. Evol. 33, 1317–1336 (2016).
dos Santos, G. et al. FlyBase: introduction of the Drosophila melanogaster Release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Res. 43, D690–D697 (2015).
Wang, M., Zhao, Y. & Zhang, B. Efficient test and visualization of multi-set intersections. Sci. Rep. 5, 16923 (2015).
Kofler, R. & Schlötterer, C. Gowinda: unbiased analysis of gene set enrichment for genome-wide association studies. Bioinformatics 28, 2084–2085 (2012).
Berriz, G. F., Beaver, J. E., Cenik, C., Tasan, M. & Roth, F. P. Next generation software for functional trend analysis. Bioinformatics 25, 3043–3044 (2009).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Thurmond, J. et al. FlyBase 2.0: the next generation. Nucleic Acids Res. 47, D759–D765 (2019).
Lack, J. B. et al. A thousand fly genomes: an expanded Drosophila genome nexus. Mol. Biol. Evol. 33, 3308–3313 (2016).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Lea, A. J., Martins, D., Kamau, J., Gurven, M. & Ayroles, J. F. Urbanization and market integration have strong, nonlinear effects on cardiometabolic health in the Turkana. Sci. Adv. 6, eabb1430 (2020).
Acknowledgements
We thank A.M. Taverner for providing the multispecies alignments, E. Gadja for helping in collecting flies from the wild, and M. Fernandez for helping in maintaining the experimental cages. We also thank members of the Ayroles and Andolfatto labs for their general support and helpful scientific discussions. A.G. Clark, M. Przeworski, G. Sella and A. Harpak provided helpful comments on the analyses. Funding: L.F.P. was funded by a Long-Term Postdoctoral Fellowship from the Human Frontiers Science Program. A.J.L. was funded by a postdoctoral fellowship from the Helen Hay Whitney Foundation. J.F.A. is funded by NIH-NIGMS R35GM124881-04 and NIH-NIEHS R01ES029929. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
L.F.P. was responsible for conceptualization, formal analysis, investigation, writing-original draft, writing-review and editing; A.J.L. was responsible for conceptualization, formal analysis, investigation, writing-original draft, writing-review and editing, and visualization; C.H. was responsible for conceptualization, formal analysis, investigation, writing-review and editing; E.V.F. was responsible for investigation; P.A. and J.F.A. Jointly supervised the research. P.A. was responsible for conceptualization, formal analysis, writing-review and editing, and funding acquisition; J.F.A. was responsible for conceptualization, formal analysis, writing-review and editing, and funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
No ethical approval was required for this work.
Peer review
Peer review information
Nature Genetics thanks David Rand and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Mean read coverage for DNA libraries and individual SNPs.
(A) The y-axis represents the number of paired end (PE) reads generated per DNA library in millions. Samples are grouped by cage and time point. (B) Average coverage per fly for each of the 268,159 SNPs used in the main analysis. Coverage is shown separately for T0, and TN ctrl and TN high sugar. Only individuals with non-zero reads per SNP were used for this plot. (Boxplot edges are the 25th and 75th percentiles, the center line is the median and whiskers are at 1.5 the interquartile range).
Extended Data Fig. 2 Sex and diet interact to affect survival.
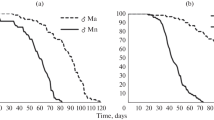
Kaplan-Meier survival curves estimated for flies in vials of (A) only females or (B) only males on a CTRL or HS diet, respectively. When survival curves where estimated for males and females in mixed sex vials on (C) CTRL or (D) HS food, females exhibited significantly reduced lifespan relative to males, but only on a CTRL diet. Full results from models testing for sex and diet effect on survival using a Cox proportional-hazards framework can be found in Supplementary Table 5. For all shaded area (band corresponds) represents to 95% confidence internal.
Extended Data Fig. 3 QQ-plots.
QQ-plots comparing the empirical null distribution of p-values (obtained via permutations of the 3-way factor representing time and diet) to the observed distribution of p-values for the genetic effect on lifespan on (A) a CTRL diet and (B) a HS diet.
Extended Data Fig. 4 Comparison of the magnitude of the genetic effect on lifespan across conditions.
(A) Across all tested sites, there is a strong correlation between the magnitude of the genetic effect on lifespan estimated on a CTRL diet (x-axis) versus a HS diet (y-axis). The R and p-value from a Pearson correlation are reported on the plot. The plot was created using the “smoothScatter” function in R with default parameters. (B) For sites with significant genetic effects on lifespan that are shared between conditions (shared) or stronger in the HS environment (GxE), the magnitude of the lifespan effect tends to be larger on a HS diet.
Extended Data Fig. 5 Allele frequency changes between T0 and TN for SNPs with significant lifespan-effects.
(A) Absolute allele frequency change (T0 vs TN) on a CTRL or HS diet for sites with lifespan effects shared between diets (dark blue solid lines) or HS-biased sites (orange line). The same distributions are also plotted for SNPs with no effects on lifespan (gray). (B) Because there are other covariates in our experiment that also influence allele frequencies (that is, sex, sequencing batch, and starting population cage), the “raw” observed allele frequency changes plotted in panel A may be downwardly biased or at the very least noisy. Therefore, we also translated our model fitted estimates of effect size (as shown in Fig. 2 C) into allele frequency changes. We used an approach similar to that of47. Specifically, for each significant site, we calculated the allele frequency at T0 by setting batch=1, sex=1, starting population pool=1, and CTRL and HS environment effects=0; we then summed the model fitted estimates of effect size multiplied by these values and passed the sum through a logistic function to obtain an allele frequency. To calculate the allele frequency at TN in the HS and CTRL environments, we performed parallel calculations where the CTRL or HS environment effects were set to 1. The distributions from these analyses are plotted here, with colors as in panel A. (C) Colored lines are the same as in panel A, but here the null distribution (gray lines) is derived by subsampling two sets of 1000 individuals with replacement at T0 and calculating the absolute difference in allele frequency across all tested sites. This null distribution is plotted for 100 subsamples.
Extended Data Fig. 6 Simulations to understand our definitions of “shared” and “GxE” SNPs.
We classify SNPs as having “shared” effect sizes between environments (defined as FDR < 10% in one environment and p < 0.05 in the other) or “GxE” effects where the lifespan effect is stronger on one diet relative to the other (“GxE” defined as FDR < 10% in one environment and p < 0.05 in the other). To understand how this thresholding affected our results, we simulated 121 datasets of 1000 loci each where the lifespan reducing allele decreased by 0-20% with identical or varied effect sizes across two environments (sample sizes = 1000 individuals at T0 and 1000 at each of the two TN end points). For each simulation, we recorded the number of loci that would be classified as (A) shared (p < 10−4.5 in one environment, similar to a 10% FDR in our real dataset, and p < 0.05 in the other environment) or (B) GxE (p < 10−4.5 in environment #2 and p > 0.05 in environment #1). See Text S6 for additional details. (C) The FDR cutoff used to define GxE effects (that is, the threshold used to consider a site significant in one environment) versus the false positive rate estimated from permutations where the TN HS and TN CTRL labels were scrambled 10 times. See Text S6 for additional details.
Extended Data Fig. 7 Power to detect a genetic effect on lifespan using different study designs.
(A) Y-axis represents the proportion of simulated true positive sites (with a significant change in allele frequency across the lifespan) that are detected at p < 10−6. Shown are results from simulations that mimic the “fly” design used here, where individuals are sampled at the beginning and end of their life, versus the “human” design of Mostafavi et al.14, where individuals were only genotyped in middle and old age. We performed simulations for the fly and human designs where true genotypes were estimated without error, as well as simulations where genotypes were derived from a low coverage (~1x) sequencing approach and therefore estimated with some error. The curves show simulation results for starting minor allele frequencies from 0.05-0.4, and effect sizes were always simulated as a 20% change in allele frequency from the youngest to the oldest age bin. Further details on the simulations and analysis of simulated data are provided in Text S11. (B) The number of lifespan-associated SNPs found by this study versus Mostafavi et al14. at different nominal p-value thresholds. We used the same binominal regression approach described in text S11. We note that the comparisons in B are likely influenced by several factors that differ between the studies, such as linkage disequilibrium, environmental conditions and covariates, and approaches to genotyping and statistical modeling. We therefore view this comparison as useful for understanding general patterns but note that future work is needed to understand the precise magnitude and specific sources of the observed power differences.
Extended Data Fig. 8 Power comparison between a Cochran‐Mantel‐Haenszel (CMH) and beta-binomial approach.
(A-B) The proportion of simulated true positives (that is, SNPs with a significant effect on lifespan) detected by a beta-binomial versus a CMH approach as a function of effect size. Different p-value thresholds are used to detect true positive SNPs in panels A and B as noted on the y-axis. Details on the simulations and analyses of simulated data are provided in the SI Methods (see Text S5. Comparison of two statistical approaches to test for allele frequency changes with age). Correlation between the -log10 p-value for a genetic effect on lifespan from the real data, estimated in (C) CTRL or (D) HS conditions using a beta-binomial model (x-axis) or a CMH test (y-axis). As described in the main text, the p-values for the beta-binomial were estimated while controlling for covariates of sex, sequencing batch, and cage. R values from a Pearson correlation are reported on each plot. Plots were created using the “smoothScatter” function in R with default parameters.
Extended Data Fig. 9 Simulations reveal that it is difficult to assign alleles to particular trajectories.
Loci were simulated to follow one of three possible trajectories: (A) allele decreases at a constant rate across all ages; (B) allele decreases in frequency early in life but increases again in late life; or (C) allele is maintained at a constant frequency up to a given age but then steadily declines. These simulations were performed for datasets with allele frequency changes between T0 and TN ranging from 0-20% (x-axis). Details of the statistical model are described under section S8. All loci were then analyzed using models that capture all three possible trajectories, and the ΔAIC value between the best fit model and the second-best fit model was calculated for each locus (y-axis). Dashed red lines represent a ΔAIC value of 2. (Boxplot edges are the 25th and 75th percentiles, the center line is the median and whiskers are at 1.5 the interquartile range).
Supplementary information
Supplementary Information
Supplementary Methods and Supplementary Figs. 1–4.
Supplementary Tables
Supplementary Tables 1–12.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pallares, L.F., Lea, A.J., Han, C. et al. Dietary stress remodels the genetic architecture of lifespan variation in outbred Drosophila. Nat Genet 55, 123–129 (2023). https://doi.org/10.1038/s41588-022-01246-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-022-01246-1
- Springer Nature America, Inc.
This article is cited by
-
Rapid seasonal changes in phenotypes in a wild Drosophila population
Scientific Reports (2023)