

Abstract
The plasma proteome can help bridge the gap between the genome and diseases. Here we describe genome-wide association studies (GWASs) of plasma protein levels measured with 4,907 aptamers in 35,559 Icelanders. We found 18,084 associations between sequence variants and levels of proteins in plasma (protein quantitative trait loci; pQTL), of which 19% were with rare variants (minor allele frequency (MAF) < 1%). We tested plasma protein levels for association with 373 diseases and other traits and identified 257,490 associations. We integrated pQTL and genetic associations with diseases and other traits and found that 12% of 45,334 lead associations in the GWAS Catalog are with variants in high linkage disequilibrium with pQTL. We identified 938 genes encoding potential drug targets with variants that influence levels of possible biomarkers. Combining proteomics, genomics and transcriptomics, we provide a valuable resource that can be used to improve understanding of disease pathogenesis and to assist with drug discovery and development.







Similar content being viewed by others


Data availability
GWAS summary statistics for all 4,907 aptamers are available at https://www.decode.com/summarydata/. Sequence variants passing GATK filters that support the findings of this study have been deposited in the European Variation Archive under accession number PRJEB15197. Other data presented in this study are included in this publication (and its Supplementary information). As we have been in the past, we are open to collaboration on this topic. The UK Biobank Resource was used under application number 56270. FinnGen and BioBank Japan data are publicly available and were downloaded from https://www.finngen.fi/ and http://jenger.riken.jp/en/, respectively.
URLs for other external data used are as follows: the GWAS Catalog (https://www.ebi.ac.uk/gwas/), the STRING database (https://string-db.org/, in the file 9606.protein.actions.v11.txt.gz), the GTEx project (https://gtexportal.org/home/), the Human Protein Atlas (https://www.proteinatlas.org/).
Code availability
We used publicly available software (URLs are listed below) in conjunction with the above described algorithms in the sequence-processing pipeline (whole-genome sequencing, association testing and RNA sequencing mapping and analysis): BWA-MEM version 0.7.10, https://github.com/lh3/bwa; GenomeAnalysisTKLite version 2.3.9, https://github.com/broadgsa/gatk/; Picard tools version 1.117, https://broadinstitute.github.io/picard/; SAMtools version 1.3, http://samtools.github.io/; bedtools version 2.25.0-76-g5e7c696z, https://github.com/arq5x/bedtools2/; Variant Effect Predictor (release 100), https://github.com/Ensembl/ensembl-vep; BOLT-LMM version 2.1, https://data.broadinstitute.org/alkesgroup/BOLT-LMM/downloads/; IMPUTE2 version 2.3.1, https://mathgen.stats.ox.ac.uk/impute/impute_v2.html; dbSNP version 140, http://www.ncbi.nlm.nih.gov/SNP/; BiNGO version 3.0.3, https://www.psb.ugent.be/cbd/papers/BiNGO/Download.html; Cytoscape version 3.7.1, https://cytoscape.org/download.html. We used R (version 3.6.0) extensively to analyze data and create plots. Figs. 1 and 2 were created using https://biorender.com/.
References
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Loos, R. J. F. 15 years of genome-wide association studies and no signs of slowing down. Nat. Commun. 11, 5900 (2020).
Suhre, K. et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 8, 14357 (2017).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Emilsson, V. et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 361, 769–773 (2018).
Folkersen, L. et al. Mapping of 79 loci for 83 plasma protein biomarkers in cardiovascular disease. PLoS Genet. 13, e1006706 (2017).
Yao, C. et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun. 9, 3268 (2018).
Folkersen, L. et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat. Metab. 2, 1135–1148 (2020).
Zheng, J. et al. Phenome-wide Mendelian randomization mapping the influence of the plasma proteome on complex diseases. Nat. Genet. 52, 1122–1131 (2020).
Suhre, K., McCarthy, M. I. & Schwenk, J. M. Genetics meets proteomics: perspectives for large population-based studies. Nat. Rev. Genet. 22, 19–37 (2020).
Rohloff, J. C. et al. Nucleic acid ligands with protein-like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol. Ther. Nucleic Acids 3, e201 (2014).
Lundberg, M., Eriksson, A., Tran, B., Assarsson, E. & Fredriksson, S. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res. 39, e102 (2011).
Nioi, P. et al. Variant ASGR1 associated with a reduced risk of coronary artery disease. N. Engl. J. Med. 374, 2131–2141 (2016).
Gudbjartsson, D. F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015).
Klarin, D. et al. Genome-wide association analysis of venous thromboembolism identifies new risk loci and genetic overlap with arterial vascular disease. Nat. Genet. 51, 1574–1579 (2019).
Sennblad, B. et al. Genome-wide association study with additional genetic and post-transcriptional analyses reveals novel regulators of plasma factor XI levels. Hum. Mol. Genet. 26, 637–649 (2017).
Law, P. J. et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun. 10, 2154 (2019).
Howe, J. R. et al. Germline mutations of the gene encoding bone morphogenetic protein receptor 1A in juvenile polyposis. Nat. Genet. 28, 184–187 (2001).
Miller, M. W. et al. Cloning of the mouse agouti gene predicts a secreted protein ubiquitously expressed in mice carrying the lethal yellow mutation. Genes Dev. 7, 454–467 (1993).
Rieder, S., Taourit, S., Mariat, D., Langlois, B. & Guérin, G. Mutations in the agouti (ASIP), the extension (MC1R), and the brown (TYRP1) loci and their association to coat color phenotypes in horses (Equus caballus). Mamm. Genome 12, 450–455 (2001).
Surendran, P. et al. Trans-ancestry meta-analyses identify rare and common variants associated with blood pressure and hypertension. Nat. Genet. 48, 1151–1161 (2016).
Surendran, P. et al. Discovery of rare variants associated with blood pressure regulation through meta-analysis of 1.3 million individuals. Nat. Genet. 52, 1314–1332 (2020).
Szklarczyk, D. et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
Ragimbeau, J. et al. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO J. 22, 537–547 (2003).
Spracklen, C. N. et al. Identification of type 2 diabetes loci in 433,540 East Asian individuals. Nature 582, 240–245 (2020).
The Haplotype Reference Consortium. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406 (2014).
Joshi, A. D. et al. Four susceptibility loci for gallstone disease identified in a meta-analysis of genome-wide association studies. Gastroenterology 151, 351–363 (2016).
Ferkingstad, E. et al. Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat. Commun. 9, 5101 (2018).
Admirand, W. H. & Small, D. M. The physicochemical basis of cholesterol gallstone formation in man. J. Clin. Invest. 47, 1043–1052 (1968).
Memon, N. et al. Developmental regulation of the gut–liver (FGF19–CYP7A1) axis in neonates. J. Matern. Fetal Neonatal Med. 33, 987–992 (2020).
Holzer, P. & Farzi, A. Neuropeptides and the microbiota–gut–brain axis. Adv. Exp. Med. Biol. 817, 195–219 (2014).
Jonsson, T. et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116 (2013).
Deming, Y. et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 11, eaau2291 (2019).
Schröder, J. M. & Harder, J. Human β-defensin-2. Int. J. Biochem. Cell Biol. 31, 645–651 (1999).
Jin, T. et al. Serum human β-defensin-2 is a possible biomarker for monitoring response to JAK inhibitor in psoriasis patients. Dermatology 233, 164–169 (2017).
Tsoi, L. C. et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 44, 1341–1348 (2012).
Wang, Y. et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 48, D1031–D1041 (2019).
Samson, M. et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382, 722–725 (1996).
Kim, M. B. et al. CCR5 receptor antagonists in preclinical to phase II clinical development for treatment of HIV. Expert Opin. Investig. Drugs 25, 1377–1392 (2016).
Parman, Y. et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr. Opin. Neurol. 29, S3–S13 (2016).
Hammarström, P., Schneider, F. & Kelly, J. W. Trans-suppression of misfolding in an amyloid disease. Science 293, 2459–2462 (2001).
Magrinelli, F. et al. Pharmacological treatment for familial amyloid polyneuropathy. Cochrane Database Syst. Rev. 4, CD012395 (2020).
Pietzner, M. et al. Genetic architecture of host proteins involved in SARS-CoV-2 infection. Nat. Commun. 11, 6397 (2020).
Rafnar, T. et al. The Icelandic Cancer Project—a population-wide approach to studying cancer. Nat. Rev. Cancer 4, 488–492 (2004).
Saevarsdottir, S. et al. FLT3 stop mutation increases FLT3 ligand level and risk of autoimmune thyroid disease. Nature 584, 619–623 (2020).
Loh, P.-R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Galili, T., O’Callaghan, A., Sidi, J. & Sievert, C. heatmaply: an R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 34, 1600–1602 (2018).
Yavorska, O. O. & Burgess, S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int. J. Epidemiol. 46, 1734–1739 (2017).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665 (2013).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Tsoi, L. C. et al. Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat. Commun. 8, 15382 (2017).
Acknowledgements
The authors received no specific funding for this work.
Author information
Authors and Affiliations
Contributions
E.F., P.S., B.A.A., G.S., M.I.M., F.Z., S.A.G., T.E., M.O.U., G.L.N., S.H.L., D.F.G., U.T. and K.S. designed the study and interpreted results. M.K.M., S.S., S.N.S., T.A.O., V.S., V.T., H.S., I.J., H.H., T.R., J.S., U.T. and K.S. carried out participant ascertainment and recruitment. E.F., P.S., B.A.A., G.S., M.I.M., E.L.S., A.O., B.V.H., B.O.J., F.Z., G.H.H., G.M., G.A.A., H.K., K.J., R.F., S.A.G., S.R., M.O.U., P.M., S.H.L., D.F.G. and U.T. performed sequencing, genotyping, expression, proteomic, statistical and/or bioinformatic analyses. K.G., O.T.M. and J.S. planned and performed the functional laboratory work. E.F., P.S., B.A.A., G.S., A.H., A.O., G.L.N., S.H.L., D.F.G., U.T. and K.S. drafted the manuscript. All authors contributed to the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
All authors are affiliated with deCODE genetics/Amgen Inc. and declare competing interests as employees.
Additional information
Peer review information Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Secondary cis and trans pQTL associations.
a) Number of secondary cis associations after conditional analysis for each of the 1,881 sentinel cis pQTL associations for a given protein, b) Number of secondary trans associations after conditional analysis for each of the 16,203 sentinel trans pQTL associations for a given protein.
Extended Data Fig. 2 Effects of cis pQTL in Olink vs SomaScan.
Cis pQTL effects (in units of standard deviations) reported in the SCALLOP study (using Olink cardiovascular 1 panel) vs effects in present study (using SomaScan).
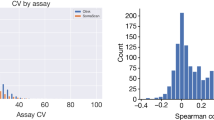
Extended Data Fig. 3 Correlations between SomaScan and Olink protein measurements.
Histogram of correlations between SomaScan and Olink protein measurements for 87 proteins measured using both SomaScan and Olink (cardivascular 1 panel).
Extended Data Fig. 4 Effects of sentinel cis pQTL subdivided by presence or absence of PAV.
Effects (absolute value, SD) of sentinel cis pQTL, subdivided by presence (n = 658) or absence (n = 1,223) of protein altering variants in high linkage disequilibrium (LD; r2 > 0.80). The boxplots show the median and lower and upper quartiles; whiskers extend to 1.5 times inter-quartile range; points beyond whiskers are plotted individually.
Extended Data Fig. 5 Heatmap based on squared correlation of effects proteins for non-specific loci.
Heatmap and non-supervised hierarchical clustering based on squared correlation (r2) of effects on all 4,907 proteins for all pairs of non-specific loci. The plot shows only the main clusters discussed in the text; detailed results are in Supplementary table 10.
Extended Data Fig. 6 Association of rs6471717-C at CYP7A1 with gallstone risk and FGF19 levels.
Diagram illustrating the association of rs6471717-C at CYP7A1 with gallstone risk and FGF19 levels. Blue arrows show reported relationships, black arrows associations seen in the present study, while the green arrow shows the inference we draw from the analysis. Plus and minus signs indicate the directions of effects.
Extended Data Fig. 7 Effect of TREM2 missense variants on TREM2 measurements by SomaScan and ELISA.
Estimate of the specificity of the sTREM2 levels in SomaScan. TREM2 levels in plasma measured by SomaScan versus TREM2 measured by ELISA on the same sample, coloured by genotype of the TREM2 R47H missense variant, rs75932628, or by genotype of the TREM2 R62H missense variant, rs143332484 (there were no compound heterozygotes). The six individuals that are homozygous for either Arg47His or Arg62His have extremely low levels of TREM2 measured by SomaScan compared to non-carriers inconsistent with the measurements by ELISA. Heterozygotes for either variant also have lower levels that non-carriers when measured by SomaScan but not when measured by ELISA. We conclude that the observed association of Arg47His allele with TREM2 levels based on SomaScan is an artefact due to altered conformation of TREM2 mediated by the coding change leading to a reduced affinity of the SomaScan aptamer targeting TREM2.
Extended Data Fig. 8 MS4A gene cluster locus and associations with Alzheimer’s disease, TREM2 protein levels and RNA expression of MS4A cluster genes.
MS4A gene cluster locus and associations with Alzheimer’s disease, TREM2 plasma protein levels, MS4A4A blood RNA epression, MS4A6A blood RNA expression, and MS4A2 blood RNA expression.
Extended Data Fig. 9 Bidirectional Mendelian Randomization analyses for Alzheimer’s disease and TREM2 level.
Mendelian Randomization (MR) analysis using a) TREM2 levels as exposure and Alzheimer’s disease risk as outcome and b) Alzheimer’s disease risk as exposure and TREM2 plasma protein levels as outcome. Blue lines are estimated MR-IVW effects, the areas between the blue dashed lines show 95% confidence regions for the MR effects.
Supplementary information
Supplementary Information
Supplementary Note and Figs. 1–4
Supplementary Tables
Supplementary Tables 1–19.
Rights and permissions
About this article

Cite this article
Ferkingstad, E., Sulem, P., Atlason, B.A. et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet 53, 1712–1721 (2021). https://doi.org/10.1038/s41588-021-00978-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-021-00978-w
- Springer Nature America, Inc.
This article is cited by
-
Prioritization of therapeutic targets for cancers using integrative multi-omics analysis
Human Genomics (2024)
-
Dissecting the shared genetic landscape of anxiety, depression, and schizophrenia
Journal of Translational Medicine (2024)
-
Proteomics for heart failure risk stratification: a systematic review
BMC Medicine (2024)
-
Proteome-wide mendelian randomization investigates potential associations in heart failure and its etiology: emphasis on PCSK9
BMC Medical Genomics (2024)
-
Proteomic associations with forced expiratory volume: a Mendelian randomisation study
Respiratory Research (2024)