

Abstract
Metal additive manufacturing (AM) enables the production of high value and high performance components1 with applications from aerospace2 to biomedical3 fields. Layer-by-layer fabrication circumvents the geometric limitations of traditional metalworking techniques, allowing topologically optimized parts to be made rapidly and efficiently4,5. Existing AM techniques rely on thermally initiated melting or sintering for part shaping, a costly and material-limited process6,7,8. We report an AM technique that produces metals and alloys with microscale resolution via vat photopolymerization (VP). Three-dimensional-architected hydrogels are infused with metal precursors, then calcined and reduced to convert the hydrogel scaffolds into miniaturized metal replicas. This approach represents a paradigm shift in VP; the material is selected only after the structure is fabricated. Unlike existing VP strategies, which incorporate target materials or precursors into the photoresin during printing9,10,11, our method does not require reoptimization of resins and curing parameters for different materials, enabling quick iteration, compositional tuning and the ability to fabricate multimaterials. We demonstrate AM of metals with critical dimensions of approximately 40 µm that are challenging to fabricate by using conventional processes. Such hydrogel-derived metals have highly twinned microstructures and unusually high hardness, providing a pathway to create advanced metallic micromaterials.
Similar content being viewed by others


Main
Metal AM is mostly achieved via powder bed fusion12 and directed energy deposition13 processes. Layer-by-layer processes enable fabrication of metal multimaterials14 and functionally graded composites15, but such laser-based processes struggle to produce materials such as copper; high thermal conductivity and low laser absorptivity cause difficulties in thermal initiation and localization of melting or sintering16. Vat photopolymerization (VP) is a promising alternative that uses light-initiated free radical polymerization to shape parts. Digital light processing (DLP) printing accomplishes this by projecting two-dimensional images of ultraviolet light into a photoresin bath to cure an entire layer of the three-dimensional (3D) structure simultaneously. DLP is capable of high print speeds17, has been demonstrated with submicrometre resolution,18 and has diverse commercial applications from the direct manufacturing of shoe soles19 to COVID-19 test swabs20. VP was developed predominantly for use with polymers21,22,23 and has also been demonstrated for glasses9 and ceramics10. However, inorganic material selection remains limited owing to challenges with incorporating appropriate precursors into photoresins as solutions24, slurries25 or inorganic–organic mixtures26. Consequently, the fabrication of metals via VP remains a challenge. Oran et al. demonstrated AM of nanoscale silver by using hydrogels as ‘nanomanufacturing reactors’27,28 in which two-photon activation guides the infiltration of precursors to volumetrically deposit 3D materials. Vyatskikh et al. demonstrated AM of nanoscale nickel by using two-photon lithography to pattern inorganic–organic resins containing nickel acrylates, followed by pyrolysis and H2 reduction26. However, these pioneering works are limited in material scope, requiring complex resin design and optimization for each new material. Other less commonly used metal AM techniques such as direct ink writing and material jetting use extrusion from a nozzle and controlled deposition of a binder, respectively, to define part shape. These methods circumvent the challenges of using heat to define part shape; copper materials have been fabricated via direct ink writing29 and material jetting30, but neither technique has produced copper parts with feature sizes under 100 µm.
Hydrogel infusion fabrication
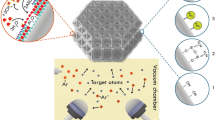
We have developed a VP-based AM technique, coined hydrogel infusion additive manufacturing (HIAM), which enables fabrication of a wide range of micro-architected metals and alloys from a single photoresin composition. We use 3D architected hydrogel scaffolds as platforms for subsequent in situ material synthesis reactions, shown schematically in Fig. 1a. To fabricate metal microlattices, we use DLP (schematic in Supplementary Fig. 1) to print N,N-dimethylformamide (DMF)/polyethylene glycol diacrylate (PEGda)-based architected organogels (see Supplementary Information Discussion 1 for resin design). The DLP printing step defines the shape of the final part; the designed octet lattice shape used throughout this work can be found in Supplementary Fig. 2. Details of the resin composition and DLP printing and swelling parameters can be found in Supplementary Tables 1 and 2. After printing, a solvent exchange replaces DMF with water, converting the organogels into hydrogels. The hydrogel structures are then soaked in a metal salt precursor solution to allow metal ions to swell the hydrogel scaffold. Calcination in air converts the metal-salt-swollen hydrogels to metal oxides, and subsequent reduction in forming gas (95% N2, 5% H2) yields metal or alloy replicas of the designed architecture. Throughout the process, the part shape, defined during DLP printing, is maintained, with each dimension undergoing approximately 60–70% linear shrinkage, with a concomitant 65–90% approximate mass loss during calcination (see Supplementary Table 3 for shrinkage and mass loss for several materials).

a, Schematic of HIAM process. A DMF/PEGda-based 3D printed organogel structure is converted to an infused hydrogel replica after leaching out photoactive compounds, solvent exchange, and infusing an appropriate aqueous precursor. Subsequent calcination in air forms metal oxide structures, which are reduced to metals in forming gas. b–e, Optical images of the HIAM process for Cu metal, showing: b, printed organogel; c, infused hydrogel; d, calcined metal oxide; and e, reduced metal. f, Additional metals fabricated via HIAM including Ag and Ni, binary alloy CuNi, high-entropy alloy CuNiCoFe and refractory alloy W–Ni. g, An octet lattice infused with Cu(NO3)2 from one end and Co(NO3)2 from the other. h, After calcination and reduction, the Cu/Co gel is transformed to a Cu/Co multimaterial. i, Parallel calcination of several different infused gels. Scale bars: b,c, 5 mm; d–f, 1 mm; g, 1 cm; h, 2 mm; i, 2 cm.
To demonstrate the versatility of HIAM compared with previous VP AM techniques24,27, we used HIAM to fabricate octet lattice structures of copper (process steps shown in Fig. 1b–e), nickel, silver and alloys thereof (Supplementary Fig. 3), as well as more complex materials such as the high-entropy alloy CuNiCoFe and the refractory alloy W–Ni (Fig. 1f). Additional development was required for these materials; fabrication and characterization of CuNiCoFe and W–Ni are described in Supplementary Figs. 4 and 5, and Supplementary Information Discussions 2 and 3, respectively. We also fabricated multimaterials such as Cu/Co (Fig. 1g,h).
HIAM is distinguished by its ability to be parallelized; several organogels can be printed simultaneously, swelled in separate solutions and then calcined/reduced together. Figure 1i shows eight hydrogel lattices (precursors of Cu, CuNi, CuNiCoFe and CuNiCoFeCr) being simultaneously calcined to form oxides. This parallelization is impossible with existing VP methods and is a direct consequence of the temporal separation of part shaping and material selection. Compared with existing techniques that include precursors in the resin or introduce precursors through chemically directed swelling, HIAM enables exploration of a much larger compositional space, including multimaterials. From here, we focus on characterization of one pure metal and one alloy: Cu and CuNi.
Structural characterization
The external and internal morphologies of metal microlattices were investigated by using a combination of scanning electron microscopy (SEM) and Ga+ source focused ion beam (FIB) milling. SEM imaging revealed that Cu and CuNi samples maintained their octet lattice shape during thermal treatment (Fig. 2a,e), with beam diameters of approximately 40 µm (Fig. 2b,f). We FIB-milled representative cross-sections at nodes and observed dense and relatively defect-free structures. The Cu showed some less than 5 µm diameter pores and a lamellar crack (Fig. 2c), whereas the CuNi alloy (Fig. 2g) exhibited a similarly dense structure with micrometre-sized spherical pores, but no observed lamellar cracks. Energy dispersive X-ray spectroscopy (EDS) mapping showed homogenous distribution of Cu in the Cu lattice (Fig. 2d), and homogenous distribution of Cu and Ni in the CuNi lattice (Fig. 2h). See Supplementary Fig. 6 for structural characterization of additional materials including Ni, Ag and CuAg alloy.

a–c,e–g, SEM images of Cu (a–c) and CuNi (e–g) octet lattices, showing multiple unit cells from the top (a,e), a single node (b,f) and a FIB-milled cross-section showing the internal structure of a node from 52° tilt (c,g). d,h, EDS elemental mapping, showing uniform distribution of Cu (d) and uniform distribution of Cu and Ni (h). Scale bars: a,e, 100 µm; b,f, 50 µm; c,g, 20 µm; d,h, 50 µm.
Chemical characterization
To understand the chemical and microstructural evolution of these materials during calcination and reduction, we investigated the chemical composition of the metal microlattices by using X-ray diffraction (XRD), EDS, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). EDS analyses (Supplementary Fig. 7) of Cu and CuNi microlattices show that these materials contain, by weight, 93% and 86% of the target materials, respectively. The balance is made up of carbon, which is difficult to accurately quantify and probably includes some adventitious carbon (Supplementary Information Discussion 4), and aluminosilicate contamination from the furnace tubes. EDS analysis shows that the atomic ratio of Cu:Ni in our CuNi material is 1.21:1, or stoichiometrically Cu55Ni45. This deviation of alloy composition from swelling solution composition is probably due to different affinities of PEGda with the metal ions31. However, by adjusting the swelling solutions to account for preferential incorporation of certain ions, target compositions can be achieved with precision. For example, to target a Cu50Ni50 alloy, we swelled a hydrogel precursor in a 1:1.21 molar ratio of Cu(NO3)2:Ni(NO3)2. After calcination and reduction, EDS analysis showed that the stoichiometry of this cupronickel alloy was within 1% of the target composition, at Cu50.5Ni49.5 (Supplementary Fig. 8).
Calcination of metal-nitrate-infused gels in air at 700 °C with gas flow rate of 50 standard cubic centimetres per minute (sccm) produces metal oxide replicas of the architectures. The XRD patterns in Fig. 3a show that the calcined Cu precursor gel, which contained Cu(NO3)2, and the CuNi precursor gel, which contained Cu(NO3)2/Ni(NO3)2, were fully converted to CuO and CuO/NiO, respectively (see Supplementary Fig. 9 for additional materials). Notably, the CuO/NiO XRD pattern shows the presence of the individual CuO and NiO phases in the calcined material. Reduction of these metal oxides in forming gas (900 °C, 150 sccm) converts the CuO and CuO/NiO lattices to Cu and a homogenous CuNi alloy, respectively (Fig. 3b). As both CuNi and Cu have face-centred cubic (FCC) crystal structures, the single set of FCC reflections in the CuNi pattern shift to higher diffraction angles, which is a result of decreased lattice spacing due to the incorporation of the smaller Ni atom into the structure.

a, XRD patterns of calcined gels: Cu(NO3)2 gel is converted to CuO and Cu(NO3)2/Ni(NO3)2 gel is converted to CuO/NiO. b, XRD patterns of oxides reduced to parent metals: CuO is converted to Cu and CuO/NiO is converted to a homogenous CuNi alloy, as evidenced by the single set of FCC reflections. c, TGA profiles of metal-ion-infused gels heated to 700 °C in air at 1 °C min−1 reveal rapid mass loss events reaching maxima at 353 °C for Cu and 331 °C for CuNi. d, DSC profiles of metal-ion-infused gels heated to 400 °C in air at 1 °C min−1 reveal exothermic (Exo) events with maximum heat flow at 308 °C for Cu and at 304 °C for CuNi. AU, arbitrary units. Ref. ICSD #, reference pattern from Inorganic Crystal Structure Database (see Methods).
Figure 3c contains TGA measurements of Cu and CuNi gels heated in air at 1 °C min−1. During calcination the Cu and CuNi precursor gels retain 12.7% and 15.8% of the original mass, respectively, reaching mass stabilization indicative of full conversion between 370 °C and 380 °C. The derivative of sample weight with respect to temperature, dW/dT, shows the regions of highest mass loss rate (approximately 1 wt% °C−1) occur at 353 °C for Cu and at 331 °C for CuNi. Guides to the eye are placed at 110 °C, where initial dW/dT peaks occur for both Cu and CuNi, and 265 °C, where a subsequent dW/dT peak occurs for only Cu. Fig. 3d contains DSC profiles of Cu and CuNi precursor gels heated in air at 1 °C min−1 (see Supplementary Fig. 10 for additional materials). Both gels exhibit similar normalized heat flow profiles; exothermic peaks begin at approximately 235 °C for both and reach a maximum heat flow of −1.5 W g−1 at 308 °C for Cu and −2.6 W g−1 at 304 °C for CuNi.
Morphology and microstructure characterization
Our HIAM-fabricated metals are microcrystalline, with randomly oriented micrograins that are densely populated by annealing twins. The presence of micrometre-scale twinned regions in Cu is seen clearly in Ga+ ion-channelling images (Fig. 4a, yellow arrows point to twins) and electron backscatter diffraction (EBSD) maps (Fig. 4b).

a,b, Ga+ ion-channelling image (a) and EBSD map of Cu (b) show annealing twins. Cu has a complex micrograined structure and multiple twinned regions denoted by yellow arrows. c, TEM images of Cu show well-formed grain boundaries and aluminosilicate inclusions. FCC copper is observed from the diffraction pattern in the inset of c. d, TEM image of a twin boundary and aluminosilicate inclusion. e, Nanoindentation hardness of HIAM-fabricated Cu and CuNi samples is higher than predicted based on Hall–Petch scaling (dashed lines show calculated twin-induced hardening). Error bars show standard deviations of grain size and nanoindentation hardness. Insets: area-weighted grain size distribution for Cu and CuNi. Sample sizes: Cu hardness, n = 22; CuNi hardness, n = 44; Cu grain size, n = 246; CuNi grain size, n = 309. Scale bars: a, 50 µm; b, 20 µm; c, 2 µm, inset 10 nm−1; d, 500 nm.
Both Cu and CuNi have high crystallographic twin densities32, defined as the twin boundary length per cross-sectional area, of 1.7 × 106 m−1 for Cu and 1.3 × 106 m−1 for CuNi. For Cu and CuNi each grain contains on average 4.8 and 3.8 twin boundaries, respectively, with 88% and 75% of all grains containing at least one twin boundary. Additional twinning statistics measured by EBSD are reported in Supplementary Table 4.
Transmission electron microscopy (TEM) analysis of HIAM-fabricated Cu (Fig. 4c) shows more microstructural detail. We observe that grain boundaries and twin boundaries are well formed, and do not observe voids at triple junctions or secondary phases (for example, unreduced CuO or amorphous carbon). However, there exist aluminosilicate inclusions (see Supplementary Fig. 11 for TEM EDS) that result from contamination from the furnace tube (Fig. 4d). Image analysis of SEM micrographs (see Supplementary Information Discussion 5 and Supplementary Figs. 12 and 13) show that Cu has an area-weighted average grain size of 13.74 ± 8.43 µm, and CuNi has an area-weighted average grain size of 9.81 ± 4.79 µm (Fig. 4e, insets).
Mechanical characterization
Nanoindentation experiments performed on HIAM-fabricated Cu and CuNi revealed the hardness of Cu to be 1.81 ± 0.37 GPa and that of CuNi to be 2.15 ± 0.22 GPa. To contextualize these results, Fig. 4e contains a plot of nanoindentation hardness versus grain size for HIAM-fabricated Cu and CuNi compared with literature data for the same metals produced via traditional processing techniques (see Supplementary Table 5 for tabulated data). The plot also contains the expected hardness based on the Hall–Petch relation33 H = H0 + kd−1/2 that relates nanoindentation hardness H to grain size d for ductile metals, in which H0 is an intrinsic hardness for a single crystalline material and k is a scaling factor related to grain-boundary-induced hardening.
Twin boundaries disrupt dislocation motion during deformation32,34, increasing measured hardness. To account for this effect, we introduce a modified Hall–Petch relation35. To calculate an upper bound on twin-induced hardening, we assume that twin boundaries and grain boundaries equally impede dislocation motion; the grain size d effectively shrinks and is replaced by d/(1+N), where N is the number of twin boundaries per grain (see Supplementary Information Discussion 6 for derivation of this expression).
Discussion
Chemical composition and processing effects
Thermogravimetric and DSC analysis of Cu and CuNi calcination reveals that the water initially bound to the hydrogel polymer network evaporates below approximately 100 °C, indicated by the endothermic heat flow of approximately 0.25 W g−1 present in the DSC profiles36. Multiple thermally induced processes occur simultaneously upon further heating. Between 100 °C and 400 °C, the Cu sample undergoes (1) dehydration of \({\rm{Cu}}{({{\rm{NO}}}_{3})}_{2}\cdot x{{\rm{H}}}_{2}{\rm{O}}\) to anhydrous Cu(NO3)2, (2) thermal decomposition of anhydrous Cu(NO3)2 and (3) an exothermic combustion reaction37 in which the nitrate salt acts as an oxidizer of the PEGda (C26H46O13) polymer through the reaction Cu(NO3)2 + η C26H46O13 + (31η − 2.5) O2 → CuO + 23η H2O + 26η CO2 + N2, for which η is the molar ratio of PEGda to nitrate salt and (4) thermal decomposition of PEGda. TGA and DSC profiles of the CuNi samples suggest a similar process, with the copper and nickel nitrate salts simultaneously decomposing and acting as oxidizers for polymer combustion.
Both Cu and CuNi precursor gels exhibit exothermic events around 300 °C that are attributed to the combustion of the PEGda polymer scaffold and oxidation of the metal ions. The rates and temperatures of maximum heat flow are −1.5 W g−1 at 308 °C for Cu, and −2.6 W g−1 at 304 °C for CuNi (Fig. 3d) and indicate that during calcination the CuNi gel releases heat more rapidly. This finding is consistent with our observation that the rate of maximum heat flow in the Ni gel is even greater, at −3.97 W g−1 at 333 °C (Supplementary Information Discussion 7 and Supplementary Table 6). The onset of this exothermic event is similar for Cu and CuNi, (approximately 235 °C), whereas the onset of the corresponding event in the Ni gel occurs at a substantially higher temperature of approximately 295 °C. This trend suggests that the heat released from the exothermic combustion of the copper nitrate salt in the CuNi gel is sufficient to quickly increase the local temperature in the gel to the point where the nickel nitrate salt also contributes to the oxidation reaction at an apparently lower temperature, as is common in combustion synthesis38.
The Cu and CuNi samples contain fewer defects and pores compared with pure Ni and Ag (Supplementary Fig. 6). The presence of defects and pores is correlated with higher maximum dW/dT. During calcination, rapid thermal decomposition inhibits global, isotropic part shrinkage and drives a kinetic competition with mass loss, resulting in internal voids and pore formation. Slowing the rate of mass loss during calcination increased HIAM part density; we achieved this through a slow ramp rate of 0.25 °C min−1 and low pressure of approximately 6 Torr.
Mechanical characterization and twinning
The high annealing twin densities we observed, formed without the ordinarily requisite melting and recrystallization39, highlight the complex interplay of kinetic processes (for example, solid-state diffusion and grain nucleation) and thermodynamic grain coalescence during calcination and reduction. However, this high boundary density does not fully explain the 47% and 15% increase in hardness for Cu and CuNi, respectively. The observed aluminosilicate nanoinclusions are heterogeneously distributed and contribute to local variations rather than an increase in average hardness. With no evidence for secondary phases or grain boundary complexions (Fig. 4c,d), the high hardness of HIAM metals probably stems from atomic-scale features (for example, dissolved carbon) formed during the HIAM process (Supplementary Information Discussion 8).
Material selection after part shaping
In nearly every AM process so far, the material is decided before part shaping, that is, the AM material feedstock is the desired material or contains precursors that are subsequently converted into the desired material. Thus, the feedstock is inseparable from the final material composition. In addition, fabricating any new material necessarily requires a different feedstock. For ceramic and metal VP with precursors incorporated into the photoresin, the fabrication of new inorganic materials requires non-trivial photoresin design and print-parameter optimization.
HIAM is fundamentally different from modern AM processes in that the material is selected after part shaping. A blank organogel structure fabricated from a single resin formulation can be transformed into a vast number of different compositions. Because the inorganic precursors are infused after part shaping, HIAM is unlike the traditional slurry or inorganic–organic hybrid resin approaches in that only a single photoresin composition needs to be designed and optimized for VP, simplifying the material development process. We have shown that this capability also allows HIAM to fabricate complex materials such as refractory metals and high-entropy alloys as well as multimaterial metal structures, which were previously impossible tasks for VP. Finally, HIAM is generalizable to other gel-producing AM processes such as direct ink writing and two-photon lithography.
Conclusion
The HIAM process enables the creation of micro-architected metal 3D structures by using a versatile VP approach. The conversion of metal salts within polymer scaffolds to metal oxides and their subsequent reduction to metals and alloys requires only that the target material has water-soluble precursors and that the intermediate oxide formed after calcination can be reduced by hydrogen gas. The ability to fabricate metals by using this accessible and high-resolution process provides new opportunities for fabrication of energy materials, micro-electromechanical systems and biomedical devices. Because the material is selected only after the part is shaped, directed infusion enables fabrication of metallic multimaterials. Unprecedented compositional flexibility enables the fabrication of multicomponent alloys such as high-entropy alloys and refractory alloys, known to have intermetallic phases that lead to superior high-temperature behaviour and enhanced yield strength40. HIAM has direct implications for industrial use, as it provides a practical and powerful capability to integrate into the burgeoning VP printing ecosystem.
Methods
Fabrication of metal microlattices
3D printing resin preparation
Here 28 ml of DMF (Sigma-Aldrich, >99.9%) was mixed with 35 ml PEGda Mn = 575 (Sigma-Aldrich). Separately, 347 mg 2-dimethylamino-2-(4-methyl-benzyl)-1-(4-morpholin-4-yl-phenyl)-butan-1-one (Irgacure 379; iGM Resins), 229 mg bis[4-(dimethylamino)phenyl]methanone (Michler’s ketone; Sigma-Aldrich) and 10.3 mg 1-(phenyldiazenyl)naphthalen-2-ol (Sudan I; Sigma-Aldrich) was stirred into 7 ml of DMF. This solution was then added to the DMF/PEGda mixture and swirled until completely homogenous.
DLP 3D printing
The resin was formed into 3D organogel structures by using a commercial 405 nm wavelength DLP 3D printer (Autodesk Ember). Lattice structures were designed to consist of octet lattices with 200 µm beam diameter and 1.5 mm unit cell size.
Post-processing washes
After printing, the organogel lattices were yellow in colour due to the presence of the ultraviolet blocker Sudan I. To remove unreacted photoresin components, each 3D printed organogel structure was soaked in DMF for 1 h on a hot plate at 70 °C. After the first DMF rinse, the DMF was decanted, and the organogel was soaked again in fresh DMF for 1 h at 70 °C. After this process, the lattice appeared clear. Subsequently, each organogel structure was soaked in deionized water for 1 h at 70 °C, followed by a second soak in fresh deionized water for 1 h at 70 °C to convert the structures from organogel lattices to hydrogel lattices. The solvent-exchange step is needed to remove residual DMF in the structure, which can lead to formation of porosity upon calcination and reduction (Supplementary Fig. 14). The presence of DMF can also cause precipitation of the metal salt during the swelling process, leading to an inhomogeneous distribution of the metal precursors.
Swelling in metal salt solutions
2 M solutions of copper nitrate, nickel nitrate, iron nitrate, cobalt nitrate, silver nitrate, chromium nitrate and ammonium metatungstate were prepared with deionized water. Hydrogel structures were immersed in the appropriate metal salt solution for 24 h at 70 °C or 2 weeks at 70 °C (only for W-Ni).
Calcination and reduction
Metal-ion-swelled hydrogel structures were calcined in a vacuum tube furnace (MTI, OTF-1500X). The samples were placed in and covered by alumina boats within an alumina tube (MTI, 80 cm length, 51 mm inner diameter) that itself sat inside a larger fused quartz tube (MTI, 1 m length, 92 mm inner diameter). The furnace was set up in this fashion to prevent copper vapours resulting from the heating process from interacting with the quartz tube, which has been shown to lead to devitrification of the amorphous quartz tube and potential deposition of SiO2(ref. 41). The addition of the inner alumina tube substantially reduced the amount of SiO2 particles observed after thermal treatment. During calcination, a 0.25 °C min−1 ramp rate was used up to a maximum temperature of 700 °C, followed by a 3 h isothermal hold and cooling at 2 °C min−1, under a compressed air flow of 50 sccm at a pressure of approximately 7 Torr. The slow ramp rate of 0.25 °C min−1 during calcination was found to be critical for controlling the highly exothermic decomposition of the nitrate salts and minimizing porosity in the fabricated samples. After calcination of metal-ion-swelled gels, metal oxide structures were produced, except for gels infused with silver nitrate salts, which produced elemental silver or silver composite lattices. All calcined structures except pure Ag and W-Ni were then reduced in forming gas at a flow rate of 150 sccm at a pressure of approximately 22 Torr by heating at 3 °C min−1 to 900 °C or 700 °C (only for CuAg) followed by a 6 h isothermal hold, resulting in copper, nickel, homogenous cupronickel alloy and heterogeneous copper–silver alloy lattices. The W-Ni structures were calcined and reduced under different conditions; W-Ni samples underwent calcination at 0.25 °C min−1 to 500 °C, then 1 °C min−1 to 700 °C, and a 3-hour isothermal hold. The W-Ni samples were reduced following a thermal profile of 3 °C min−1 to 1,200 °C and a 1-hour isothermal hold, under forming gas at a flow rate of 500 sccm at atmospheric pressure (through a gas bubbler).
Characterization and measurement
SEM and FIB milling
Samples were imaged via SEM (FEI Versa 3D DualBeam) at an accelerating voltage of 10–20 kV. Elemental analysis was performed in the same instrument by using EDS (Bruker Quantax 200, XFlash 6|60 detector), with an applied voltage of 20 kV or 5 kV. The applied voltage was selected to ensure the greatest accuracy when quantifying metal elements (Supplementary Information Discussion 4 and Supplementary Fig. 15). Gallium FIB milling was performed in the same instrument to mill lattice cross-sections by using an accelerating voltage of 30 kV and a current of 50 nA. FIB cleaning of the cross-sections was performed by using an accelerating voltage of 16 kV and a current of 25 nA.
Electron backscatter diffraction
With the z direction and build direction aligned, metal lattice samples were loaded into an Oxford EBSD System in a Zeiss 11550VP SEM and were imaged by using a 120 µm aperture at 20 kV. Data analysis for the Kikuchi maps was done in AZtecHKL software. All maps display the inverse pole figure in the z direction.
TEM
Lamellae with thicknesses of less than 100 nm were prepared for TEM by using a liftout procedure in an SEM (FEI Versa 3D DualBeam). The top surface of the liftout region was protected with an approximately 100 nm thick layer of Pt deposited via a gas injection system, followed by an approximately 400 nm thick layer of Ga+ FIB-deposited Pt in the same chamber. Next, a Ga+ ion beam was used to carve out trenches into the node of the lattice, forming a U-cut to free the approximately 1 µm thick metal lamella base from the rest of the lattice. A tungsten needle (EZlift program) was attached to the lamella with FIB-deposited Pt before being cut free of the sample and transferred to a copper halfmoon grid. The lamella was attached to the Cu grid with FIB-deposited Pt and the tungsten needle was cut away to free the sample. After detaching the tungsten needle, a series of FIB cuts with a decreasing Ga+ voltage/current (30 kV/100 pA; 30 kV/10 pA; 16 kV/23 pA) were used to progressively thin the cross-section of the lamella structure to less than 100 nm. TEM imaging was performed in a Jeol JEM-2800 transmission electron microscope with a 200 kV beam.
XRD
Powder XRD (PANalytical X’Pert Pro) patterns were collected by using a Cu Kα1 source at 45 kV and 40 mA. Samples were either ground into powders or flattened and attached to an amorphous zero-background sample holder by using clay before XRD analysis. Experimental patterns were compared to references from the Inorganic Crystal Structure Database42.
TGA
TGA (TA Instruments TGA 550A) was performed by heating samples to 700 °C at a rate of 1 °C min−1 in an air flow of 25 ml min−1 while the mass of the sample was continuously measured.
DSC
DSC (TA Instruments DSC 25) was performed by heating samples to 700 °C at a rate of 1 °C min−1 in an air flow of 25 ml min−1 while heat flow to the sample was continuously measured.
Nanoindentation
Samples were prepared for nanoindentation by mounting them in acrylic (Beuhler SamplKwik) and curing for 12 h in silicone moulds. The samples were polished first with 300 grit until the metal structure was exposed. The samples then were polished with 600 grit, followed by 1200 grit and subsequently a 0.25 µm grit suspension (Beuhler MetaDi Polycrystalline Diamond Slurry). Indentation was performed by using an Agilent G200 Nano Indenter with XP module by using a Berkovich tip with an area function given by A = 24.5h2 + 688h, where A is the tip contact area and h is the contact depth. Samples were indented at 10−3 strain rate to a maximum depth of 1 µm, followed by a 2 s hold and subsequent unloading. Grain sizes were determined via SEM image analysis that consisted of tracing grains on the surface of the sample and by using ImageJ to analyse the resulting shapes. These values were corroborated by EBSD mapping analysis of grain size. Experimental data was compared to a Hall–Petch fit of reference data from the literature43,44,45,46,47.
Data availability
The data generated and/or analysed during the current study are available from the corresponding authors on reasonable request.
Code availability
The code generated during the current study is available from the corresponding authors on reasonable request.
References
Vafadar, A., Guzzomi, F., Rassau, A. & Hayward, K. Advances in metal additive manufacturing: a review of common processes, industrial applications, and current challenges. Appl. Sci. 11, 1213 (2021).
Mohd Yusuf, S., Cutler, S. & Gao, N. Review: the impact of Mmtal additive manufacturing on the aerospace industry. Metals 9, 1286 (2019).
Velásquez-García, L. F. & Kornbluth, Y. Biomedical applications of metal 3D printing. Annu. Rev. Biomed. Eng. 23, 307–338 (2021).
Laureijs, R. E. et al. Metal additive manufacturing: cost competitive beyond low volumes. J. Manuf. Sci. Eng. 139, 81010 (2017).
Plocher, J. & Panesar, A. Review on design and structural optimisation in additive manufacturing: towards next-generation lightweight structures. Mater. Des. 183, 108164 (2019).
DebRoy, T. et al. Additive manufacturing of metallic components – process, structure and properties. Prog. Mater. Sci. 92, 112–224 (2018).
Svetlizky, D. et al. Directed energy deposition (DED) additive manufacturing: physical characteristics, defects, challenges and applications. Materials Today 49, 271–295 (2021).
Bartlett, J. L. & Li, X. An overview of residual stresses in metal powder bed fusion. Addit. Manuf. 27, 131–149 (2019).
Zhang, D., Liu, X. & Qiu, J. 3D printing of glass by additive manufacturing techniques: a review. Front. Optoelectron. 14, 263–277 (2021).
Chen, Z. et al. 3D printing of ceramics: a review. J. Eur. Ceram. Soc. 39, 661–687 (2019).
Yee, D. W. & Greer, J. R. Three-dimensional chemical reactors: in situ materials synthesis to advance vat photopolymerization. Polym. Int. 70, 964–976 (2021).
King, W. E. et al. Laser powder bed fusion additive manufacturing of metals; physics, computational, and materials challenges. Appl. Phys. Rev. 2, 041304 (2015).
Dass, A. & Moridi, A. State of the art in directed energy deposition: from additive manufacturing to materials design. Coatings 9, 418 (2019).
Bandyopadhyay, A. & Heer, B. Additive manufacturing of multi-material structures. Mater. Sci. Eng. R Reports 129, 1–16 (2018).
Li, Y. et al. A review on functionally graded materials and structures via additive manufacturing: from multi-scale design to versatile functional properties. Adv. Mater. Technol. 5, 1900981 (2020).
Jadhav, S. D. et al. Surface modified copper alloy powder for reliable laser-based additive Mmanufacturing. Addit. Manuf. 35, 101418 (2020).
Tumbleston, J. R. et al. Continuous liquid interface production of 3D objects. Science 347, 1349–1352 (2015).
Sun, C., Fang, N., Wu, D. M. & Zhang, X. Projection micro-stereolithography using digital micro-mirror dynamic mask. Sens. Actuator A Phys. 121, 113–120 (2005).
Kajtaz, M., Subic, A., Brandt, M. & Leary, M. in Materials in Sports Equipment (ed. Subic, A.) 161–198 (Elsevier, 2019).
Manoj, A., Bhuyan, M., Raj Banik, S. & Ravi Sankar, M. 3D printing of nasopharyngeal swabs for COVID-19 diagnose: past and current trends. Mater. Today Proc. 44, 1361–1368 (2021).
Ligon, S. C., Liska, R., Stampfl, J., Gurr, M. & Mülhaupt, R. Polymers for 3D printing and customized additive manufacturing. Chem. Rev. 117, 10212–10290 (2017).
Herzberger, J., Meenakshisundaram, V., Williams, C. B. & Long, T. E. 3D Printing all-aromatic polyimides using stereolithographic 3D printing of polyamic acid salts. ACS Macro Lett. 7, 493–497 (2018).
Zhang, B. et al. Highly stretchable hydrogels for UV curing based high-resolution multimaterial 3D printing. J. Mater. Chem. B 6, 3246–3253 (2018).
Yee, D. W. et al. Hydrogel-based additive manufacturing of lithium cobalt oxide. Adv. Mater. Technol. 6, 2000791 (2021).
Nguyen, H. X., Suen, H., Poudel, B., Kwon, P. & Chung, H. Development of an innovative, high speed, large-scaled, and affordable metal additive manufacturing process. CIRP Ann. 69, 177–180 (2020).
Vyatskikh, A. et al. Additive manufacturing of 3D nano-architected metals. Nat. Commun. 9, 593 (2018).
Oran, D. et al. 3D nanofabrication by volumetric deposition and controlled shrinkage of patterned scaffolds. Science 362, 1281–1285 (2018).
Long, T. E. & Williams, C. B. Printing nanomaterials in shrinking gels. Science 362, 1244–1245 (2018).
Mooraj, S. et al. Three-dimensional hierarchical nanoporous copper via direct ink writing and dealloying. Scr. Mater. 177, 146–150 (2020).
Miyanaji, H. et al. Binder jetting additive manufacturing of copper foam structures. Addit. Manuf. 32, 100960 (2020).
Sari, N., Kahraman, E., Sari, B. & Özgün, A. Synthesis of some polymer-metal complexes and elucidation of their structures. J. Macromol. Sci. A 43, 1227–1235 (2006).
Roy, B. & Das, J. Strengthening face centered cubic crystals by annealing induced nano-twins. Sci. Rep. 7, 1–8 (2017).
Hall, E. O. The deformation and ageing of mild steel: III discussion of results. Proc. Phys. Soc. B 64, 747–753 (1951).
Lu, L., Shen, Y., Chen, X., Qian, L. & Lu, K. Ultrahigh strength and high electrical conductivity in copper. Science 304, 422–426 (2004).
Pande, C. S., Rath, B. B. & Imam, M. A. Effect of annealing twins on Hall–Petch relation in polycrystalline materials. Mater. Sci. Eng. A 367, 171–175 (2004).
Gun’ko, V. M., Savina, I. N. & Mikhalovsky, S. V. Properties of water bound in hydrogels. Gels 3, 37 (2017).
Danks, A. E., Hall, S. R. & Schnepp, Z. The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis. Mater. Horiz. 3, 91–112 (2016).
Moore, J. J. & Feng, H. J. Combustion synthesis of advanced materials: part I. Reaction parameters. Prog. Mater. Sci. 39, 243–273 (1995).
Bahl, S. et al. Non-equilibrium microstructure, crystallographic texture and morphological texture synergistically result in unusual mechanical properties of 3D printed 316L stainless steel. Addit. Manuf. 28, 65–77 (2019).
Senkov, O. N., Miracle, D. B., Chaput, K. J. & Couzinie, J. P. Development and exploration of refractory high entropy alloys - A review. J. Mater. Res. 33, 3092–3128 (2018).
Lisi, N. et al. Contamination-free graphene by chemical vapor deposition in quartz furnaces. Sci. Rep. 7, 1–11 (2017).
Bergerhoff, G. & Brown, I. D. in Crystallographic Databases (eds Allen, F. H. et al.) (International Union of Crystallography, 1987; https://icsd.fiz-karlsruhe.de/).
Wang, C., Hossain Bhuiyan, M. E., Moreno, S. & Minary-Jolandan, M. Direct-write printing copper-nickel (Cu/Ni) alloy with controlled composition from a single electrolyte using co-electrodeposition. ACS Appl. Mater. Interfaces 12, 18683–18691 (2020).
Emeis, F., Peterlechner, M., Divinski, S. V. & Wilde, G. Grain boundary engineering parameters for ultrafine grained microstructures: proof of principles by a systematic composition variation in the Cu-Ni system. Acta Mater. 150, 262–272 (2018).
Chang, S. Y. & Chang, T. K. Grain size effect on nanomechanical properties and deformation behavior of copper under nanoindentation test. J. Appl. Phys. 101, 033507 (2007).
Bansal, S., Toimil-Molares, E., Saxena, A. & Tummala, R. R. Nanoindentation of single crystal and polycrystalline copper nanowires. in Proceedings Electronic Components and Technology, 2005. ECTC ’05. Vol. 1, 71–76 (IEEE, 2005).
Bahr, D. F. & Vasquez, G. Effect of solid solution impurities on dislocation nucleation during nanoindentation. J. Mater. Res. 20, 1947–1951 (2005).
Acknowledgements
This work was supported by the US Department of Energy, Office of Science, Basic Energy Sciences under award no. DE-SC0016945. We thank C. Ma for support and assistance with instruments in the Geological and Planetary Sciences Division Analytical Facility at Caltech, as well as M. Xu and X. Pan for assistance with TEM experiments at UC Irvine Materials Research Institute. M.A.S. acknowledges a graduate fellowship from the Resnick Sustainability Institute at Caltech. R.A.G. acknowledges the AI4SCIENCE graduate fellowship at Caltech. K.N. acknowledges a fellowship from the Masason Foundation.
Author information
Authors and Affiliations
Contributions
M.A.S, D.W.Y, and J.R.G. conceived of and designed the experiments. M.A.S. and D.W.Y. designed the photoresin, printing parameters and swelling protocol and fabricated samples. M.A.S., D.W.Y., and K.N. performed the thermal treatments and FIB/SEM/EDS experiments. M.A.S. performed the DSC, TGA and XRD experiments. R.A.G. performed the nanoindentation and EBSD experiments and prepared TEM samples. All authors analysed data and discussed the findings. M.A.S., R.A.G., D.W.Y. and J.R.G. wrote the manuscript. All authors edited and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
K.N. founded the company 3D Architech, Inc., which has an option and right to acquire an exclusive license to US Patent 11318435B2.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–15, Tables 1–6 and Discussions 1–8.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Saccone, M.A., Gallivan, R.A., Narita, K. et al. Additive manufacturing of micro-architected metals via hydrogel infusion. Nature 612, 685–690 (2022). https://doi.org/10.1038/s41586-022-05433-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05433-2
- Springer Nature Limited
This article is cited by
-
Seven technologies to watch in 2024
Nature (2024)
-
Roll-to-roll, high-resolution 3D printing of shape-specific particles
Nature (2024)
-
High-throughput printing of combinatorial materials from aerosols
Nature (2023)
-
Metal 3D nanoprinting with coupled fields
Nature Communications (2023)
-
A Generalized Polymer Precursor Ink Design for 3D Printing of Functional Metal Oxides
Nano-Micro Letters (2023)