

Abstract
Human topoisomerases comprise a family of six enzymes: two type IB (TOP1 and mitochondrial TOP1 (TOP1MT), two type IIA (TOP2A and TOP2B) and two type IA (TOP3A and TOP3B) topoisomerases. In this Review, we discuss their biochemistry and their roles in transcription, DNA replication and chromatin remodelling, and highlight the recent progress made in understanding TOP3A and TOP3B. Because of recent advances in elucidating the high-order organization of the genome through chromatin loops and topologically associating domains (TADs), we integrate the functions of topoisomerases with genome organization. We also discuss the physiological and pathological formation of irreversible topoisomerase cleavage complexes (TOPccs) as they generate topoisomerase DNA–protein crosslinks (TOP-DPCs) coupled with DNA breaks. We discuss the expanding number of redundant pathways that repair TOP-DPCs, and the defects in those pathways, which are increasingly recognized as source of genomic damage leading to neurological diseases and cancer.
Similar content being viewed by others


Introduction
DNA topoisomerases are present in all domains of life to resolve a wide variety of topological problems arising from the length of the human double-helix DNA polymer (about 3 × 109 bp) as it is folded, bent and highly compacted into the cell nucleus while remaining accessible to RNA and DNA polymerases. In addition, each human cell contains 100–1,000 copies of circular, ~16,000 bp mitochondrial DNA (mtDNA), as well as long and folded RNAs that are even more abundant than DNA and present in all subcellular compartments.
Human topoisomerases and the associated repair enzymes tyrosyl-DNA phosphodiesterases (TDPs; TDP1 and TDP2) are located both in the nucleus and in mitochondria; in addition, cytoplasmic RNAs are handled by TOP3B and TDP2 (refs1,2) (Supplementary Table 1).
In this Review, we first provide basic information on the six human topoisomerases. We discuss their specific and overlapping roles as regulators of nucleic acid topology and metabolism, thereby complementing other recent reviews1,3,4,5,6,7,8. We emphasize the need to further define the roles of topoisomerases in genome organization and stability, as well as the increasingly recognized implication of topoisomerases in generating deleterious genomic lesions associated with irreversible topoisomerase cleavage complexes (TOPccs). We describe the molecular mechanisms by which abortive TOPccs damage the genome and the multiple pathways that repair those cellular lesions and how they relate to genomic instability.
DNA and RNA topological problems
To manage the topology of the long, folded and intertwined DNA and RNA polymers that are attached to scaffolding structures and are metabolically and dynamically processed by large molecular machines (such as transcription, DNA replication, chromatin remodelling and DNA repair complexes), human cells use their six topoisomerases often redundantly, but also in specific ways depending on the topological problem, the surrounding cellular structures and the differentiation status of the cell. This section outlines topological problems and the molecular solutions provided by each of the topoisomerases. Additional details are provided for the TOP3 enzymes, which were not covered in depth in our previous Review1.
DNA and RNA topological conversions by topoisomerases
One of the most common changes in DNA topology is the opening of the double helix by helicases during transcription and replication and by ATPase translocase ‘motors’ during chromatin remodelling (Fig. 1a,b). In addition, each nucleosome absorbs about two left-handed (negative) supercoils. Due to the length of the genome, its attachment to the nuclear scaffold and the viscosity of the nuclear milieu, DNA cannot freely rotate on its axis and the opening of the double helix generates DNA overtwisting (positive DNA supercoiling (Sc+)) in front (downstream) of the helicase or other motor proteins, and DNA under-twisting (negative supercoiling (Sc–)) behind (upstream) them. Excessive torsional strain first changes the helical twist (number of crossovers of the two strands across each other) until the entire duplex of DNA (or RNA) winds around itself (writhe) and flips into alternative plectonemic structures with crossover segments and entanglements. TOP1, TOP1MT, TOP2A and TOP2B readily remove both Sc+ and Sc– (Fig. 1; Supplementary Fig. 1) until the torsional strain is eliminated. The other two topoisomerases, TOP3A and TOP3B, just relax hyper-negative supercoiling, as they can only form cleavage complexes in single-stranded nucleic acids (Fig. 1a,e–g).

Sites of action of topoisomerases (red, blue and green circles), duplex nucleic acids (parallel lines) without their double-helix structure and DNA segments whose ends are not free to rotate on their helical axis due to steric and physical constraints (parallel pegs) representing topological domains. a | Twin supercoiled domain model54. Opening the DNA duplex by helicases and ATPase translocases within a topological domain during transcription and chromatin remodelling generates positive DNA supercoiling (Sc+) ahead of the moving helicase (or translocase) and negative supercoiling (Sc–) behind it. Excessive supercoiling generates writhe that brings together distant regions of DNA that form crossovers. Topoisomerase 1 (TOP1), mitochondrial TOP1 (TOP1MT), TOP2A and TOP2B remove both Sc+ and Sc– by incising double-stranded DNA; TOP3B (and TOP3A) relax hyper-negative supercoiling by nicking and closing single-stranded DNA segments. b | Replication forks generate Sc+ in front of the translocating replisome, which is removed by TOP1 and TOP2A. If the replisome swivels due to the twisting force, Sc+ diffuses behind the replisome and generates precatenanes, which are removed by TOP2A37. TOP3A may also remove precatenanes if they include single-stranded DNA segments. c | TOP2A and TOP2B decatenate topological domains by passing one DNA molecule through the double-stranded DNA break made in the other DNA molecule (double-strand passage). d | TOP2A and TOP2B resolve DNA knots by double-strand passage. e | TOP3A in association with the Bloom syndrome protein (BLM)–TOP3A–RecQ-mediated genome instability proteins (RMI1/2) (BTR) dissolvasome complex (not shown) resolves DNA hemicatenanes arising during replication and recombination by passing a single strand of DNA through a break made in another DNA strand (single-strand passage)6,357. f | TOP3B is the only RNA-only topoisomerase14; it resolves intramolecular RNA intertwines (knots) by single-strand passage. g | TOP3B can also resolve RNA catenanes by single-strand passage197. See Supplementary Fig. 1 for biochemical, molecular and structural details.
Other topological difficulties arise during replication and chromatin loop formation when duplex DNA molecules form catenanes. Decatenation reactions are carried out by TOP2 enzymes1 (Fig. 1c; Supplementary Fig. 1d,m). TOP2 enzymes can also resolve precatenanes formed behind replication forks (Fig. 1b) and unknot duplex DNA molecules (Fig. 1d). Such knots have been proposed to contribute to chromatin organization9,10 and recombination11 in yeast models.
The eukaryotic type IA topoisomerases, TOP3A and TOP3B, catalyse a unique range of topological changes requiring the passage of a single strand of nucleic acid through another (Fig. 1e–g). DNA hemicatenanes are the preferential substrate for TOP3A6 (Fig. 1e). A transcript isoform of TOP3A is also essential for mtDNA replication and segregation12,13 (Box 1). Unique among the human topoisomerases, TOP3B — similar to its yeast and bacterial paralogs Top3 and Topo III, respectively — acts as a dual DNA and RNA topoisomerase14,15. RNA knots and catenanes are the likely cellular substrates for TOP3B14 (Fig. 1f,g).
Topoisomerase biochemistry
All topoisomerases act by cleaving and rejoining the nucleic acid backbone using a tyrosine nucleophilic residue1 (Supplementary Fig. 1a,b). Each break results from the formation of a covalent bond between the topoisomerase catalytic Tyr residue and one end of the broken nucleic acid. The polarity is specific for each topoisomerase: 3′ end bonding for TOP1 and TOP1MT, and 5′ end bonding for the other topoisomerases. The covalent catalytic intermediates are referred to as the TOPccs. Vertebrate TOP2A and TOP2B and yeast Top2 (type II topoisomerases) function as homodimers and generate DNA double-strand breaks (DSBs) with a canonical 5′ base overhang1,16,17 (Supplementary Fig. 1d).
Four main biochemical characteristics differentiate topoisomerases: polarity (3′ versus 5′ tyrosyl linkage); substrate specificity (single-stranded versus double-stranded DNA or RNA); nucleic acid relaxation mechanisms (DNA strand rotation in the case of TOP1 and TOP1MT versus crossover inversion in the case of TOP2A, TOP2B, TOP3A and TOP3B, and RNA crossover inversion in the case of TOP3B); and cofactor requirements (ATP and/or magnesium) (Supplementary Fig. 1d,e; Supplementary Table 1).
TOP1 and TOP1MT (type IB topoisomerases) cleave only one strand of double-stranded DNA by forming the 3′-phosphotyrosyl linkage (3′ DNA–protein crosslinks (DPCs)) (Supplementary Fig. 1a; Supplementary Table 1), which relates them to the prokaryotic Tyr recombinases18,19. Unlike type IA topoisomerases (TOP3A and TOP3B) (see below), TOP1 and TOP1MT only process double-stranded DNA and relax DNA supercoils processively and without metal or protein cofactor by controlled rotation of the broken strand around the intact strand20,21, with a preference for nucleosome-free DNA10,22 (Supplementary Fig. 1c). Hence, TOP1 and TOP1MT are also referred to as ‘swivelases’ or ‘DNA untwisting enzymes’23.
TOP2A and TOP2B (type IIA topoisomerases) act as ‘writhases’ or ‘crossover invertases’, which enables them to resolve not only supercoils but also catenanes and knots by passing one DNA duplex through the DSB made in another duplex at crossover points between the two duplexes (Fig. 1c,d; Supplementary Fig. 1d,m; Supplementary Table 1). Note that TOP2A and TOP2B also generate DNA single-strand breaks (SSBs) when only one protein of the dimer breaks the DNA1. The main differences between TOP2A and TOP2B are in the polypeptide sequence of their carboxy-terminal domains (CTDs) (Supplementary Fig. 1g), their cell cycle regulation (TOP2A increases in abundance throughout S phase, is degraded at the end of mitosis and is dispensable in terminally differentiated cells)8,24 and the fact that TOP2A preferentially relaxes Sc+ whereas TOP2B relaxes Sc– and Sc+ similarly17,25.
Both the type IIA topoisomerases and the type IA topoisomerases — TOP3A and TOP3B — cleave nucleic acids by forming 5′ DPCs with the help of magnesium positioning nucleic acids in their active site3,6 (Supplementary Fig. 1g,h). However, type IA topoisomerases differ from type IIA topoisomerases in their selectivity for binding single-stranded rather than double-stranded nucleic acids, their lack of ATP requirement and the activity of TOP3B as a dual DNA and RNA topoisomerase6,14,26 (Fig. 1e–g; Supplementary Table 1). TOP3 enzymes are also dependent on protein scaffolding cofactors: RecQ-mediated genome instability protein 1 (RMI1) and RMI2 for TOP3A, and Tudor domain-containing protein 3 (TDRD3) for TOP3B6,15,27. TOP3A, RMI1 and RIM2 form the heterotrimeric Bloom syndrome protein (BLM)–TOP3A–RMI1/2 (BTR) dissolvasome complex for recombination28 and resolution of double Holliday junctions associated with DNA replication6. TOP3A can also associate with other helicases and translocases including the helicase Fanconi anaemia group M protein (FANCM) to suppress sister chromatid exchanges and promote replication restart29. The TOP3B partner TDRD3 forms a multimeric RNA regulator complex with fragile X mental retardation protein15. In addition to its role in TOP3B recruitment, TDRD3 has also been shown to increase the processivity of TOP3B27,30 (Supplementary Fig. 1n).
TOPccs are normally transient and ‘self-reversible’, as the deoxyribose hydroxyl ends of the cleaved DNA act as nucleophiles towards the tyrosyl–DNA phosphodiester bonds1. Efficient substrate resealing requires the alignment of the ends of the broken DNA inside the TOPcc (Supplementary Fig. 1a,b). Such alignment is provided both by the base pairing and stacking of duplex DNA and by atomic interactions with the topoisomerase. If the TOPccs fail to reseal, they become topoisomerase DNA–protein crosslinks (TOP-DPCs): stalled (abortive or irreversible) TOPccs consisting of a DNA–protein (topoisomerase) crosslink at the end of a break, which require DNA repair for their resolution (see below). Trapping of TOP-DPCs is the mechanism of action of widely used anticancer and antibacterial chemotherapies17,31,32 (Supplementary Box 1). Multiple endogenous and environmental factors also induce TOP-DPCs1,17.
Roles in replication and transcription
Transcription and replication constantly change DNA topology, and thus require the activities of topoisomerases.
Replication of the nuclear and mitochondrial genomes
As expected for genes involved in regulating DNA replication, TOP1, TOP2A and TOP3A transcript levels are linked with cell proliferation6,33,34,35, and their expression is correlated with the proliferation marker Ki67 and replicative polymerases and helicases in the cancer cell line encyclopaedia database36 (Supplementary Fig. 1).
During replication fork progression, topoisomerases dissipate the supercoiling generated by DNA unwinding37. Without topoisomerase activity, the formation of Sc+ in front of the replication fork and the resulting tightening of the DNA duplex with the potential formation of plectonemes would block replisome movement. Being a highly processive swivelase, TOP1 can remove supercoils in short DNA segments devoid of nucleosomes, whereas TOP2A, acting as a writhase, likely acts at crossover points where the DNA enters and exits nucleosomes22,38 (Fig. 1b). Yeast Top2 has also been shown to efficiently remove Sc+ in single-molecule model systems39, and yeast Top1 was shown to act at replication pause sites in ribosomal gene arrays40. Unexpectedly, yeast Top1 (and Top2 when acting as a Top1 backup) enforces replication pausing by the topoisomerase 1-associated factor (Tof1) at replication fork barriers by binding to Tof1 and preventing head-on collisions between replication and transcription machineries40. Whether the vertebrate paralog of Tof1 has the same coordinated functions with TOP1 and TOP2A requires testing41.
The topology of DNA behind replisomes depends on whether the frontal Sc+ forces the replisome to rotate around the DNA axis. Such rotation diffuses the Sc+ behind the replisome, generating braided daughter strands that are referred to as precatenanes37 (Fig. 1b), and can only be resolved by TOP2 (or potentially by TOP3 if the precatenanes contain a single-stranded segment of DNA)42. Rotation of the replisome is believed to be rare during replication due to the large protein complexes assembled around the replisome and the removal of Sc+ by TOP1 and TOP2. However, rotation has been reported in yeast43,44 as replisomes converge or in mammalian cells at common fragile sites, which are chromosomal regions that are sensitive to replication stress and prone to DNA breaks45,46. In addition to its role in replication fork pausing, Tof1 can also stabilize replisomes by preventing fork rotation43.
The role of TOP2A in replication termination appears to be limited, as newly replicated DNA is decatenated during replication (Fig. 1b), and most decatenation is complete upon entry into prophase8. Nevertheless, Top2 and TOP2A may be crucial for replication termination in highly repetitive sequences, such as telomeres, centromeres and ribosomal DNA, where Top2 and TOP2A activity may maintain segments of sister chromosomes catenated to assist in their condensation and cohesion, while subsequently allowing segregation at anaphase8.
As replication proceeds within replication domains (of 400–800 kb) that match topologically associating domains (TADs)47, daughter DNA molecules are catenated due to the helical structure of the replicating DNA. TOP2A is essential for decatenating daughter DNA molecules and resolving sister chromatid intertwines1,8,48 (Fig. 1c). In Xenopus laevis oocytes, following dissociation of the replicative CMG helicase at converging replisomes, catenated DNA loops are disentangled at their crossover points by TOP2A49.
TOP3A is a key enzyme for the completion of replication because it is the sole topoisomerase capable of removing hemicatenanes arising from converging replication forks42 and from recombination intermediates1,6 (Fig. 1e). The BTR dissolvasome complex has been shown to sense replication protein A (RPA)-coated single-stranded DNA, which may explain its role in restarting stalled replication forks50,51. At the end of S phase, in addition to its role in resolving ultra-fine DNA bridges between sister chromatids52, TOP3A in coordination with the helicase Plk1-interacting checkpoint helicase (PICH; also known as ERCC6L) has been shown to generate replicated DNA Sc+ segments, which are preferential substrates for TOP2A-mediated decatenation, thereby enabling the segregation of chromatin fibres during anaphase53. These observations suggest that TOP3A can couple its activity with different helicases: BLM in the dissolvasome, FANCM at stalled replication forks and PICH during mitosis. The potential activity of TOP3A for removing hemicatenanes during replication in human cells needs to be further established.
In mitochondria, TOP3A acts as a hemicatenane resolvase; in its absence, newly replicated mtDNA molecules remain intertwined at the mtDNA replication origin, a phenotype which is further accentuated in the absence of TOP1MT.12 (Box 1). Notably, TOP3A acts independently of the BTR dissolvasome (as BLM, RMI1 and RMI2 do not enter mitochondria), which could imply that a mitochondrial helicase replaces the dissolvasome in working together with TOP3A.
Topoisomerases and transcription
Topoisomerases are needed to resolve the topological tensions arising during transcription. The classical twin domain model predicts a requirement for topoisomerase activity to relax Sc+ ahead of the RNA polymerase and Sc– behind it54,55 (Fig. 1a). Both TOP1 and TOP2 can act behind transcription complexes to remove Sc– (Fig. 2a): TOP1 in underwound duplex DNA segments and TOP2 at crossovers between DNA duplexes. Whereas the Sc+ generated ahead of the RNA polymerase II (Pol II) complex is readily removed by TOP1 (ref.56), the Sc– behind Pol II (Fig. 2a) appears to be less efficiently processed. This disparity provides an explanation for the generation of ‘transcription-induced supercoiling’57. Transcription-induced Sc– has been mapped genome-wide using psoralen, which binds DNA proportionally to Sc– levels58,59,60,61. It is plausible, but not formally demonstrated, that the accumulation of Sc– could be attenuated when transcription complexes follow each other in tandem, as the Sc– generated by a transcription complex could be absorbed by (and facilitate the translocation of) the following transcription complex (Fig. 2b).

a | Transcription induces positive DNA supercoiling (Sc+) ahead of RNA polymerase II (Pol II) and negative supercoiling (Sc–) behind it54. Topoisomerase 1 (TOP1) directly binds the carboxyl terminus domain (CTD) of Pol II (dashed arrows) and is activated by bromodomain-containing protein 4 (BRD4), which phosphorylates the Pol II CTD (P)56. This interaction can efficiently remove Sc+ and allow translocation of Pol II. Behind the transcription complex, excessive Sc– must be removed to prevent formation of R-loops and alternative DNA structures169 (not shown). Recruitment of TOP3B by Tudor domain-containing protein 3 (TDRD3), which interacts with the Pol II CTD, suppresses R-loops27. TOP1 deficiency also leads to increased levels of R-loops owing to Sc– accumulation behind Pol II169. b | In theory, polymerases transcribing in tandem could cancel supercoiling between them, which would facilitate transcript elongation. c–f | Removal of topological constraints by TOP2 and TOP1 (part c) facilitates close interactions between enhancers and promoters: enhancer–promoter interaction within a topologically associating domain (TAD) (part d); a single enhancer activating two promoters (P1 and P2) in the same TAD (part e); and activation of two promoters (P1 and P3) in different TADs (part f).
TOP1 has recently been shown to bind the heptad repeats of the Pol II CTD in association with the chromatin regulator bromodomain-containing protein 4 (BRD4)56 (Fig. 2a). Based on these data, a model was proposed in which TOP1 is recruited to promoters, but kept inactive until phosphorylation of Pol II CTD Ser2 by BRD4 releases Pol II and TOP1 from promoter-proximal pausing into productive transcript elongation56. The TOP1 requirement in front of transcription complexes to remove Sc+ (Fig. 2a) may account for its role in activating long genes such as neuronal and inflammatory genes62,63,64,65. Accordingly, TOP1 inhibition by topotecan (Supplementary Box 1) has been reported to suppress transcription-mediated lethal inflammation during SARS-CoV-2 infection66. TOP2 also appears to be required for the transcription of long genes, both in mammals62 and in yeast7.
Behind the transcription complex, hyper-negative supercoiling resulting from insufficient TOP1 activity has been proposed as a source of R-loops67,68,69, G quadruplex structures70, left-handed Z-DNA71 and nucleosome destabilization59,61. The accumulation of R-loops in the context of TOP1 deficiency is a potential source of DNA breaks and genomic instability72,73. Excessive Sc– — similar to excessive Sc+ — can block transcription55, suggesting that TOP1, in addition to its role in removing Sc+, has a preferential role for removing Sc– behind transcription complexes (Fig. 2a,b).
TOP3B is also likely to suppress excessive transcription-induced Sc– and R-loops by passing a single strand of DNA through a break made in another DNA strand (single-strand passage). The recruitment of TOP3B has been attributed to its interaction with TDRD3, which binds methylated Arg residues: on histones H3 and H4 (H3R17me2a and H4R3me2a) at active promoters, and on the Pol II CTD (R1810me2a)27,74 through its Tudor domains (Fig. 2a,b). However, it is not known whether the potential role of TOP3B in resolving R-loops is related to its dual DNA and RNA topoisomerase activities.
In addition to its topological activity, TOP2 has been shown to regulate neuronal immediate-early response genes75,76 and the transcription of hormone-responsive genes (including genes activated by androgens, oestrogens, corticoid hormones, retinoic acid and more) by forming TOP2Bccs at promoters75,76,77,78,79,80,81,82,83. Both TOP2B and TOP2A have been proposed to regulate the promoter-proximal pause and release of Pol II at immediate-early response genes84,85,86,87, as does TOP1 (ref.56). TOP2 has also been shown to activate the transcription of MYC by oestrogens, which has been linked with oncogenesis80,85,86,87,88. Further research is needed to determine why TOP2ccs tend to be stalled at promoters and whether the TOP2 cleavage-rejoining activity responds to post-translational modifications, such as phosphorylation and oxidation. The potential role of DNA repair factors (including TDPs, ataxia telangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PK)) in ligand-induced gene activation also remains to be elucidated.
Because genes are activated by enhancers that are generally located quite a distance in cis from promoters, enhancer–promoter interactions require the formation of chromatin loops89. Such chromatin loops generate DNA topological and torsional constraints, which are substrates for topoisomerases (Fig. 2c–f).
Roles in genome organization
Advanced microscopy methods and chromosome conformation capture-based techniques that identify DNA sequences in close proximity, owing to chromatin folding, have revealed the high-order organization and dynamic folding of the genome in interphase cells and led to the identification of chromatin loops and TADs89,90,91. Structural maintenance of chromosomes (SMC) complexes are crucial for the formation of chromatin loops and TADs. They comprise condensins, cohesins and the SMC5–SMC6 complexes89,92,93,94. Cohesin and condensin are loaded onto DNA in nucleosome-depleted promoter regions during interphase, and were proposed to move in the same direction as transcription93. Whereas condensins organize chromatin loops and provide compaction and elasticity to chromosomes during mitosis, cohesins establish chromatin loops in interphase and sister chromatid cohesion from S phase to mitosis (Fig. 3). The functions of the SMC5–SMC6 complex are less known and are discussed only briefly here. SMC5–SMC6 has been proposed to function in promoting Top2-mediated resolution of sister chromatid intertwines, and for recruiting the yeast dissolvasome complex94,95. The human SMC5–SMC6 complex has also been proposed to resolve TOP2A-mediated DSBs during replication96, and to promote homologous recombination between sister chromatids by recruiting the cohesin SMC1–SMC3 complex to DSBs97.

a–d | The loop extrusion model, with proposed roles for topoisomerase 1 (TOP1) and TOP2B.57: cohesin holds two ends of a chromatin loop containing an enhancer, TOP1 preferentially removes positive DNA supercoiling induced by enhancer RNA (eRNA) synthesis, the resulting negative DNA supercoiling is proposed to pull the ends of DNA through the cohesin complex, and TOP2B bound to CCCTC-binding factor (CTCF) may allow this translocation by removing topological obstacles such as knots and supercoils (part a); as eRNA transcription continues, the extruded loop increases in size, and extrusion of one end (left) is arrested when cohesin encounters CTCF (part b); following further loop extrusion, the enhancer comes into contact with a promoter, and mRNA synthesis begins (part c); CTCF at the second end of the loop comes into contact with the cohesin complex and the chromatin loop is fully extruded, with TOP1 shown acting next to the promoter and TOP2 at DNA crossovers (part d). e–g | Proposed roles of cohesins, TOP1 and TOP2B in assembly of chromatin loops and topologically associating domains (TADs): cohesin and condensin complexes are loaded onto DNA during G1 phase of cell cycle, along with transcription resumption after mitosis (part e); transcription-driven negative DNA supercoiling is proposed to extrude chromatin loops (parts a–d) and form TADs, with TOP2B removing associated topological barriers such as DNA crossovers and catenanes (part f), resulting in TAD formation (part g). h | During mitosis, TOP2A is part of the chromosome scaffold comprising condensin complexes, whereas TOP1 is present in loop domains. i | Transversal axial view of chromatin scaffolded around TOP2A and condensins, with TOP1 in loop domains to remove supercoiling tension. Pol II, polymerase II. Sc, supercoiling.
Because TOPccs are protein-associated DNA strand breaks, they can be mapped by pulling down the topoisomerase polypeptides to retrieve the bound DNA segments or by directly mapping the DNA breaks (the different techniques used to map TOPccs are summarized in Supplementary Box 2). Genome-wide mapping techniques relying on massive parallel sequencing have been used to locate the sites of action of topoisomerases in relationship to SMC complexes and high-order chromatin structures.
TADs and roles of TOP1 and TOP2 in loop extrusion
Cohesins extrude chromatin into loops that are delineated by CCCTC-binding factor (CTCF) sites (Fig. 3a–d). Loop extrusion requires the ATPase activity and the translocation of cohesins, which stops upon cohesin encountering a CTCF protein bound to DNA in a convergent orientation98,99,100,101. When CTCF sites are convergent, the C termini of their bound CTCFs are positioned to interact with the cohesins at TAD borders57 (Fig. 3d). The tightness of the cohesin central pore may not allow free axial rotation of the DNA102, and the two DNA segments at the basis of TADs were proposed to be held together by two cohesin rings in a ‘handcuff-like’ arrangement103 (Figs 2c–f,3a–g). Hence, CTCF and cohesin define the borders of TADs104.
TOP2 chromatin binding is dependent on and proportional to cohesin binding100. However, cohesin binding is not TOP2-dependent, indicating that TOP2 is recruited to chromatin downstream of cohesins100. Genome-wide mappings of TOP2B sites by chromatin immunoprecipitation followed by sequencing (ChIP–seq) and End-seq (Supplementary Box 2) show a significant overlap between TOP2B, CTCF and cohesin binding sites75,99,100,101,105,106,107. These sites co-localize with the ChIP–seq peaks of the cohesin subunit SCC1 (also known as RAD21)89. The coincidence of TOP2B locations with TAD borders has been related to an interaction of TOP2B with the amino terminus of CTCF99,100,106 (Fig. 3a–d). Although the TOP2Bccs at CTCF–cohesin sites may form independently of transcription, their conversion to DSBs appears to be enhanced by transcription, and by the ‘debulking’ of the TOP2ccs by the ubiquitin–proteasome system (UPS) and TOP2 excision by TDP2 (ref.100). The preferential location of TOP2B at TAD boundaries coincides with chromosomal translocations100,101, and the repair of aborted TOP2cc by TDP2 was shown to suppress genomic breaks induced by androgens in prostate epithelial cells108.
It is plausible that the presence of TOP2B at TAD borders resolves the topological issues arising from loop extrusion57. As the DNA is threaded through the cohesin complex, TOP2B would be strategically positioned to eliminate topological barriers and DNA entanglements such as knots, plectonemes and catenanes before the DNA can translocate through the cohesin complexes (Fig. 3a–d). Transcription-induced Sc– within TADs has been proposed to be generated by RNA production at enhancers57,58,109,110,111 (Fig. 3a,b). Sc– and plectonemes can be viewed as facilitators of promoter–enhancer contacts within the same TAD57,112,113 (Fig. 3c,d), as observed in bacterial systems57.
Mitotic chromosomes and the decatenation checkpoint
The removal of cohesin at prophase through the so-called ‘prophase pathway’ is crucial for successful mitosis as condensins replace cohesins93. In fission yeast, this pathway depends on the Smc5–SMC6 complex, replication signals and Top2 activity114. The prophase pathway is poorly understood in human cells. TOP2A dysfunction has been proposed to activate a decatenation checkpoint that arrests cells at G2–M phase transition to protect them against chromosomal damage8.
Top2 activity is required during mitosis115 and vertebrate cells treated with the TOP2 inhibitor ICRF-193 (Supplementary Box 1) die with severe chromosomal alterations116. TOP2A, but not TOP2B, is cell cycle regulated1,16,24,34: it increases sharply from mid S phase through mitosis through transcriptional activation of TOP2A, stabilization of TOP2A mRNA and activation of the deubiquitylase USP15, which prevents the targeting of TOP2A for proteasomal degradation8,117. Following mitosis, TOP2A is rapidly degraded following its ubiquitylation by the anaphase promoting complex and its activator CDH1, and USP15 is inactivated8,117.
During prophase, condensin is the primary driver of chromosome condensation as it scaffolds DNA loops (Fig. 3h,i). Condensin also generates Sc+ in an ATP-dependent manner within those loops, which further contributes to chromosome compaction. TOP2A, but not TOP2B, readily relaxes this Sc+25, suggesting the existence of a fine-tuned balance between TOP2A and condensins8. TOP2A may minimize DNA entanglements as condensin organizes mitotic DNA loops9,118,119,120,121. However, yeast Top2 has recently been shown to promote sister chromatid intertwines during prophase while removing them at the onset of anaphase122. Whereas TOP2A and Top2 are essential components of the chromosome mitotic scaffold, TOP1 has also been observed within mitotic chromosomal loops, where it may act to dissipate excessive supercoiling123,124 (Fig. 3h,i).
Centromeric and telomeric regions and ribosomal DNA appear to retain catenated segments until anaphase8. In addition to TOP2A and condensins, resolution of ribosomal DNA regions requires the activity of the helicase PICH, of the poly(ADP-ribose) polymerase tankyrase 1 and of TOP3A125. The translocase activity of PICH was recently reported to extrude Sc– DNA loops, which are relaxed by TOP3A to provide Sc+ substrates for sister chromatid disjunction by TOP2A and for the rapid disjunction of sister centromeres at anaphase53.
Recent studies provide evidence that, as part of the dissolvasome complex, the helicase activity of BLM coupled with the single-strand passage activity of TOP3A ensures telomere integrity both in alternative lengthening of telomeres cells, which represent approximately 10–15% of cancers, and in normal cells that use telomerase to maintain their telomeres126,127. These data are consistent with the prior finding that TOP3A is involved in the alternative lengthening of telomeres pathway of telomere maintenance128. TOP2A has also been shown to cooperate with telomeric repeat-binding factor 2 and Apollo to protect telomeres from replicative damage129. The roles of topoisomerases in centromeres and telomeres warrant further investigations, as these regions consist of DNA repeats prone to recombination and formation of non-canonical DNA structures130,131.
It has been proposed that cells possess a ‘decatenation checkpoint’ that limits chromosomal instability and aneuploidy in response to TOP2A dysfunction8. However, the molecular pathways that activate this checkpoint are poorly understood; they may be related to abnormal regulation of TOP2A by SUMOylation, ubiquitylation and protein kinases that modify its C terminus8,132,133,134.
Chromatin remodelling and heterochromatin
In addition to their role in organizing chromatin and chromosomes and minimizing DNA entanglements, topoisomerases associate with chromatin remodelling complexes. TOP2A was found associated with the SWI/SNF complex BRG1-associated factor to facilitate decatenation135 and chromatin remodelling for both resolution and formation of facultative heterochromatin136. TOP2A has also been found in association with cohesin and BAZ2A, an essential component of the chromatin remodeller nucleolar remodelling complex, to regulate chromatin accessibility and expression of developmental genes in mouse embryonic stem cells137.
Studies with TOP2B-deficient cells34 are warranted to explore the potential connection between chromatin remodelling and TOP2B activity and the role of TOP2B in the formation of chromatin loops and TADs.
TOP3B has also been implicated in heterochromatin formation by interacting with the RNAi machinery to promote transcriptional gene silencing in Drosophila melanogaster4,138, and TOP3A has been suggested to act with PICH to condense chromatin at centromeres53. BRG1 (also known as SMARCA4) itself has also been shown to recruit TOP1 to suppress transcription-associated genomic instability139.
It is tempting to speculate that persistent TOPccs may provide anchors for tethering DNA segments to nuclear structures. Such ‘functional’ DPCs would be self-reversible, and if not reversible they would be removed by the repair pathways described below. Post-translational modifications of topoisomerases such as phosphorylation133,140, oxidation141,142, acidification143 and acetylation133 could regulate the stability and reversibility of the TOPcc, and thus maintain such genomic anchor points.
Topoisomerases and genome instability
Topoisomerases can both damage the DNA when they abort their catalytic cycle in response to anticancer drugs, environmental agents or reactive metabolites1,17 and contribute to DNA repair. Consequently, topoisomerases modulate genome stability, and their dysfunction causes chromosomal rearrangements and contributes to cancer.
Roles in DNA repair and genome stability
TOP1 is a potent ribonuclease at single ribonucleotides or short stretches of ribonucleotides incorporated into the genome by DNA polymerases144,145. The canonical pathway of ribonucleotide excision repair is primarily carried out by RNAse H2 (ref.146). However, the frequency of ribonucleotide misincorporation is so high147,148 that, when embedded ribonucleotides are not removed by RNAse H2, TOP1 converts them into SSBs with 3′ ends bearing a 2′,3′-cyclophosphate145 (Figs 4,5). Such blocked ends are processed by apurinic endonuclease 2 (APE2), as a potential backup gap repair pathway for the ribonucleotide excision repair pathway149,150. Subsequently, a TOP1cc formed 5′ adjacent to the ribonucleotide (possibly by the same TOP1 molecule sliding back on the DNA) (Fig. 5a) can release a short 2–5 bp DNA fragment and form a single-stranded gap (Fig. 5a). Following the excision of this secondary TOP1-DPC by TDP1 or APE2, gap filling has been suggested to restore the DNA1,146,149,151,152,153. However, the flexibility of the single-stranded DNA segment, especially in DNA sequences with short repeats, may also allow the 5′-hydroxyl (5′-OH) to attack the TOP1–tyrosyl–DNA bond and reverse the TOP1cc144 (Fig. 5a). Although this reaction is primarily viewed as a source of genomic instability151,152, the ribonuclease activity of TOP1 may serve as a backup pathway for the removal of ribonucleotides in DNA152,153,154.

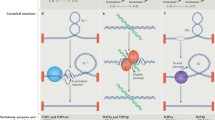
Catalytic intermediates of topoisomerases are normally transient because topoisomerase cleavage complexes (TOPccs) are self-reversible (Supplementary Fig. 1). Irreversible TOPccs are generated by trapping of TOPccs by anticancer drugs (Supplementary Box 1) and by pre-existing DNA alterations1,17. TOPccs produce complex nucleic acid alterations, including DNA–protein crosslinks (DPCs) and RNA–protein crosslinks (RPCs), DNA breaks and topological defects. a | DPCs form either at 3′ DNA ends (topoisomerase 1 (TOP1) or mitochondrial TOP1 (TOP1MT)) or 5′ DNA ends (TOP2A, TOP2B, TOP3A or TOP3B). They also form at 5′ RNA ends for TOP3B2. TOP1, TOP1MT, TOP2A and TOP2B bind double-stranded DNA; TOP3A and TOP3B bind single-stranded DNA or RNA. DPCs and RPCs generally need to be proteolysed or debulked (denatured) before their excision201,244. b | DNA single-strand breaks (SSBs) formed by stalled or irreversible TOP1ccs (top). TOP1 can also generate SSBs by converting ribonucleotides incorporated by DNA polymerases into nicks with 2′,3′-cyclophosphate blocking ends (red triangle) (middle)144,145,211. Cleavage of DNA by TOP2 can be asymmetrical, with only one component of the TOP2 homodimer forming a TOP2cc (bottom). This situation is commonly observed following treatment with etoposide227,358. c | DNA double-strand breaks (DSBs) formed by trapping of TOP2A and TOP2B1,32,359 following their proteasomal degradation (top)201,249. TOP1 can also generate DSBs, when it nicks the DNA opposite to a nick211,360 or when collision with a replisome produces ‘replication run-off’ with a single-ended DSB (seDSB) (middle)221. R-loops forming due to insufficient TOP1 activity induce DSBs (bottom)72. d | Insufficient topoisomerase activity can result in excessive positive DNA supercoiling (Sc+) that arrests transcription and replication, and in negative supercoiling (Sc–) that induces formation of R-loops and alternative DNA structures, including G quadruplexes (G4) and Z-DNA67,165. Catenanes and knots, which also stop DNA (and possibly RNA) transactions, increase in conditions of TOP2 and TOP3 deficiency. Unresolved recombination intermediates owing to TOP3A deficiency lead to sister chromatid exchanges and genomic instability6.

a | Model for formation of topoisomerase 1 (TOP1)-mediated deletions at sites of ribonucleotide incorporation within short tandem repeats. Incorporation of ribonucleotides by DNA polymerases is one of the most common abnormalities in the DNA218. A ‘primary’ TOP1 cleavage complex (TOP1cc) forms on a ribonucleotide. The 2′-hydroxyl of the ribose sugar reverses the bond with TOP1 (not shown) and generates a nick with a 2′,3′-cyclophosphate end. A ‘secondary’ TOP1 forms a TOP1cc 5′ of the nick, and the resulting short oligonucleotide bearing the 2′,3′-cyclophosphate is released. The 5′ end of DNA is captured by TOP1, which is followed by rejoining of the two ends and release of TOP1, thus generating a short deletion144,151,152. The ribonuclease activity of TOP1 has been linked with the ‘Indel Signature 4′ (ID4) in the Catalogue of Somatic Mutations in Cancer (COSMIC) database361, which consists of 2–5 base pair deletions. The ID4 signature has been proposed to be named the ID-TOP1 mutational signature362. b | TOP1-mediated large deletions. A replication fork collides with a TOP1cc on the leading (bottom) strand (Y represents the covalently linked catalytic Tyr at the 3′ end of the break). Replication fork regression induced by poly(ADP-ribose) polymerase 1 (PARP1)363 promotes TOP1cc self-reversal or fork stabilization and replication restart with RAD51 and breast cancer-associated type 2 (BRCA2). Alternative to fork regression, ‘replication run-off’221 generates a single-ended DNA double-strand break (seDSB), and ligation of two distant seDSBs by non-homologous end joining (NHEJ) produces large deletions226. c | TOP2-mediated short duplications203. Processing of TOP2cc by proteolysis and tyrosyl-DNA phosphodiesterase 2 (TDP2) produce DSBs. The 3′ ends of the break can undergo resection followed by gap filling. Ligation through NHEJ results in 4-bp duplications. An indel signature consisting of 2–4 base pair duplications and due to a TOP2A mutation (K473N) that traps TOP2A has been found in patient tumours and proposed to be named as the ID-TOP2A signature364. d | Simplified model of TOP2B-mediated chromosomal rearrangements based on the loop extrusion model100. Schematic depicts chromatin loops with cohesin, CCCTC-binding factor (CTCF) and TOP2B at base of loops. Stalled or irreversible TOP2Bccs generate DSBs that disjoin the loops. Rejoining of two adjacent DSBs produces a translocation.
The above TOP1 sequential cleavage model has also been invoked for TOP1-mediated repair of ultraviolet-induced DNA damage in nucleotide excision repair-deficient cells and for base excision repair and removal of 6–4 photoproducts (pyrimidine dimers)155. Because of its potent ligase activity across non-canonical and non-homologous structures152,156,157, including Holliday junctions158, TOP1 may have additional DNA repair and rejoining activities, warranting further investigations. In addition, a recent study showed that TOP1 interacts with cyclic cGMP–AMP synthase and promotes its binding to DNA, thereby eliciting innate immune responses and cell senescence159.
TOP3A, in addition to its roles in resolving double Holliday junctions and suppressing sister chromatid exchanges, is involved in DNA end resection160 and homology-directed repair (HDR)6,28,161,162. Recent studies suggest that TOP3A senses RPA-coated single-stranded DNA and contributes to the restart of replication forks50. In addition, by resolving recombination intermediates, TOP3A may prevent telomere exchanges and crossovers, leading to telomere erosion126,127.
To our knowledge, there is no evidence for a direct repair role of TOP2 enzymes. Yet their association with chromatin remodelling complexes could contribute to DNA repair135,136,137. Moreover, TOP2B binds to DSB sites163 and its activity at chromatin loops could serve to regulate DNA repair domains164.
Genome instability related to insufficient topoisomerase activity
Insufficient activity of the topoisomerases has profound effects on DNA and RNA metabolism (transcription and replication), and on the accumulation of non-canonical DNA structures such as R-loops, G-quadruplexes, Z-DNA, catenanes and knots67,165 (Fig. 4d).
TOP1 is essential both for early development in mice and for viability of vertebrate cells166 (Table 1; Supplementary Table 1). Studies in mice with neuron-specific TOP1 inactivation demonstrate the crucial roles of TOP1 in postmitotic cells65. The cerebral cortex develops normally, but neurons exhibit reduced transcription of long genes and, ultimately, undergo degeneration at postnatal day 7, which is accompanied by accumulation of the DNA damage response biomarker γH2AX167. DNA breakage results, at least in part, from the formation of R-loops72,168. Indeed, TOP1 depletion increases R-loop abundance in heterochromatin domains of human HEK293 cells169 and at transcription termination sites of highly expressed genes in HeLa cells69. Although R-loops induce DSBs mainly by interfering with DNA replication forks170, they can also generate DSBs in postmitotic cells72,73,171,172.
Unlike TOP1, TOP1MT is dispensable for mouse development173 (Table 1). Yet knocking out TOP1MT impairs liver regeneration and tumour growth by reducing the translation of genes encoded in mitochondria174,175,176,177. Notably, TOP1MT is transcriptionally activated by the oncogene MYC178.
Genetic studies show that TOP2 is required for chromosome condensation and segregation and for heterochromatin assembly in cell line models179. The essential role of TOP2A in mitosis is consistent with the embryogenic lethality of TOP2A-deficient mice at the four-cell or eight-cell stages180.
TOP2B-deficient mice develop in utero, as TOP2A can compensate for the loss of transcriptional gene regulation in cycling cells33,181. However, TOP2B-deficient mice die at birth because the nerves that innervate their diaphragm do not form dendrites to connect to the muscles. The mice also fail to form laminar structures in their cortex182,183 (Table 1).
TOP3A is essential for the nuclear and mitochondrial genomes184,185,186 (Box 1; Table 1). Bloom syndrome has recently been linked with genetic alterations in TOP3A187 and in the dissolvasome component RMI2 (ref.188), which is consistent with the fact that TOP3A and BLM cooperate in the dissolvasome complex6,189 to suppress sister chromatid exchanges161,190 and HDR191 and contribute to the resolution of stalled replication forks192. The mitochondrial isoform of TOP3A is crucial for mtDNA replication12, fertility186,193 and, owing to its mitochondrial functions, the viability of postmitotic cells185 (Box 1; Table 1).
TOP3B is not essential, but mice lacking TOP3B have a shortened lifespan, a higher incidence of aneuploidy in germ cells, increased autoimmunity194,195,196, abnormal synapse formation197 and behavioural impairments198 (Table 1). Individuals with genetic inactivation of TOP3B suffer from severe neurological symptoms, including cognitive impairment, which have been related to the RNA topoisomerase activity of TOP3B26,197. TOP3B inactivation has also been reported in a person with renal carcinoma199. The molecular mechanisms underlying these phenotypes and how they are related to the RNA and DNA functions of TOP3B2,4,15,138,198,200 are important issues for future studies.
Topoisomerase trapping by metabolic and environmental perturbations and drugs
Many environmental and metabolic perturbations induce TOPcc trapping1,17,201, which produces highly deleterious DNA lesions emanating from TOP-DPCs associated with SSBs and DSBs (Fig. 4). Studies with anticancer drugs that target TOPccs and with self-poisoning topoisomerases provide complementary approaches to elucidate the DNA damaging effects of trapped TOPccs and the repair pathways of and cellular responses to TOP1ccs31, TOP1MTccs202, TOP2ccs203,204 and TOP3Bccs2.
Trapping of TOP1ccs is the mechanism of action of the anticancer drugs derived from camptothecins and indenoisoquinolines205,206 (Supplementary Box 1), which are classically used to study the DNA damaging effects of and cellular responses to TOP1ccs in cellular models. Topotecan and irinotecan are widely used for cancer therapy and tumour-targeted TOP1 inhibitors are promising novel chemotherapies31. Local structural DNA perturbations can trap TOP1ccs as well207,208. These include the incorporation of ribonucleotides and of anticancer nucleoside analogues such as cytarabine (Ara-C) and gemcitabine, and DNA adducts by platinum derivatives such as cisplatin and carboplatin209,210,211,212. TOP1cc can also be stabilized near DNA mismatches, abasic sites and 7,8-dihydro-8-oxoguanine (8-oxoG) base oxidation, ultraviolet damage and single-strand nicks in the strand not cleaved by TOP1 (refs1,155,201,213,214,215,216,217) (Fig. 4c).
Trapped TOP1ccs are a well-established source of mutations and genomic instability (Figs 4,5). TOP1-mediated deletions at short repeated sequences (Fig. 5a) can be initiated by ribonucleotides incorporated into the DNA by replicative polymerases148,218 (Fig. 5a). TOP1 converts them to SSBs (Fig. 4b), and following the formation of a second TOP1cc on the same DNA strand (Fig. 5a), TOP1 readily generates short deletions144,151,152 due to its potent DNA ligation activity154,213,219,220. This recombination property is routinely used for molecular cloning with TOP1 (ref.145). The occurrence of TOP1ccs across nicks can also induce recombinogenic DSBs1,211,213 (Fig. 4c). Another mutagenic consequence of irreversible TOP1ccs results from their collisions with replication forks (Figs 4c,6). When the TOP1cc is on the leading strand, it produces ‘replication run-off’221 resulting in the formation of a single-ended DSB (seDSB) (Figs 4c,5b). Such seDSBs are highly cytotoxic204, unless the replication fork reverses (Fig. 5b), and can produce large deletions and chromosomal translocations through non-homologous end joining (NHEJ)222,223,224,225,226 (Fig. 5b).

a | Overall scheme for conversion of topoisomerase DNA–protein crosslinks (TOP-DPCs) into protein-free DNA breaks201. Association of TOP-DPCs with replication or transcription complexes and phase of the cell cycle (S phase versus G1 phase) are likely determinants of pathway choice. Debulking of TOP-DPCs by the three ubiquitin–proteasome pathways includes the conserved SUMO-targeted ubiquitin ligase (STUbL) pathway, in which RNF4 is the human E3 ubiquitin ligase for TOP1-DPCs, TOP2A-DPCs and TOP2B-DPCs249; the TRIM41 E3 ligase pathway for crosslinks between TOP3B and DNA or RNA2; and the cullin pathway for TOP1-DPCs365,366 (step 1). Non-proteasomal TOP-DPC proteolytic pathways245. The proteases Spartan (SPRTN), GCNA (also known as ACRC), FAM111A and DDI debulk TOP1-DPCs and TOP2-DPCs (step 2). Non-proteolytic pathway for TOP2, driven by SUMO E3 ligase ZNF451 (ref.259) (step 3). Nucleic-acid excision pathways for TOP1 and/or TOP2 include excision by the endonucleases MRE11, CtIP and XPF–ERCC1, or excision by tyrosyl-DNA phosphodiesterase 1 (TDP1) and TDP2 (step 4). b | TDP1 is activated by poly(ADP-ribose) polymerase 1 (PARP1)268,269,275, and upon cleaving DNA leaves a 3′-phosphate that is further processed by polynucleotide kinase phosphatase (not shown). TDP2 leaves a 5′-phosphate that can be directly ligated or extended by DNA polymerases (not shown). Both TDP1 and TDP2 require debulking of TOP-DPCs to gain access to the tyrosyl–DNA links. Additional excision pathways involve endonucleases. c | Differential roles of non-homologous end joining (NHEJ) and homology-directed repair (HDR) in repair of TOP1-induced single-ended DNA double-strand breaks (seDSBs) and TOP2-DPC-induced DSBs. Whereas seDSB repair by NHEJ is toxic222, possibly by inducing large deletions owing to illegitimate end joining of distant seDSBs (Fig. 5b), NHEJ is crucial for repair of TOP2-DPCs. TOP1cc, TOP1 cleavage complex.
TOP2ccs are also a well-established source of DNA damage and carcinogenic mutations by endogenous and environmental agents17. Their trapping by TOP2 poisons is the target of the anticancer drugs etoposide, doxorubicin, mitoxantrone and amsacrine (Supplementary Box 1), which generate TOP2-DPCs coupled with DSBs and SSBs. These drugs are classically used to map TOP2ccs and study their DNA damaging effects and repair pathways32,48,206,227,228,229,230. In addition, the TOP2 catalytic inhibitors merbarone and ICRF-193 (or ICRF-187) prevent the enzymatic activity of TOP2 by stabilizing topological TOP2–DNA complexes32,231, which are highly cytotoxic or require the intervention of DNA repair pathways196,222,231. The conformation of TOP2 is sensitive to temperature and biochemical perturbations141,142,143,232. Exposure to etoposide either at an elevated temperature (39 °C) or together with an inhibitor of heat shock protein 90 (HSP90) enhances the abundance of TOP2-DPCs233,234. Thus, HSP90 appears to be required for maintaining the conformation of TOP2 and avoiding TOP2 misfolding and the stalling of TOP2-DPCs. In addition to misfolding, TOP2 trapping can occur at sites of endogenous base damage17, including abasic sites, oxidized and alkylated bases and base mismatches235,236,237,238, and at secondary DNA structures239. This relation with endogenous base damage suggests that physiological TOP2cc trapping occurs more frequently than is commonly appreciated240. This conclusion is supported by the finding that loss of MRE11, the nuclease that removes TOP2 adducts (Fig. 6a,b), causes the endogenous accumulation of TOP2cc80,241,242,243.
Genomic damage owing to inaccurate repair of TOP1ccs and TOP2ccs
Due to the inaccessible location of the covalent tyrosyl–DNA bonds inside the TOPccs (Supplementary Fig. 1i–k), the removal of topoisomerase adducts requires a debulking step201 so that excision repair enzymes can access the topoisomerase tyrosyl–DNA junctions201,244 (Fig. 6; Supplementary Fig. 1a,b). To that effect, TOP-DPCs are subjected to proteolytic degradation2,201,244,245 (Fig. 6a). The UPS is highly active in cancer cells, which is exploited for the treatment of multiple myeloma with proteasome inhibitors. Moreover, it has recently become possible to examine proteolysed TOP1-DPCs using a specific antibody that recognizes the TOP1–DNA cleavage junctions246,247,248,249.
The endogenous accumulation of TOP1-DPCs has been detected in mice deficient in the kinase ATM or in the protease Spartan (SPRTN)247,250. Cancer cells treated with camptothecin or etoposide undergo rapid ubiquitin-dependent degradation of TOP1 and TOP2 (refs249,251,252). Likewise, human embryonic kidney cells proteolyse more than 50% of their cellular TOP1, TOP2A and TOP2B upon exposure to camptothecin or etoposide249. Accordingly, proteasome and ubiquitin inhibitors delay TOP-DPC removal2,249,253. The UPS-mediated removal of TOP1-DPCs is variable in different cell lines and tends to be more robust in drug-resistant cancer cells than in normal cells251,252. Recently, we showed that TOP3B-DPCs are also removed by the UPS pathway2.
TOP2-DPCs can also be proteolysed by non-proteasomal pathways245,248,254,255,256, and non-proteolytically by TDP2 following their SUMOylation and unfolding by ZNF451 (refs257,258,259) (Fig. 6). Non-proteasomal proteolysis by SPRTN repairs replication-associated TOP1-DPCs and TOP2-DPCs245, and genetic inactivation of SPRTN causes Ruijs–Aalfs syndrome with progeroid features and hepatocellular carcinomas245,247. Further studies are warranted to explore whether the sensitivity of malignant tumours to camptothecin and etoposide and the carcinogenic consequence of TOPccs are related to the UPS or SPRTN pathways, and whether the genomic instability of Ruijs–Aalfs syndrome is related to the accumulation of unrepaired TOP-DPCs.
Figure 6a outlines TDP1-dependent, TDP2-dependent and exonuclease-dependent repair of TOP1-DPCs, TOP2-DPCs and TOP3B-DPCs, and Table 1 lists the disorders and cellular defects related to TDP1 and TDP2 deficiencies. Human and yeast recombinant TDP1 proteins hydrolyse phosphodiester linkages between Tyr residues and 3′-phosphates of DNA207,260,261,262 (Fig. 6b; Supplementary Fig. 1a,b). Although purified TDP2 protein has weak activity as a 3′ TDP263,264, human TDP2 contributes to the removal of TOP1-DPCs in vivo. Recent analyses in human B cells show that more than 90% of TOP1-DPCs are removed during a 15-min repair time in wild type and TDP2–/– cells, 70% in TDP1–/– cells and only 40% in TDP1–/–TDP2–/– cells253. Thus, both TDP1 and TDP2 are important for TOP1-DPC removal. The stability of TDP1 is regulated by the deubiquitylase UCHL3 (ref.265). TDP1 activity is also controlled by its SUMOylation266, its phosphorylation by ATM and DNA-PK267, and its recruitment by poly(ADP-ribose) polymerase 1 (PARP1)268,269. ATM-deficient mice exhibit endogenous accumulation of TOP1-DPCs and neurodegeneration250.
TOP2-DPCs can also be directly excised by endonucleases100,241,270 (Fig. 6a). Additionally, the endonuclease complexes MUS81–EME1 (refs271,272) and XPF–ERCC1 (refs273,274,275) can excise TOP1-DPCs (Fig. 6). Hence, cancer and genetic deficiencies of such nucleases or their scaffolding proteins (for example, SLX4 in the case of XPF–ERCC1) may predispose to genomic instability by defective excision of TOP-DPCs275,276. Somatic mutations in tumours may also provide therapeutic opportunities related to synthetic lethality, such as the use of PARP inhibitors to block the TDP1 repair pathway in ERCC1-deficient cancers treated with TOP1 inhibitors275 (Fig. 6b).
Following the excision of TOP-DPCs, cells restore the DNA by filling the gaps and resealing the DNA breaks. Camptothecin generates DSBs in cycling cells when replication forks encounter TOP1-DPC (Figs 4c,6c). HDR, but not NHEJ, repairs the DSBs using the intact sister chromatid as a template. However, error-prone NHEJ frequently occurs in camptothecin-treated cells upon inhibition of ATM226,277, suggesting that genome instability resulting from endogenous TOP1cc is accentuated in the context of ATM and HDR deficiencies.
In contrast to the dominant role for HDR in repairing TOP1-induced DSBs, NHEJ is preferentially used for TOP2-induced DSBs even in cycling cells222 (Fig. 6c). Whereas DSB end resection by HDR efficiently removes TOP2-DPCs278, NHEJ can only ligate TOP2-DPCs after the removal of 5′ TOP2 adducts, which restores the 3′-OH and 5′-phosphate ends279,280,281. Abnormal rejoining of TOP2-induced DSBs has recently been reported to account for short duplications203 (Fig. 5c). Following hydrolysis of the 5′-tyrosyl bond by TDP2 (Tdp1 in yeast), gap filling by DNA polymerase generates a blunt-ended DSB, which is ligated by NHEJ.
Both TDP2 and MRE11 remove TOP2 adducts (Fig. 6b), and their relative contribution varies in different conditions. TDP2 removes 50–70% of TOP2 adducts generated by 2 h of etoposide exposure in mouse primary B cells. In mouse embryonic fibroblasts, loss of ATM and TDP2 together (but not separately) causes a significant delay in the repair of DSBs induced by etoposide282. ATM may phosphorylate MRE11 and its cofactor, CtIP (also known as RBBP8), as these proteins are known to be phosphorylated by ATM and promote NHEJ of etoposide-induced TOP2-DPCs in G1 phase283. More than 99% depletion of MRE11 and inactivation of its nuclease activity is required for the accumulation of endogenous TOP2ccs in human B cells242. Thus, a residual amount of MRE11 activity appears sufficient for the repair of TOP2ccs. Similar to the complete loss of MRE11, neuron-specific disruption of Nijmegen breakage syndrome 1 (NBS1), an essential cofactor of MRE11 (with RAD50), also causes the accumulation of endogenous TOP2cc242. Overexpression of TDP2 partially rescues the lethality of MRE11-deficient cells, indicating that stalled TOP2ccs generate lethal DSBs in cycling cells. Considering the viability of TDP2-deficient mice280 and the pivotal role of TDP2 in the repair of etoposide-induced TOP2ccs264,280,284, future studies are warranted to understand why TDP2 fails to suppress the endogenous accumulation of lethal TOP2ccs. Another unsolved question is the molecular mechanism(s) linking MRE11 with NHEJ to ensure the accurate repair of TOP2ccs, as NHEJ cannot accurately rejoin DSBs carrying the 3′ overhangs generated by the endonucleolytic removal of TOP2 adducts.
DSB end resection during HDR generates 3′ single-strand tails, and thereby removes 5′ blocking adducts such as TOP2-DPCs. A recent study shows that breast cancer-associated type 1 (BRCA1) and the ubiquitin-associated proteins RAP80 and UBC13 (also known as UBE2N), which are involved in DSB end resection, contribute to the removal of etoposide-induced 5′ TOP2-DPCs independently of their function in HDR243,281. RAP80 and UBC13 promote the physical interaction between BRCA1 and MRE11 in G1 phase as well as in S/G2 phase285. These findings suggest that BRCA1 and ubiquitylation promote MRE11-dependent removal of TOP2ccs prior to NHEJ during subsequent G0 or G1 phases.
The redundancy and complexity of the pathways that repair irreversible TOPccs and their associated DNA breaks are consistent with the occurrence of such lesions under physiological conditions. Somatic mutations in the pathways that repair these lesions (including in XPF, ERCC1, BRCA1 and BRCA2), in MRE11 and in other factors, may provide opportunities for synthetic lethal interaction treatment of cancers.
Chromosomal translocations and therapy-related acute myeloid leukaemia
Anticancer drugs targeting TOP2 enzymes (Supplementary Box 1) are associated with long-term undesirable side effects including therapy-related acute myeloid leukaemia (t-AML)17,286,287. Although rare compared with the high lethality of the initial cancers, t-AML can be devastating for those individuals affected. t-AML occurs following treatment for a wide range of primary neoplasia. Breast cancer accounts for about 35% of t-AML cases, presumably owing to the relative number of breast cancer cases and the age profile of the individuals, and prior haematological malignancies account for approximately 30% of t-AML cases288. As cancer survival statistics have improved, t-AML cases have increased; up to 15% of all acute myeloid leukaemia (AML) cases can be classified as t-AML289.
Therapy-related leukaemia following treatment with TOP2 poisons is presumed to result from genetic damage to haematopoietic progenitor cells leading to the emergence of clones exhibiting a differentiation block and/or an increase in self-renewal and proliferation. The precise point or points during haematopoietic differentiation where progenitors can be transformed in this way is open to debate, but the ‘window of opportunity’ is likely between multipotential progenitor cells and granulocyte–macrophage progenitors290,291,292,293,294.
Therapy-associated AML appearing after exposure to TOP2 poisons often contains one of a set of recurrent chromosomal translocations giving rise to fusion genes that disrupt blood cell development and differentiation17,228,295. Thus, errors in the cellular processing and repair of TOP2-DPCs in haematopoietic cells are viewed as a common cause of t-AML. Indeed, translocation junction sequences are consistent with the erroneous joining of heterologous chromosome segments by NHEJ, or by the variant pathway alternative end joining296,297. The most frequent translocations observed in t-AML involve fusion between the gene myeloid/lymphoid or mixed-lineage leukaemia (MLL; also known as KMT2A) at 11q23 and the genes AF9 (MLLT3) at 9p21, AF4 (AFF1) at 4q21 or, less frequently, numerous other genes17,101,295,298. Breakage at the MLL–AF9 or MLL–AF4 fusion genes is associated with TOP2 cleavage sites99. Other recurrent translocations include t(15;17)(PML-RARA), t(8;21)(AML-ETO), t(11q23,var) and inv(16)(p13;q22)(CBFB-MYH11)295,299,300. Thus, a simple model for the aetiology of t-AML associated with TOP2 poisons involves illegitimate end joining between DNA repair intermediates, resulting in the formation of leukaemogenic chromosomal translocations292,295,299 (Fig. 5d). In addition, there appears to be some preference for which translocation (and therefore which AML subtype) is induced by different TOP2 poisons. For example, etoposide is associated with MLL translocations, whereas t(15,17)(PML-RARA) translocations resulting in treatment-associated acute promyelocytic leukaemia have been associated with mitoxantrone and epirubicin300,301,302,303,304,305.
Chromosome break points involved in MLL translocations in AML fall within an 8 kb break-point cluster region, and those derived from t-AML are over-represented in the distal 1 kb of this break-point cluster region. This break-point clustering suggests that a specific mechanism is associated with therapy-related, but not de novo, AML translocations. The co-localization of TOP2B and CTCF99,106,306 provides one possible explanation for the over-representation of t-AML translocation break sites in the distal 1 kb of the MLL break-point cluster region, as this region is adjacent to a CTCF binding site within the MLL gene299,307. The proximity to nascent mRNAs suggests that the translocations occur within transcription hubs228,299,308. Furthermore, exposure of cells to large doses of TOP2 poisons results in chromosomal breakage in this 1 kb region that can be detected by Southern blotting309,310,311,312, which strongly implicates TOP2 either directly or indirectly313,314,315,316,317 in the generation of DSBs that are the precursor of the chromosomal translocations.
Conclusions and future perspective
Topoisomerases are magicians of DNA and RNA15,318, and their full range of functions remain to be discovered. Unknown functions such as the manipulation of RNA topology and how cells use and resolve knots, and the roles of topoisomerases in metabolism and mitochondria, are partially understood. Because topoisomerases are exploited as targets for a wide range of anticancer and antibacterial drugs, understanding the repair pathways of TOPccs can provide opportunities for improving the rational use of topoisomerase inhibitors and their combination with other drugs in cancer treatment.
Topoisomerases are an increasingly recognized source of genome instability. In this Review we have discussed the emerging range of (redundant) repair pathways that handle the failures of topoisomerases in physiological conditions and following their therapeutic targeting. These pathways have to repair highly damaging lesions, consisting of DPCs, SSBs and DSBs. The existence of redundant mechanisms that repair topoisomerase-induced genomic damage begs the question of repair pathway choice and selection. It is likely that the sites of damage and whether topoisomerases are trapped during transcription or replication, or whether the damage occurs in highly proliferative cells such as haematopoietic progenitors or in highly differentiated cells such as neurons, are germane to repair pathway choice.
Because of the potential danger of topoisomerases, it is likely that their activity is controlled and restricted to proper sites of action. In that context, further studies are warranted to elucidate the molecular partners of topoisomerases within molecular machines and their regulation by post-translational modifications.
Finally, the detailed location and roles of topoisomerases at centromeres and telomeres, and how topoisomerases are coordinated with chromatin remodelling factors and the architecture of chromatin and chromosomes, represent promising areas of investigation.
References
Pommier, Y., Sun, Y., Huang, S. N. & Nitiss, J. L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 17, 703–721 (2016). This prior review complements the current Review with detailed biochemical and pharmacological information.
Saha, S. et al. DNA and RNA cleavage complexes and repair pathway for TOP3B RNA- and DNA–protein crosslinks. Cell Rep. 33, 108569 (2020). This work presents the first evidence for cellular TOP3B RNA cleavage complexes and repair pathways.
Capranico, G., Marinello, J. & Chillemi, G. Type I DNA topoisomerases. J. Med. Chem. 60, 2169–2192 (2017). This work presents a detailed overview of the type I topoisomerases, which complements the present Review.
Lee, S. K. & Wang, W. Roles of topoisomerases in heterochromatin, aging, and diseases. Genes 10, 884 (2019).
Bjornsti, M. A. & Kaufmann, S. H. Topoisomerases and cancer chemotherapy: recent advances and unanswered questions. F1000Res. 8, 1704 (2019).
Bizard, A. H. & Hickson, I. D. The many lives of type IA topoisomerases. J. Biol. Chem. 295, 7138–7153 (2020). This comprehensive review complements the present Review.
Joshi, R. S., Nikolaou, C. & Roca, J. Structure and chromosomal organization of yeast genes regulated by topoisomerase II. Int. J. Mol. Sci. 19, 134 (2018).
Lee, J. H. & Berger, J. M. Cell cycle-dependent control and roles of DNA topoisomerase II. Genes 10, 859 (2019).
Dyson, S., Segura, J., Martinez-Garcia, B., Valdes, A. & Roca, J. Condensin minimizes topoisomerase II-mediated entanglements of DNA in vivo. EMBO J. 40, e105393 (2021). This article provides new insights into the coordination of SMCs and TOP2 in yeast.
Valdes, A., Segura, J., Dyson, S., Martinez-Garcia, B. & Roca, J. DNA knots occur in intracellular chromatin. Nucleic Acids Res. 46, 650–660 (2018).
Griffith, J. D. & Nash, H. A. Genetic rearrangement of DNA induces knots with a unique topology: implications for the mechanism of synapsis and crossing-over. Proc. Natl Acad. Sci. USA 82, 3124–3128 (1985).
Nicholls, T. J. et al. Topoisomerase 3α is required for decatenation and segregation of human mtDNA. Mol. Cell 69, 9–23.e6 (2018).
Jiang, W. et al. Predominant cellular mitochondrial dysfunction in the TOP3A gene-caused Bloom syndrome-like disorder. Biochim. Biophys. Acta 1867, 166106 (2021).
Ahmad, M. et al. RNA topoisomerase is prevalent in all domains of life and associates with polyribosomes in animals. Nucleic Acids Res. 44, 6335–6349 (2016).
Ahmad, M., Xu, D. & Wang, W. Type IA topoisomerases can be “magicians” for both DNA and RNA in all domains of life. RNA Biol. 14, 854–864 (2017).
Nitiss, J. L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 9, 327–337 (2009).
Vann, K. R., Oviatt, A. A. & Osheroff, N. Topoisomerase II poisons: converting essential enzymes into molecular scissors. Biochemistry 60, 1630–1641 (2021). This work presents a complementary review of TOP2 in oncology.
Champoux, J. J. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70, 369–413 (2001).
Yang, W. Topoisomerases and site-specific recombinases: similarities in structure and mechanism. Crit. Rev. Biochem. Mol. Biol. 45, 520–534 (2010).
Seol, Y., Zhang, H., Pommier, Y. & Neuman, K. C. A kinetic clutch governs religation by type IB topoisomerases and determines camptothecin sensitivity. Proc. Natl Acad. Sci. USA 109, 16125–16130 (2012).
Stewart, L., Redinbo, M. R., Qiu, X., Hol, W. G. & Champoux, J. J. A model for the mechanism of human topoisomerase I. Science 279, 1534–1541 (1998).
Salceda, J., Fernandez, X. & Roca, J. Topoisomerase II, not topoisomerase I, is the proficient relaxase of nucleosomal DNA. EMBO J. 25, 2575–2583 (2006).
Champoux, J. J. Evidence for an intermediate with a single-strand break in the reaction catalyzed by the DNA untwisting enzyme. Proc. Natl. Acad. Sci. USA 73, 3488–3491 (1976).
Woessner, R. D., Mattern, M. R., Mirabelli, C. K., Johnson, R. K. & Drake, F. H. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 2, 209–214 (1991).
McClendon, A. K., Rodriguez, A. C. & Osheroff, N. Human topoisomerase IIα rapidly relaxes positively supercoiled DNA: implications for enzyme action ahead of replication forks. J. Biol. Chem. 280, 39337–39345 (2005).
Stoll, G. et al. Deletion of TOP3β, a component of FMRP-containing mRNPs, contributes to neurodevelopmental disorders. Nat. Neurosci. 16, 1228–1237 (2013). This article provides the first evidence that TOP3B genetic defects lead to neurological disorder and that TOP3B acts as a dual RNA and DNA topoisomerase.
Yang, Y. et al. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol. Cell 53, 484–497 (2014).
Wright, W. D., Shah, S. S. & Heyer, W. D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 293, 10524–10535 (2018).
Hoadley, K. A. et al. Defining the molecular interface that connects the Fanconi anemia protein FANCM to the Bloom syndrome dissolvasome. Proc. Natl Acad. Sci. USA 109, 4437–4442 (2012).
Siaw, G. E., Liu, I. F., Lin, P. Y., Been, M. D. & Hsieh, T. S. DNA and RNA topoisomerase activities of Top3β are promoted by mediator protein Tudor domain-containing protein 3. Proc. Natl Acad. Sci. USA 113, E5544–5551 (2016).
Thomas, A. & Pommier, Y. Targeting topoisomerase I in the era of precision medicine. Clin. Cancer Res. 25, 6581–6589 (2019). This article provides an up-to-date review of TOP1 inhibitors approved and in clinical development for cancer treatments.
Nitiss, J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350 (2009). This article provides an extensive review of topoisomerase inhibitors.
Thakurela, S. et al. Gene regulation and priming by topoisomerase IIα in embryonic stem cells. Nat. Commun. 4, 2478 (2013).
Austin, C. A. et al. TOP2B: the first thirty years. Int. J. Mol. Sci. 19, 2765 (2018). This excellent article reviews the discovery and biology of TOP2B.
Pommier, Y. DNA Topoisomerases and Cancer (Springer, 2012).
Rajapakse, V. N. et al. CellMinerCDB for integrative cross-database genomics and pharmacogenomics analyses of cancer cell lines. iScience 10, 247–264 (2018).
Keszthelyi, A., Minchell, N. E. & Baxter, J. The causes and consequences of topological stress during DNA replication. Genes 7, 134 (2016).
Capranico, G., Jaxel, C., Roberge, M., Kohn, K. W. & Pommier, Y. Nucleosome positioning as a critical determinant for the DNA cleavage sites of mammalian DNA topoisomerase II in reconstituted simian virus 40 chromatin. Nucleic Acids Res. 18, 4553–4559 (1990).
Le, T. T. et al. Synergistic coordination of chromatin torsional mechanics and topoisomerase activity. Cell 179, 619–631.e15 (2019).
Servettaz, A. et al. Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J. Immunol. 182, 5855–5864 (2009).
Larcher, M. V. & Pasero, P. Top1 and Top2 promote replication fork arrest at a programmed pause site. Genes Dev. 34, 1–3 (2020).
Lee, C. M., Wang, G., Pertsinidis, A. & Marians, K. J. Topoisomerase III acts at the replication fork to remove precatenanes. J. Bacteriol. 201, e00563-18 (2019).
Schalbetter, S. A., Mansoubi, S., Chambers, A. L., Downs, J. A. & Baxter, J. Fork rotation and DNA precatenation are restricted during DNA replication to prevent chromosomal instability. Proc. Natl Acad. Sci. USA 112, E4565–4570 (2015).
Minchell, N. E., Keszthelyi, A. & Baxter, J. Cohesin causes replicative DNA damage by trapping DNA topological stress. Mol. Cell 78, 739–751.e8 (2020).
Boteva, L. et al. Common fragile sites are characterized by faulty condensin loading after replication stress. Cell Rep. 32, 108177 (2020).
Sarni, D. et al. 3D genome organization contributes to genome instability at fragile sites. Nat. Commun. 11, 3613 (2020).
Sima, J. et al. Identifying cis elements for spatiotemporal control of mammalian DNA replication. Cell 176, 816–830.e18 (2019).
Nitiss, J. L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 9, 327–337 (2009).
Dewar, J. M., Budzowska, M. & Walter, J. C. The mechanism of DNA replication termination in vertebrates. Nature 525, 345–350 (2015).
Shorrocks, A. K. et al. The Bloom syndrome complex senses RPA-coated single-stranded DNA to restart stalled replication forks. Nat. Commun. 12, 585 (2021).
Davies, S. L., North, P. S. & Hickson, I. D. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 14, 677–679 (2007).
Chan, K. L., North, P. S. & Hickson, I. D. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 26, 3397–3409 (2007).
Bizard, A. H. et al. PICH and TOP3A cooperate to induce positive DNA supercoiling. Nat. Struct. Mol. Biol. 26, 267–274 (2019). This article reveals a new function of TOP3A as reverse gyrase.
Liu, L. F. & Wang, J. C. Supercoiling of the DNA template during transcription. Proc. Natl Acad. Sci. USA 84, 7024–7027 (1987).
Ma, J. & Wang, M. D. DNA supercoiling during transcription. Biophys. Rev. 8, 75–87 (2016). This article provides evidence for the selective activity of yeast TOP2 positive supercoils using single-molecule techniques.
Baranello, L. et al. RNA polymerase II regulates topoisomerase 1 activity to favor efficient transcription. Cell 165, 357–371 (2016). This article provides evidence and a model for the role of TOP1 and BRD4 in transcription regulation.
Racko, D., Benedetti, F., Dorier, J. & Stasiak, A. Transcription-induced supercoiling as the driving force of chromatin loop extrusion during formation of TADs in interphase chromosomes. Nucleic Acids Res. 46, 1648–1660 (2018). This article provides a model for the role of TOP1 and supercoiling in generating loop extrusion.
Kouzine, F., Levens, D. & Baranello, L. DNA topology and transcription. Nucleus 5, 195–202 (2014).
Naughton, C. et al. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 20, 387–395 (2013).
Naughton, C., Corless, S. & Gilbert, N. Divergent RNA transcription: a role in promoter unwinding? Transcription 4, 162–166 (2013).
Teves, S. S. & Henikoff, S. Transcription-generated torsional stress destabilizes nucleosomes. Nat. Struct. Mol. Biol. 21, 88–94 (2014).
King, I. F. et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 501, 58–62 (2013).
Meng, L. et al. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518, 409–412 (2015).
Rialdi, A. et al. Topoisomerase 1 inhibition suppresses inflammatory genes and protects from death by inflammation. Science 352, aad7993 (2016).
Fragola, G. et al. Deletion of topoisomerase 1 in excitatory neurons causes genomic instability and early onset neurodegeneration. Nat. Commun. 11, 1962 (2020).
Ho, J. S. Y. et al. TOP1 inhibition therapy protects against SARS-CoV-2-induced lethal inflammation. Cell 184, 2618–2632.e17 (2021).
Tuduri, S. et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 11, 1315–1324 (2009).
Garcia-Muse, T. & Aguilera, A. R loops: from physiological to pathological roles. Cell 179, 604–618 (2019).
Promonet, A. et al. Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat. Commun. 11, 3940 (2020).
Miglietta, G., Russo, M. & Capranico, G. G-quadruplex–R-loop interactions and the mechanism of anticancer G-quadruplex binders. Nucleic Acids Res. 48, 11942–11957 (2020).
Wells, R. D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 32, 271–278 (2007).
Sordet, O. et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 10, 887–893 (2009).
Cristini, A. et al. Dual processing of R-loops and topoisomerase I induces transcription-dependent DNA double-strand breaks. Cell Rep. 28, 3167–3181.e6 (2019).
Sims, R. J. 3rd et al. The C-terminal domain of RNA polymerase II is modified by site-specific methylation. Science 332, 99–103 (2011).
Madabhushi, R. et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161, 1592–1605 (2015).
Madabhushi, R. The roles of DNA topoisomerase IIβ in transcription. Int. J. Mol. Sci. 19, 1917 (2018).
Haffner, M. C. et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 42, 668–675 (2010).
Haffner, M. C., De Marzo, A. M., Meeker, A. K., Nelson, W. G. & Yegnasubramanian, S. Transcription-induced DNA double strand breaks: both oncogenic force and potential therapeutic target? Clin. Cancer Res. 17, 3858–3864 (2011).
Hedayati, M. et al. Androgen deprivation followed by acute androgen stimulation selectively sensitizes AR-positive prostate cancer cells to ionizing radiation. Clin. Cancer Res. 22, 3310–3319 (2016).
Morimoto, S. et al. Type II DNA topoisomerases cause spontaneous double-strand breaks in genomic DNA. Genes 10, 868 (2019).
McNamara, S., Wang, H., Hanna, N. & Miller, W. H. Jr. Topoisomerase IIβ negatively modulates retinoic acid receptor α function: a novel mechanism of retinoic acid resistance. Mol. Cell Biol. 28, 2066–2077 (2008).
Perillo, B. et al. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206 (2008).
Ju, B. G. et al. A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science 312, 1798–1802 (2006).
Calderwood, S. K. A critical role for topoisomerase IIb and DNA double strand breaks in transcription. Transcription 7, 75–83 (2016).
Bunch, H. et al. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 6, 10191 (2015).
Buchel, G. et al. Association with Aurora-A controls N-MYC-dependent promoter escape and pause release of RNA polymerase II during the cell cycle. Cell Rep. 21, 3483–3497 (2017).
Herrero-Ruiz, A. et al. Topoisomerase IIα represses transcription by enforcing promoter-proximal pausing. Cell Rep. 35, 108977 (2021).
Itou, J. et al. Estrogen induces mammary ductal dysplasia via the upregulation of myc expression in a DNA-repair-deficient condition. iScience 23, 100821 (2020).
Davidson, I. F. & Peters, J. M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 22, 445–464 (2021). This work reviews chromatin loops and TADs.
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Misteli, T. The self-organizing genome: principles of genome architecture and function. Cell 183, 28–45 (2020).
Waldman, T. Emerging themes in cohesin cancer biology. Nat. Rev. Cancer 20, 504–515 (2020).
Uhlmann, F. SMC complexes: from DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 17, 399–412 (2016). This work is a detailed review of SMC complexes.
Aragon, L. The Smc5/6 complex: new and old functions of the enigmatic long-distance relative. Annu. Rev. Genet. 52, 89–107 (2018).
Bonner, J. N. et al. Smc5/6 mediated sumoylation of the Sgs1–Top3–Rmi1 complex promotes removal of recombination intermediates. Cell Rep. 16, 368–378 (2016).
Verver, D. E. et al. Non-SMC element 2 (NSMCE2) of the SMC5/6 complex helps to resolve topological stress. Int. J. Mol. Sci. 16, 368–378 (2016).
Potts, P. R., Porteus, M. H. & Yu, H. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 25, 3377–3388 (2006).
Vian, L. et al. The energetics and physiological impact of cohesin extrusion. Cell 175, 292–294 (2018).
Canela, A. et al. Genome organization drives chromosome fragility. Cell 170, 507–521.e18 (2017). This article uses End-seq to map TOP2 sites with cohesin and CTCF.
Canela, A. et al. Topoisomerase II-induced chromosome breakage and translocation is determined by chromosome architecture and transcriptional activity. Mol. Cell 75, 252–266 (2019). This article uses the End-seq method developed by the authors to map TOP2 sites and their processing to frank breaks and translocations.
Gothe, H. J. et al. Spatial chromosome folding and active transcription drive DNA fragility and formation of oncogenic MLL translocations. Mol. Cell 75, 267–283.e12 (2019).
Stigler, J., Camdere, G. O., Koshland, D. E. & Greene, E. C. Single-molecule imaging reveals a collapsed conformational state for DNA-bound cohesin. Cell Rep. 15, 988–998 (2016).
Schwarzer, W. et al. Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56 (2017).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Martinez-Garcia, P. M. et al. Genome-wide prediction of topoisomerase IIβ binding by architectural factors and chromatin accessibility. PLoS Comput. Biol. 17, e1007814 (2021).
Uuskula-Reimand, L. et al. Topoisomerase IIβ interacts with cohesin and CTCF at topological domain borders. Genome Biol. 17, 182 (2016).
Manville, C. M. et al. Genome-wide ChIP–seq analysis of human TOP2B occupancy in MCF7 breast cancer epithelial cells. Biol. Open. 4, 1436–1447 (2015).
Al Mahmud, M. R. et al. TDP2 suppresses genomic instability induced by androgens in the epithelial cells of prostate glands. Genes. Cell 25, 450–465 (2020). This article suggests the role of abortive TOP2ccs and defective repair as a source of prostate cancer.
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014).
Hirabayashi, S. et al. NET-CAGE characterizes the dynamics and topology of human transcribed cis-regulatory elements. Nat. Genet. 51, 1369–1379 (2019).
Li, W., Notani, D. & Rosenfeld, M. G. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat. Rev. Genet. 17, 207–223 (2016).
Tang, Z. et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163, 1611–1627 (2015).
Smith, E. M., Lajoie, B. R., Jain, G. & Dekker, J. Invariant TAD boundaries constrain cell-type-specific looping interactions between promoters and distal elements around the CFTR locus. Am. J. Hum. Genet. 98, 185–201 (2016).
Lin, S. J. & O’Connell, M. J. DNA topoisomerase II modulates acetyl-regulation of cohesin-mediated chromosome dynamics. Curr. Genet. 63, 923–930 (2017).
Holm, C., Goto, T., Wang, J. C. & Bolstein, D. DNA topoisomerase II is required at the time of mitosis in yeast. Cell. 41, 553–563 (1985).
Ishida, R. et al. Inhibition of intracellular topoisomerase II by antitumor bis(2,6-dioxopiperazine) derivatives: mode of cell growth inhibition distinct from that of cleavable complex-forming type inhibitors. Cancer Res. 51, 4909–4916 (1991).
Fielding, A. B. et al. The deubiquitylase USP15 regulates topoisomerase IIα to maintain genome integrity. Oncogene 37, 2326–2342 (2018).
Nielsen, C. F. et al. PICH promotes sister chromatid disjunction and co-operates with topoisomerase II in mitosis. Nat. Commun. 6, 8962 (2015).
Shintomi, K., Takahashi, T. S. & Hirano, T. Reconstitution of mitotic chromatids with a minimum set of purified factors. Nat. Cell Biol. 17, 1014–1023 (2015).
Ono, T., Sakamoto, C., Nakao, M., Saitoh, N. & Hirano, T. Condensin II plays an essential role in reversible assembly of mitotic chromosomes in situ. Mol. Biol. Cell 28, 2875–2886 (2017).
Piskadlo, E. & Oliveira, R. A. A topology-centric view on mitotic chromosome architecture. Int. J. Mol. Sci. 18, 2751 (2017).
Sen, N. et al. Physical proximity of sister chromatids promotes Top2-dependent intertwining. Mol. Cell 64, 134–147 (2016).
Earnshaw, W. C., Halligan, B., Cooke, C. A., Heck, M. M. & Liu, L. F. Topoisomerase II is a structural component of mitotic chromosome scaffolds. J. Cell. Biol. 100, 1706–1715 (1985).
Gasser, S. M., Laroche, T., Falquet, J., Boy de la Tour, E. & Laemmli, U. K. Metaphase chromosome structure. Involvement of topoisomerase II. J. Mol. Biol. 188, 613–629 (1986).
Daniloski, Z., Bisht, K. K., McStay, B. & Smith, S. Resolution of human ribosomal DNA occurs in anaphase, dependent on tankyrase 1, condensin II, and topoisomerase IIα. Genes. Dev. 33, 276–281 (2019).
Loe, T. K. et al. Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 34, 650–662 (2020).
Sobinoff, A. P. et al. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 36, 2907–2919 (2017).
Temime-Smaali, N. et al. Topoisomerase IIIα is required for normal proliferation and telomere stability in alternative lengthening of telomeres. EMBO J. 27, 1513–1524 (2008).
Ye, J. et al. TRF2 and apollo cooperate with topoisomerase 2α to protect human telomeres from replicative damage. Cell 142, 230–242 (2010).
Zhang, T. et al. Looping-out mechanism for resolution of replicative stress at telomeres. EMBO Rep. 18, 1412–1428 (2017).
Clarke, D. J. & Azuma, Y. Non-catalytic roles of the topoisomerase IIα C-terminal domain. Int. J. Mol. Sci. 18, 2438 (2017).
Deiss, K. et al. A genome-wide RNAi screen identifies the SMC5/6 complex as a non-redundant regulator of a Topo2a-dependent G2 arrest. Nucleic Acids Res. 47, 2906–2921 (2019).
Bedez, C. et al. Post-translational modifications in DNA topoisomerase 2α highlight the role of a eukaryote-specific residue in the ATPase domain. Sci. Rep. 8, 9272 (2018).
Vanden Broeck, A. et al. Structural basis for allosteric regulation of human topoisomerase IIα. Nat. Commun. 12, 2962 (2021).
Dykhuizen, E. C. et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 497, 624–627 (2013).
Miller, E. L. et al. TOP2 synergizes with BAF chromatin remodeling for both resolution and formation of facultative heterochromatin. Nat. Struct. Mol. Biol. 24, 344–352 (2017).
Dalcher, D. et al. BAZ2A safeguards genome architecture of ground-state pluripotent stem cells. EMBO J. 39, e105606 (2020).
Lee, S. K. et al. Topoisomerase 3β interacts with RNAi machinery to promote heterochromatin formation and transcriptional silencing in Drosophila. Nat. Commun. 9, 4946 (2018).
Husain, A. et al. Chromatin remodeller SMARCA4 recruits topoisomerase 1 and suppresses transcription-associated genomic instability. Nat. Commun. 7, 10549 (2016).
Vanden Broeck, A. et al. Structural basis for allosteric regulation of human topoisomerase IIα. Nat. Commun. 12, 2962 (2021).
Li, T. K. et al. Activation of topoisomerase II-mediated excision of chromosomal DNA loops during oxidative stress. Genes Dev. 13, 1553–1560 (1999).
Wang, H. et al. Stimulation of topoisomerase II-mediated DNA damage via a mechanism involving protein thiolation. Biochemistry 40, 3316–3323 (2001).
Xiao, H., Li, T. K., Yang, J. M. & Liu, L. F. Acidic pH induces topoisomerase II-mediated DNA damage. Proc. Natl Acad. Sci. USA 100, 5205–5210 (2003).
Kim, N. et al. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332, 1561–1564 (2011).
Sekiguchi, J. & Shuman, S. Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol. Cell 1, 89–97 (1997).
Sparks, J. L. et al. RNase H2-initiated ribonucleotide excision repair. Mol. Cell 47, 980–986 (2012).
Reijns, M. A. & Jackson, A. P. Ribonuclease H2 in health and disease. Biochemical Soc. Trans. 42, 717–725 (2014).
Williams, J. S., Lujan, S. A. & Kunkel, T. A. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat. Rev. Mol. Cell Biol. 17, 350–363 (2016).
Alvarez-Quilon, A. et al. Endogenous DNA 3′ blocks are vulnerabilities for BRCA1 and BRCA2 deficiency and are reversed by the APE2 nuclease. Mol. Cell 78, 1152–1165.e8 (2020).
Lin, Y. et al. APE2 promotes DNA damage response pathway from a single-strand break. Nucleic Acids Res. 46, 2479–2494 (2018).
Kim, N. & Jinks-Robertson, S. The Top1 paradox: friend and foe of the eukaryotic genome. DNA Repair. 56, 33–41 (2017).
Huang, S. Y., Ghosh, S. & Pommier, Y. Topoisomerase I alone is sufficient to produce short DNA deletions and can also reverse nicks at ribonucleotide sites. J. Biol. Chem. 290, 14068–14076 (2015).
Williams, J. S. et al. Topoisomerase 1-mediated removal of ribonucleotides from nascent leading-strand DNA. Mol. Cell 49, 1010–1015 (2013).
Sparks, J. L. & Burgers, P. M. Error-free and mutagenic processing of topoisomerase 1-provoked damage at genomic ribonucleotides. EMBO J. 34, 1259–1269 (2015).
Saha, L. K. et al. Topoisomerase I-driven repair of UV-induced damage in NER-deficient cells. Proc. Natl Acad. Sci. USA 117, 14412–14420 (2020).
Pourquier, P., Jensen, A. D., Gong, S. S., Pommier, Y. & Rogler, C. E. Human DNA topoisomerase I-mediated cleavage and recombination of duck hepatitis B virus DNA in vitro. Nucleic Acids Res. 27, 1919–1925 (1999).
Shuman, S. Polynucleotide ligase activity of eukaryotic topoisomerase I. Mol. Cell 1, 741–748 (1998).
Sekiguchi, J., Seeman, N. C. & Shuman, S. Resolution of Holliday junctions by eukaryotic DNA topoisomerase I. Proc. Natl Acad. Sci. USA 93, 785–789 (1996).
Zhao, B. et al. Topoisomerase 1 cleavage complex enables pattern recognition and inflammation during senescence. Nat. Commun. 11, 908 (2020).
Gravel, S., Chapman, J. R., Magill, C. & Jackson, S. P. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22, 2767–2772 (2008).
Wu, L. & Hickson, I. D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426, 870–874 (2003).
Piazza, A. & Heyer, W. D. Homologous recombination and the formation of complex genomic rearrangements. Trends Cell Biol. 29, 135–149 (2019).
Morotomi-Yano, K., Saito, S., Adachi, N. & Yano, K. I. Dynamic behavior of DNA topoisomerase IIβ in response to DNA double-strand breaks. Sci. Rep. 8, 10344 (2018).
Arnould, C. et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 590, 660–665 (2021).
Miao, Z. H. et al. Nonclassic functions of human topoisomerase I: genome-wide and pharmacologic analyses. Cancer Res. 67, 8752–8761 (2007).
Morham, S. G., Kluckman, K. D., Voulomanos, N. & Smithies, O. Targeted disruption of the mouse topoisomerase I gene by camptothecin selection. Mol. Cell. Biol. 16, 6804–6809 (1996).
Bonner, W. M. et al. γH2AX and cancer. Nat. Rev. Cancer 8, 957–967 (2008). This landmark review is by William Bonner, who discovered γH2AX.
Belotserkovskii, B. P., Tornaletti, S., D’Souza, A. D. & Hanawalt, P. C. R-loop generation during transcription: formation, processing and cellular outcomes. DNA Repair. 71, 69–81 (2018).
Manzo, S. G. et al. DNA topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 19, 100–100 (2018).
Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T. & Cimprich, K. A. Transcription–replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170, 774–786.e19 (2017).
Cristini, A., Gromak, N. & Sordet, O. Transcription-dependent DNA double-strand breaks and human disease. Mol. Cell Oncol. 7, 1691905 (2020).
Sordet, O., Nakamura, A. J., Redon, C. E. & Pommier, Y. DNA double-strand breaks and ATM activation by transcription-blocking DNA lesions. Cell Cycle 9, 274–278 (2010).
Zhang, H. et al. Increased negative supercoiling of mtDNA in TOP1mt knockout mice and presence of topoisomerases IIα and IIβ in vertebrate mitochondria. Nucleic Acids Res. 42, 7259–7267 (2014).
Khiati, S. et al. Lack of mitochondrial topoisomerase I (TOP1mt) impairs liver regeneration. Proc. Natl Acad. Sci. USA 112, 11282–11287 (2015).
Rosa, I. D., Zhang, H., Khiati, S., Wu, X. & Pommier, Y. Transcription profiling suggests that mitochondrial topoisomerase IB acts as a topological barrier and regulator of mitochondrial DNA transcription. J. Biol. Chem. 292, 20162–20172 (2017).
Baechler, S. A. et al. The mitochondrial type IB topoisomerase drives mitochondrial translation and carcinogenesis. Nat. Commun. 10, 83–83 (2019).
Baechler, S. A., Dalla Rosa, I., Spinazzola, A. & Pommier, Y. Beyond the unwinding: role of TOP1MT in mitochondrial translation. Cell Cycle 18, 2377–2384 (2019).
Zoppoli, G. et al. Coordinated regulation of mitochondrial topoisomerase IB with mitochondrial nuclear encoded genes and MYC. Nucleic Acids Res. 39, 6620–6632 (2011).
Adachi, Y., Luke, M. & Laemmli, U. K. Chromosome assembly in vitro: topoisomerase II is required for condensation. Cell 64, 137–148 (1991).
Akimitsu, N. et al. Enforced cytokinesis without complete nuclear division in embryonic cells depleting the activity of DNA topoisomerase IIα. Genes Cell 8, 393–402 (2003).
Tiwari, V. K. et al. Target genes of topoisomerase IIβ regulate neuronal survival and are defined by their chromatin state. Proc. Natl Acad. Sci. USA 109, E934–E943 (2012).
Lyu, Y. L. & Wang, J. C. Aberrant lamination in the cerebral cortex of mouse embryos lacking DNA topoisomerase IIβ. Proc. Natl Acad. Sci. USA 100, 7123–7128 (2003).
Yang, X., Li, W., Prescott, E. D., Burden, S. J. & Wang, J. C. DNA topoisomerase IIβ and neural development. Science 287, 131–134 (2000).
Li, W. & Wang, J. C. Mammalian DNA topoisomerase IIIα is essential in early embryogenesis. Proc. Natl. Acad. Sci. USA 95, 1010–1013 (1998).
Plank, J. L., Chu, S. H., Pohlhaus, J. R., Wilson-Sali, T. & Hsieh, T. S. Drosophila melanogaster topoisomerase IIIα preferentially relaxes a positively or negatively supercoiled bubble substrate and is essential during development. J. Biol. Chem. 280, 3564–3573 (2005).
Wu, J., Feng, L. & Hsieh, T. S. Drosophila topo IIIα is required for the maintenance of mitochondrial genome and male germ-line stem cells. Proc. Natl Acad. Sci. USA 107, 6228–6233 (2010).
Martin, C. A. et al. Mutations in TOP3A cause a Bloom syndrome-like disorder. Am. J. Hum. Genet. 103, 456 (2018).
Hudson, D. F. et al. Loss of RMI2 increases genome instability and causes a Bloom-like syndrome. PLoS Genet. 12, e1006483 (2016).
Manthei, K. A. & Keck, J. L. The BLM dissolvasome in DNA replication and repair. Cell Mol. Life Sci. 70, 4067–4084 (2013).
Lee, S. H., Siaw, G. E. L., Willcox, S., Griffith, J. D. & Hsieh, T. S. Synthesis and dissolution of hemicatenanes by type IA DNA topoisomerases. Proc. Natl Acad. Sci. USA 110, E3587–3594 (2013).
Tubbs, A. & Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656 (2017).
Deans, A. J. & West, S. C. FANCM connects the genome instability disorders Bloom’s syndrome and fanconi anemia. Mol. Cell 36, 943–953 (2009).
Tsai, H. Z., Lin, R. K. & Hsieh, T. S. Drosophila mitochondrial topoisomerase IIIα affects the aging process via maintenance of mitochondrial function and genome integrity. J. Biomed. Sci. 23, 38 (2016).
Kwan, K. Y. & Wang, J. C. Mice lacking DNA topoisomerase IIIβ develop to maturity but show a reduced mean lifespan. Proc. Natl Acad. Sci. USA 98, 5717–5721 (2001). This article demonstrates the viability of Top3b-knockout mice and their phenotype.
Kwan, K. Y., Moens, P. B. & Wang, J. C. Infertility and aneuploidy in mice lacking a type IA DNA topoisomerase IIIβ. Proc. Natl Acad. Sci. USA 100, 2526–2531 (2003).
Kwan, K. Y. et al. Development of autoimmunity in mice lacking DNA topoisomerase 3β. Proc. Natl Acad. Sci. USA 104, 9242–9247 (2007).
Xu, D. et al. Top3β is an RNA topoisomerase that works with fragile X syndrome protein to promote synapse formation. Nat. Neurosci. 16, 1238–1247 (2013). This article provides the first evidence that TOP3B acts as a dual RNA and DNA topoisomerase.
Joo, Y. et al. Topoisomerase 3β knockout mice show transcriptional and behavioural impairments associated with neurogenesis and synaptic plasticity. Nat. Commun. 11, 3143 (2020). This article reveals the importance of TOP3B for neuronal functions.
Zhang, T. et al. Loss of TOP3B leads to increased R-loop formation and genome instability. Open Biol. 9, 190222 (2019).
Ahmad, M. et al. Topoisomerase 3β is the major topoisomerase for mRNAs and linked to neurodevelopment and mental dysfunction. Nucleic Acids Res. 45, 2704–2713 (2017).
Sun, Y., Saha, L. K., Saha, S., Jo, U. & Pommier, Y. Debulking of topoisomerase DNA–protein crosslinks (TOP-DPC) by the proteasome, non-proteasomal and non-proteolytic pathways. DNA Repair 94, 102926 (2020).
Dalla Rosa, I. et al. Mapping topoisomerase sites in mitochondrial DNA with a poisonous mitochondrial topoisomerase I (Top1mt). J. Biol. Chem. 289, 18595–18602 (2014).
Stantial, N. et al. Trapped topoisomerase II initiates formation of de novo duplications via the nonhomologous end-joining pathway in yeast. Proc. Natl Acad. Sci. USA 117, 26876–26884 (2020).
Pommier, Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 8, 82–95 (2013).
Hsiang, Y. H., Hertzberg, R., Hecht, S. & Liu, L. F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 260, 14873–14878 (1985).
Pommier, Y. & Marchand, C. Interfacial inhibitors: targeting macromolecular complexes. Nat. Rev. Drug Discov. 11, 25–36 (2011). This review describes the landmark discovery of topoisomerase inhibitors and interfacial inhibitors and a paradigm for pharmacology.
Pommier, Y. et al. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair. 19, 114–129 (2014).
Comeaux, E. Q. & van Waardenburg, R. C. Tyrosyl-DNA phosphodiesterase I resolves both naturally and chemically induced DNA adducts and its potential as a therapeutic target. Drug Metab. Rev. 46, 494–507 (2014).
Pourquier, P. et al. Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin. Cancer Res. 8, 2499–2504 (2002).
Pourquier, P. et al. Induction of topoisomerase I cleavage complexes by 1-β-d-arabinofuranosylcytosine (ara-C) in vitro and in ara-C-treated cells. Proc. Natl Acad. Sci. USA 97, 1885–1890 (2000).
Huang, S. N., Williams, J. S., Arana, M. E., Kunkel, T. A. & Pommier, Y. Topoisomerase I-mediated cleavage at unrepaired ribonucleotides generates DNA double-strand breaks. EMBO J. 36, 361–373 (2017).
Zimmermann, M. et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289 (2018).
Pourquier, P. et al. Trapping of mammalian topoisomerase I and recombinations induced by damaged DNA containing nicks or gaps: importance of DNA end phosphorylation and camptothecin effects. J. Biol. Chem. 272, 26441–26447 (1997).
Pourquier, P. et al. Induction of reversible complexes between eukaryotic DNA topoisomerase I and DNA-containing oxidative base damages: 7,8-dihydro-8-oxoguanine and 5-hydroxycytosine. J. Biol. Chem. 274, 8516–8523 (1999).
Pourquier, P. et al. Effects of uracil incorporation, DNA mismatches, and abasic sites on cleavage and religation activities of mammalian topoisomerase I. J. Biol. Chem. 272, 7792–7796 (1997).
Pourquier, P. et al. Topoisomerase I-mediated cytotoxicity of N-methyl-N′-nitro-N-nitrosoguanidine: trapping of topoisomerase I by the O6-methylguanine. Cancer Res. 61, 53–58 (2001).
Van Waardenburg, R. C. A. M. et al. Platinated DNA adducts enhance poisoning of DNA topoisomerase I by camptothecin. J. Biol. Chem. 279, 54502–54509 (2004).
Reijns, M. A. et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022 (2012).
Pommier, Y., Jenkins, J., Kohlhagen, G. & Leteurtre, F. DNA recombinase activity of eukaryotic DNA topoisomerase I; effects of camptothecin and other inhibitors. Mutat. Res. 337, 135–145 (1995).
Shuman, S. Vaccinia DNA topoisomerase I promotes illegitimate recombination in Escherichia coli. Proc. Natl Acad. Sci. USA 86, 3489–3493 (1989).
Strumberg, D. et al. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol. 20, 3977–3987 (2000).
Maede, Y. et al. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Ther. 13, 214–220 (2014).
Britton, S. et al. ATM antagonizes NHEJ proteins assembly and DNA-ends synapsis at single-ended DNA double strand breaks. Nucleic Acids Res. 48, 9710–9723 (2020).
Nakamura, K. et al. Proteome dynamics at broken replication forks reveal a distinct ATM-directed repair response suppressing DNA double-strand break ubiquitination. Mol. Cell (2021).
Whelan, D. et al. Super-resolution visualization of distinct stalled and broken replication fork structures. PLoS Genetics 16, e1009256 (2020).
Balmus, G. et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 10, 87 (2019).
Muslimovic, A., Nystrom, S., Gao, Y. & Hammarsten, O. Numerical analysis of etoposide induced DNA breaks. PLoS ONE 4, e5859 (2009).
Cowell, I. G. & Austin, C. A. Mechanism of generation of therapy related leukemia in response to anti-topoisomerase II agents. Int. J. Environ. Res. Public Health 9, 2075–2091 (2012).
Chen, G. L. et al. Nonintercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem. 259, 13560–13566 (1984).
Tewey, K. M., Chen, G. L., Nelson, E. M. & Liu, L. F. Intercalative antitumor drugs interfere with the breakage-reunion reaction of mammalian DNA topoisomerase II. J. Biol. Chem. 259, 9182–9187 (1984).
Xiao, H. et al. The topoisomerase IIβ circular clamp arrests transcription and signals a 26S proteasome pathway. Proc. Natl Acad. Sci. USA 100, 3239–3244 (2003).
Hsiang, Y. H. & Liu, L. F. Evidence for the reversibility of cellular DNA lesion induced by mammalian topoisomerase II poisons. J. Biol. Chem. 264, 9713–9715 (1989).
Karras, G. I. et al. HSP90 shapes the consequences of human genetic variation. Cell 168, 856–866.e12 (2017).
Lai, C. H. et al. HSP-90 inhibitor ganetespib is synergistic with doxorubicin in small cell lung cancer. Oncogene 33, 4867–4876 (2014).
Kingma, P. S., Greider, C. A. & Osheroff, N. Spontaneous DNA lesions poison human topoisomerase IIα and stimulate cleavage proximal to leukemic 11q23 chromosomal breakpoints. Biochemistry 36, 5934–5939 (1997).
Cline, S. D., Jones, W. R., Stone, M. P. & Osheroff, N. DNA abasic lesions in a different light: solution structure of an endogenous topoisomerase II poison. Biochemistry 38, 15500–15507 (1999).
Bigioni, M. et al. Position-specific effects of base mismatch on mammalian topoisomerase II DNA cleaving activity. Biochemistry 35, 153–159 (1996).
Sabourin, M. & Osheroff, N. Sensitivity of human type II topoisomerases to DNA damage: stimulation of enzyme-mediated DNA cleavage by abasic, oxidized and alkylated lesions. Nucleic Acids Res. 28, 1947–1954 (2000).
Szlachta, K. et al. Topoisomerase II contributes to DNA secondary structure-mediated double-stranded breaks. Nucleic Acids Res. 48, 6654–6671 (2020).
Deweese, J. E. & Osheroff, N. The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 37, 738–748 (2009).
Lee, K. C. et al. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol. Open. 1, 863–873 (2012).
Hoa, N. N. et al. Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Mol. Cell 64, 580–592 (2016).
Sasanuma, H. et al. BRCA1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase II–DNA complexes. Proc. Natl Acad. Sci. USA 115, E10642–E10651 (2018). This article reveals the role of TOP2-DPCs as a source of breast cancers in patients deficient in BRCA1.
Sun, Y. et al. Excision repair of topoisomerase DNA–protein crosslinks (TOP-DPC). DNA Repair. 89, 102837 (2020).
Ruggiano, A. & Ramadan, K. DNA–protein crosslink proteases in genome stability. Commun. Biol. 4, 11 (2021).
Patel, A. G. et al. Immunodetection of human topoisomerase I–DNA covalent complexes. Nucleic Acids Res. 44, 2816–2826 (2016).
Maskey, R. S. et al. Spartan deficiency causes accumulation of topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res. 45, 4564–4576 (2017).
Kojima, Y. et al. FAM111A protects replication forks from protein obstacles via its trypsin-like domain. Nat. Commun. 11, 1318 (2020).
Sun, Y. et al. A conserved SUMO pathway repairs topoisomerase DNA–protein cross-links by engaging ubiquitin-mediated proteasomal degradation. Sci. Adv. 6, eaba6290 (2020). Together with Sun, Y. et al. (2020) (ref. 201), this review provides details and references for the redundant pathways that repair abortive TOPccs, complementary to the current Review.
Katyal, S. et al. Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat. Neurosci. 17, 813–21 (2014).
Desai, S. D., Liu, L. F., Vazquez-Abad, D. & D’Arpa, P. Ubiquitin-dependent destruction of topoisomerase I is stimulated by the antitumor drug camptothecin. J. Biol. Chem. 272, 24159–24164 (1997).
Mao, Y., Desai, S. D., Ting, C. Y., Hwang, J. L. & Liu, L. F. 26S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 276, 40652–40658 (2001).
Tsuda, M. et al. Tyrosyl-DNA phosphodiesterase 2 (TDP2) repairs topoisomerase 1 DNA–protein crosslinks and 3-blocking lesions in the absence of tyrosyl-DNA phosphodiesterase 1 (TDP1). DNA Repair. 91-92, 102849–102849 (2020).
Kojima, Y. & Machida, Y. J. DNA–protein crosslinks from environmental exposure: mechanisms of formation and repair. Env. Mol. Mutagen. 61, 716–729 (2020).
Dokshin, G. A. et al. GCNA interacts with spartan and topoisomerase II to regulate genome stability. Dev. Cell 52, 53–68.e6 (2020).
Serbyn, N. et al. The aspartic protease Ddi1 contributes to DNA–protein crosslink repair in yeast. Mol. Cell 77, 1066–1079 (2020).
Schellenberg, M. J. et al. ZATT (ZNF451)-mediated resolution of topoisomerase 2 DNA–protein cross-links. Science 357, 1412–1416 (2017).
Schellenberg, M. J. et al. Ubiquitin stimulated reversal of topoisomerase 2 DNA–protein crosslinks by TDP2. Nucleic Acids Res. 48, 6310–6325 (2020).
Riccio, A. A., Schellenberg, M. J. & Williams, R. S. Molecular mechanisms of topoisomerase 2 DNA–protein crosslink resolution. Cell Mol. Life Sci. 77, 81–91 (2020).
Yang, S.-W. et al. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. USA 93, 11534–11539 (1996).
Interthal, H., Chen, H. J. & Champoux, J. J. Human Tdp1 cleaves a broad spectrum of substrates including phosphoamide linkages. J. Biol. Chem. 280, 36518–36528 (2005).
Nitiss, K. C., Malik, M., He, X., White, S. W. & Nitiss, J. L. Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage. Proc. Natl Acad. Sci. USA 103, 8953–8958 (2006).
Gao, R., Huang, S. Y., Marchand, C. & Pommier, Y. Biochemical characterization of human tyrosyl-DNA phosphodiesterase 2 (TDP2/TTRAP): a Mg2+/Mn2+-dependent phosphodiesterase specific for the repair of topoisomerase cleavage complexes. J. Biol. Chem. 287, 30842–30852 (2012).
Ledesma, F. C., El Khamisy, S. F., Zuma, M. C., Osborn, K. & Caldecott, K. W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461, 674–678 (2009).
Liao, C. et al. UCHL3 regulates topoisomerase-induced chromosomal break repair by controlling TDP1 proteostasis. Cell Rep. 23, 3352–3365 (2018).
Hudson, J. J., Chiang, S. C., Wells, O. S., Rookyard, C. & El-Khamisy, S. F. SUMO modification of the neuroprotective protein TDP1 facilitates chromosomal single-strand break repair. Nat. Commun. 3, 733 (2012).
Das, B. B. et al. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 28, 3667–3680 (2009).
Das, B. B. et al. PARP1–TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 42, 4435–4449 (2014).
Sun, Y. et al. PARylation prevents the proteasomal degradation of topoisomerase I DNA–protein crosslinks and induces their deubiquitylation. Nat. Commun. 12, 5010 (2021).
Lee, K. C., Bramley, R. L., Cowell, I. G., Jackson, G. H. & Austin, C. A. Proteasomal inhibition potentiates drugs targeting DNA topoisomerase II. Biochem. Pharmacol. 103, 29–39 (2016).
Pardo, B., Moriel-Carretero, M., Vicat, T., Aguilera, A. & Pasero, P. Homologous recombination and Mus81 promote replication completion in response to replication fork blockage. EMBO Rep. 21, e49367 (2020).
Regairaz, M. et al. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I–DNA complexes. J. Cell Biol. 195, 739–749 (2011).
Ganguly, A. et al. Tdp1 processes chromate-induced single-strand DNA breaks that collapse replication forks. PLoS Genet. 14, e1007595 (2018).
Kikuchi, K. et al. Structure-specific endonucleases Xpf and Mus81 play overlapping but essential roles in DNA repair by homologous recombination. Cancer Res. 73, 4362–4371 (2013).
Zhang, Y. W. et al. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 39, 3607–3620 (2011).
Sousa, F. G. et al. Alterations of DNA repair genes in the NCI-60 cell lines and their predictive value for anticancer drug activity. DNA Repair. 28, 107–115 (2015).
Alagoz, M., Chiang, S.-C., Sharma, A. & El-Khamisy, S. F. ATM deficiency results in accumulation of DNA–topoisomerase I covalent intermediates in neural cells. PLoS ONE 8, e58239 (2013).
Neale, M. J., Pan, J. & Keeney, S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436, 1053–1057 (2005).
Sunter, N. J., Cowell, I. G., Willmore, E., Watters, G. P. & Austin, C. A. Role of topoisomerase II β in DNA damage response following IR and etoposide. J. Nucleic Acids 2010, 710589–710589 (2010).
Gomez-Herreros, F. et al. TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo. PLoS Genet. 9, e1003226 (2013).
Akagawa, R. et al. UBC13-mediated ubiquitin signaling promotes removal of blocking adducts from DNA double-strand breaks. iScience 23, 101027–101027 (2020).
Álvarez-Quilón, A. et al. ATM specifically mediates repair of double-strand breaks with blocked DNA ends. Nat. Commun. 5, 3347–3347 (2014).
Quennet, V., Beucher, A., Barton, O., Takeda, S. & Löbrich, M. CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic Acids Res. 39, 2144–2152 (2011).
Zagnoli-Vieira, G. & Caldecott, K. W. TDP2, TOP2, and SUMO: what is ZATT about? Cell Res. 27, 1405–1406 (2017).
Zhao, G. Y. et al. A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol. Cell 25, 663–675 (2007).
Allan, J. M. & Travis, L. B. Mechanisms of therapy-related carcinogenesis. Nat. Rev. Cancer 5, 943–955 (2005).
Leone, G., Fianchi, L., Pagano, L. & Voso, M. T. Incidence and susceptibility to therapy-related myeloid neoplasms. Chem. Biol. Interact. 184, 39–45 (2010).
Kayser, S. et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood 117, 2137–2145 (2011).
Mauritzson, N. et al. Pooled analysis of clinical and cytogenetic features in treatment-related and de novo adult acute myeloid leukemia and myelodysplastic syndromes based on a consecutive series of 761 patients analyzed 1976–1993 and on 5098 unselected cases reported in the literature 1974–2001. Leukemia 16, 2366–2378 (2002).
Chen, W. et al. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer Cell 13, 432–440 (2008).
Krivtsov, A. V. et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822 (2006).
Cozzio, A. et al. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 17, 3029–3035 (2003).
Grimwade, D. & Enver, T. Acute promyelocytic leukemia: where does it stem from? Leukemia 18, 375–384 (2004).
Seita, J. & Weissman, I. L. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. 2, 640–653 (2010).
Rowley, J. D. & Olney, H. J. International workshop on the relationship of prior therapy to balanced chromosome aberrations in therapy-related myelodysplastic syndromes and acute leukemia: overview report. Genes Chromosomes Cancer 33, 331–345 (2002).
Lieber, M. R. NHEJ and its backup pathways: relation to chromosomal translocations. Nat. Struct. Mol. Biol. 17, 393–395 (2010).
Lieber, M. R. Mechanisms of human lymphoid chromosomal translocations. Nat. Rev. Cancer 16, 387–398 (2016).
Meyer, C. et al. The MLL recombinome of acute leukemias in 2013. Leukemia 27, 2165–2176 (2013).
Cowell, I. G. et al. Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity. Proc. Natl Acad. Sci. USA 109, 8989–8994 (2012).
Felix, C. A. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim. Biophys. Acta 1400, 233–255 (1998).
Libura, J., Slater, D. J., Felix, C. A. & Richardson, C. Therapy-related acute myeloid leukemia-like MLL rearrangements are induced by etoposide in primary human CD34+ cells and remain stable after clonal expansion. Blood 105, 2124–2131 (2005).
Hasan, S. K. et al. Molecular analysis of t(15;17) genomic breakpoints in secondary acute promyelocytic leukemia arising after treatment of multiple sclerosis. Blood 112, 3383–3390 (2008).
Hasan, S. K. et al. Analysis of t(15;17) chromosomal breakpoint sequences in therapy-related versus de novo acute promyelocytic leukemia: association of DNA breaks with specific DNA motifs at PML and RARA loci. Genes Chromosomes Cancer 49, 726–732 (2010).
Mays, A. N. et al. Evidence for direct involvement of epirubicin in the formation of chromosomal translocations in t(15;17) therapy-related acute promyelocytic leukemia. Blood 115, 326–330 (2010).
Mistry, A. R. et al. DNA topoisomerase II in therapy-related acute promyelocytic leukemia. N. Engl. J. Med. 352, 1529–1538 (2005).
Uuskula-Reimand, L. & Wilson, M. D. Break check: transcription-driven topoisomerase II collisions near chromatin loop anchors are hotspots for DNA damage and translocations. Mol. Cell 75, 203–205 (2019).
Scharf, S. et al. Transcription linked to recombination: a gene-internal promoter coincides with the recombination hot spot II of the human MLL gene. Oncogene 26, 1361–1371 (2007).
Smith, K. A., Cowell, I. G., Zhang, Y., Sondka, Z. & Austin, C. A. The role of topoisomerase IIβ on breakage and proximity of RUNX1 to partner alleles RUNX1T1 and EVI1. Genes Chromosomes Cancer 53, 117–128 (2014).
Ploski, J. E. & Aplan, P. D. Characterization of DNA fragmentation events caused by genotoxic and non-genotoxic agents. Mutat. Res. 473, 169–180 (2001).
Aplan, P. D., Chervinsky, D. S., Stanulla, M. & Burhans, W. C. Site-specific DNA cleavage within the MLL breakpoint cluster region induced by topoisomerase II inhibitors. Blood 87, 2649–2658 (1996).
Ng, A., Taylor, G. M. & Eden, O. B. Genotoxicity of etoposide: greater susceptibility of MLL than other target genes. Cancer Genet. Cytogenet. 164, 164–167 (2006).
Strissel, P. L., Strick, R., Rowley, J. D. & Zeleznik-Le, N. J. An in vivo topoisomerase II cleavage site and a DNase I hypersensitive site colocalize near exon 9 in the MLL breakpoint cluster region. Blood 92, 3793–3803 (1998).
Tang, H. L. et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 23, 2240–2252 (2012).
Betti, C. J., Villalobos, M. J., Diaz, M. O. & Vaughan, A. T. M. Apoptotic triggers initiate translocations within the MLL gene involving the nonhomologous end joining repair system. Cancer Res. 61, 4550–4555 (2001).
Betti, C. J. et al. Cleavage of the MLL gene by activators of apoptosis is independent of topoisomerase II activity. Leukemia 19, 2289–2295 (2005).
Sim, S.-P. & Liu, L. F. Nucleolytic cleavage of the mixed lineage leukemia breakpoint cluster region during apoptosis. J. Biol. Chem. 276, 31590–31595 (2001).
Hars, E. S., Lyu, Y. L., Lin, C.-P. & Liu, L. F. Role of apoptotic nuclease caspase-activated DNase in etoposide-induced treatment-related acute myelogenous leukemia. Cancer Res. 66, 8975–8979 (2006).
Wang, J. C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 3, 430–440 (2002). This work is a landmark review for topoisomerase discovery and biology by a pioneer in the topoisomerase field.
Lee, M. P., Brown, S. D., Chen, A. & Hsieh, T. S. DNA topoisomerase I is essential in Drosophila melanogaster. Proc. Natl Acad. Sci. USA 90, 6656–6660 (1993).
Zhang, C. X., Chen, A. D., Gettel, N. J. & Hsieh, T. S. Essential functions of DNA topoisomerase I in Drosophila melanogaster. Dev. Biol. 222, 27–40 (2000).
Brill, S. J., DiNardo, S., Voelkel-Meiman, K. & Sternglanz, R. Need for DNA topoisomerase activity as a swivel for DNA replication for transcription of ribosomal RNA. Nature 326, 414–416 (1987).
Zhang, H., Seol, Y., Agama, K., Neuman, K. C. & Pommier, Y. Distribution bias and biochemical characterization of TOP1MT single nucleotide variants. Sci. Rep. 7, 8614 (2017).
Chiang, S.-C. et al. Mitochondrial protein-linked DNA breaks perturb mitochondrial gene transcription and trigger free radical-induced DNA damage. Sci. Adv. 3, e1602506 (2017).
Sobek, S. et al. Negative regulation of mitochondrial transcription by mitochondrial topoisomerase I. Nucleic Acids Res. 41, 9848–9857 (2013).
Douarre, C. et al. Mitochondrial topoisomerase I is critical for mitochondrial integrity and cellular energy metabolism. PLoS ONE 7, e41094 (2012).
Khiati, S. et al. Mitochondrial topoisomerase I (Top1mt) is a novel limiting factor of doxorubicin cardiotoxicity. Clin. Cancer Res. 20, 4873–4881 (2014).
Morimoto, H. et al. An interplay of NOX1-derived ROS and oxygen determines the spermatogonial stem cell self-renewal efficiency under hypoxia. Genes Dev. 35, 250–260 (2021).
Hoffmann, A., Heck, M. M., Bordwell, B. J., Rothfield, N. F. & Earnshaw, W. C. Human autoantibody to topoisomerase II. Exp. Cell Res. 180, 409–418 (1989).
Imai, H. et al. Autoantibody to DNA topoisomerase II in primary liver cancer. Clin. Cancer Res. 1, 417–424 (1995).
Akimitsu, N. et al. Induction of apoptosis by depletion of DNA topoisomerase IIα in mammalian cells. Biochem. Biophys. Res. Commun. 307, 301–307 (2003).
Broderick, L. et al. Mutations in topoisomerase IIβ result in a B cell immunodeficiency. Nat. Commun. 10, 3644 (2019).
Hiraide, T. et al. A de novo TOP2B variant associated with global developmental delay and autism spectrum disorder. Mol. Genet. Genom. Med. 8, e1145 (2020).
Lam, C. W., Yeung, W. L. & Law, C. Y. Global developmental delay and intellectual disability associated with a de novo TOP2B mutation. Clin. Chim. Acta 469, 63–68 (2017).
Dereuddre, S., Delaporte, C. & Jacquemin-Sablon, A. Role of topoisomerase IIβ in the resistance of 9-OH-ellipticine-resistant Chinese hamster fibroblasts to topoisomerase II inhibitors. Cancer Res. 57, 4301–4308 (1997).
Edmond, M., Hanley, O. & Philippidou, P. Topoisomerase IIβ selectively regulates motor neuron identity and peripheral connectivity through Hox/Pbx-dependent transcriptional programs. eNeuro 4, ENEURO.0404-17.2017 (2017).
Zhang, Y. L. et al. TOP2βeta is essential for ovarian follicles that are hypersensitive to chemotherapeutic drugs. Mol. Endocrinol. 27, 1678–1691 (2013).
Navabpour, S., Rogers, J., McFadden, T. & Jarome, T. J. DNA double-strand breaks are a critical regulator of fear memory reconsolidation. Int. J. Mol. Sci. 21, 8995 (2020).
Chapman, J., Ng, Y. S. & Nicholls, T. J. The maintenance of mitochondrial DNA integrity and dynamics by mitochondrial membranes. Life 10, 164 (2020).
Herbaux, C. et al. TOP3A, a new partner gene fused to MLL in an adult patient with de novo acute myeloid leukaemia. Br. J. Haematol. 157, 128–131 (2012).
Wallis, J. W., Chrebet, G., Brodsky, G., Rolfe, M. & Rothstein, R. A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 58, 409–419 (1989).
Kim, R. A. & Wang, J. C. Identification of the yeast TOP3 gene product as a single strand-specific DNA topoisomerase. J. Biol. Chem. 267, 17178–17185 (1992).
Gangloff, S., MacDonald, J. P., Bendixen, C., Arthur, L. & Rothstein, R. The yeast type 1 topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol. Cell. Biol. 14, 8391–8398 (1994).
Rosato, M. et al. Combined cellomics and proteomics analysis reveals shared neuronal morphology and molecular pathway phenotypes for multiple schizophrenia risk genes. Mol. Psychiatry 26, 784–799 (2021).
El-Khamisy, S. F. et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 434, 108–113 (2005).
Takashima, H. et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 32, 267–272 (2002).
Murai, J. et al. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J. Biol. Chem. 287, 12848–12857 (2012).
Katyal, S. et al. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 26, 4720–4731 (2007).
Guo, D., Dexheimer, T. S., Pommier, Y. & Nash, H. A. Neuroprotection and repair of 3′-blocking DNA ends by glaikit (gkt) encoding Drosophila tyrosyl-DNA phosphodiesterase 1 (TDP1). Proc. Natl Acad. Sci. USA 111, 15816–15820 (2014).
Pouliot, J. J., Robertson, C. A. & Nash, H. A. Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes. Cell 6, 677–687 (2001).
Pouliot, J. J., Yao, K. C., Robertson, C. A. & Nash, H. A. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topo I covalent complexes. Science 286, 552–555 (1999).
Zoghi, S. et al. A novel non-sense mutation in TDP2 causes spinocerebellar ataxia autosomal recessive 23 accompanied by bilateral upward gaze; report of a case and review of the literature. Eur. J. Med. Genet. 64, 104348 (2021).
Zeng, Z. et al. TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1. Nucleic Acids Res. 40, 8371–8380 (2012).
Maciejewski, S., Ullmer, W. & Semler, B. L. VPg unlinkase/TDP2 in cardiovirus infected cells: re-localization and proteolytic cleavage. Virology 516, 139–146 (2018).
Virgen-Slane, R. et al. An RNA virus hijacks an incognito function of a DNA repair enzyme. Proc. Natl Acad. Sci. USA 109, 14634–14639 (2012).
Gomez-Herreros, F. et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat. Genet. 46, 516–521 (2014).
Alvarez-Quilon, A. et al. Endogenous topoisomerase II-mediated DNA breaks drive thymic cancer predisposition linked to ATM deficiency. Nat. Commun. 11, 910 (2020).
Bakx, J. A. M. et al. Duplex DNA and BLM regulate gate opening by the human TopoIIIα-RMI1-RMI2 complex. Nat. Commun. 13, 584 (2022).
Long, B. H., Musial, S. T. & Brattain, M. G. Single- and double-strand DNA breakage and repair in human lung adenocarcinoma cells exposed to etoposide and teniposide. Cancer Res. 45, 3106–3112 (1985).
Pommier, Y., Leo, E., Zhang, H. & Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17, 421–433 (2010).
Pourquier, P. & Pommier, Y. Topoisomerase I-mediated DNA damage. Adv. Cancer Res. 80, 189–216 (2001).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Reijns, M. A. M. et al. Signatures of TOP1 transcription-associated mutagenesis in cancer and germline. Nature https://doi.org/10.1038/s41586-022-04403-y (2022).
Berti, M. et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 20, 347–354 (2013).
Boot, A. et al. Recurrent mutations in topoisomerase IIα cause a previously undescribed mutator phenotype in human cancers. Proc. Natl Acad. Sci. USA 119, e2114024119, (2022).
Zhang, H. F. et al. Cullin 3 promotes proteasomal degradation of the topoisomerase I–DNA covalent complex. Cancer Res. 64, 1114–1121 (2004).
Kerzendorfer, C. et al. Mutations in Cullin 4B result in a human syndrome associated with increased camptothecin-induced topoisomerase I-dependent DNA breaks. Hum. Mol. Genet. 19, 1324–1334 (2010).
Falkenberg, M. & Gustafsson, C. M. Mammalian mitochondrial DNA replication and mechanisms of deletion formation. Crit. Rev. Biochem. Mol. Biol. 55, 509–524 (2020).
Wang, Y., Lyu, Y. L. & Wang, J. C. Dual localization of human DNA topoisomerase IIIα to mitochondria and nucleus. Proc. Natl Acad. Sci. USA 99, 12114–12119 (2002).
Zhang, H. et al. Human mitochondrial topoisomerase I. Proc. Natl Acad. Sci. USA 98, 10608–10613 (2001).
Zhang, H., Meng, L. H., Zimonjic, D. B., Popescu, N. C. & Pommier, Y. Thirteen-exon-motif signature for vertebrate nuclear and mitochondrial type IB topoisomerases. Nucleic Acids Res. 32, 2087–2092 (2004).
Low, R. L., Orton, S. & Friedman, D. B. A truncated form of DNA topoisomerase IIβ associates with the mtDNA genome in mammalian mitochondria. Eur. J. Biochem. 270, 4173–4186 (2003).
Das, B. B., Dexheimer, T. S., Maddali, K. & Pommier, Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc. Natl Acad. Sci. USA 107, 19790–19795 (2010).
Huang, S. N. et al. Mitochondrial tyrosyl-DNA phosphodiesterase 2 and its TDP2(S) short isoform. EMBO Rep 19, e42139 (2018).
Acknowledgements
The authors’ understanding of topoisomerases stems from the dedication and constant discussions of members of the Laboratory of Molecular Pharmacology, Developmental Therapeutics Branch, Center for Cancer Research, the Intramural Program of the National Cancer Institute, National Institutes of Health (NIH) (Z01-BC-006161). They thank K. W. Kohn, who first hypothesized that doxorubicin and other anthracycline anticancer drugs act by forming protein-associated DNA breaks, which are now referred to as TOPccs. Y.P. and A.N. are supported by the Center for Cancer Research of the US National Cancer Institute (CCR-NCI) (Z01 BC 006161 and Z01 BC 006150 to Y.P.). This work was also supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (16H12595 and 16H06306) (to S.T.) and JSPS Core-to-Core Program, A. Advanced Research Networks (to S.T.). The authors thank X. Yang, postdoctoral fellow in the Pommier group, for his contribution to Supplementary Fig. 1. They are also grateful to the three reviewers who provided multiple suggestions and careful corrections.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Molecular Cell Biology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Tyrosyl-DNA phosphodiesterases
-
(TDPs). Referring to TDP1 and TDP2, enzymes that excise topoisomerase–DNA crosslinks by hydrolysing 3′-phosphodiester and 5′-phosphodiester bonds, respectively. TDP2 also excises TOP3–RNA crosslinks.
- DNA supercoiling
-
The amount of DNA twist, which is the number of crossovers of the two strands across each other; writhe is a measure of the double helix winding around itself. Positive DNA supercoiling (Sc+) is defined by increased twist and/or writhe; negative DNA supercoiling (Sc–) is the opposite.
- Hemicatenanes
-
Crossovers of two strands of DNA originating from different DNA molecules.
- Holliday junctions
-
Branched DNA structures consisting of four double-stranded arms joined together. Double Holliday junctions form hemicatenanes that are resolved by the Bloom syndrome protein (BLM)–topoisomerase 3A (TOP3A)–RecQ-mediated genome instability proteins (RMI1/2) (BTR) dissolvasome complex.
- Topologically associating domains
-
(TADs). In interphase chromosomes, genomic regions in which interactions between loci are enriched compared with interactions with loci outside the domain.
- Structural maintenance of chromosomes
-
(SMC). SMC complexes are ATPase complexes that tether and organize chromatin by forming chromatin loops and ensuring sister chromatid cohesion.
- Homology-directed repair
-
(HDR). A replication-associated DNA double-strand break (DSB) repair pathway that uses a homologous sequence as a template for resynthesizing a missing DNA segment. Its classical form is homologous recombination and its common effector is RAD51.
- γH2AX
-
Histone H2AX phosphorylated at Ser139; a sensitive biomarker of DNA double-strand breaks (DSBs).
- TOPcc trapping
-
Stabilization of topoisomerase cleavage complexes (TOPccs) by inhibition of DNA end rejoining by a drug molecule bound at the interface of the DNA break and the enzyme or by DNA lesions that misalign the broken DNA ends, thereby preventing the release of the topoisomerase.
- Non-homologous end joining
-
(NHEJ). The prominent DNA double-strand break (DSB) repair pathway, which rapidly joins adjacent DNA ends. Its main effectors are Ku70–Ku80 and DNA-dependent protein kinase (DNA-PK).
Rights and permissions
About this article

Cite this article
Pommier, Y., Nussenzweig, A., Takeda, S. et al. Human topoisomerases and their roles in genome stability and organization. Nat Rev Mol Cell Biol 23, 407–427 (2022). https://doi.org/10.1038/s41580-022-00452-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41580-022-00452-3
- Springer Nature Limited
This article is cited by
-
The nucleolar shell provides anchoring sites for DNA untwisting
Communications Biology (2024)
-
Chromatin dynamics and RNA metabolism are double-edged swords for the maintenance of plant genome integrity
Nature Plants (2024)
-
Tumor growth-arrest effect of tetrahydroquinazoline-derivative human topoisomerase II-alpha inhibitor in HPV-negative head and neck squamous cell carcinoma
Scientific Reports (2024)
-
MTHFD2 in healthy and cancer cells: Canonical and non-canonical functions
npj Metabolic Health and Disease (2024)
-
Dimerization-dependent serine protease activity of FAM111A prevents replication fork stalling at topoisomerase 1 cleavage complexes
Nature Communications (2024)