

Abstract
Bacterial chemotaxis requires bidirectional flagellar rotation at different rates. Rotation is driven by a flagellar motor, which is a supercomplex containing multiple rings. Architectural uncertainty regarding the cytoplasmic C-ring, or ‘switch’, limits our understanding of how the motor transmits torque and direction to the flagellar rod. Here we report cryogenic electron microscopy structures for Salmonella enterica serovar typhimurium inner membrane MS-ring and C-ring in a counterclockwise pose (4.0 Å) and isolated C-ring in a clockwise pose alone (4.6 Å) and bound to a regulator (5.9 Å). Conformational differences between rotational poses include a 180° shift in FliF/FliG domains that rotates the outward-facing MotA/B binding site to inward facing. The regulator has specificity for the clockwise pose by bridging elements unique to this conformation. We used these structures to propose how the switch reverses rotation and transmits torque to the flagellum, which advances the understanding of bacterial chemotaxis and bidirectional motor rotation.
Similar content being viewed by others


Main
The biased random walk of chemotaxis is essential for bacterial survival and pathogenesis1,2,3. This process relies on bidirectional flagellar rotation1,2,3 (Fig. 1a) by a motor composed of four rings. The C-ring (C = cytoplasmic), which contains multiple copies of protein subunits called FliG, FliM and FliN, switches the rotation of the flagellum between counterclockwise (CCW) and clockwise (CW). For this reason, it is termed the ‘switch’. CCW rotation allows bacteria such as Escherichia coli and Salmonella enterica to swim straight. Conversely, CW rotation induces tumbling and reorientation with a new trajectory1,2,3. The switch is also the site of torque generation for the flagellum, through an electrostatic interaction with a stator called MotA/B4. The switch also connects to the MS-ring (MS = membrane–supramembrane), which transmits both the direction and the speed of rotation to the flagellar rod. Finally, the P- (P = peptidoglycan) and L-rings (L = lipid), which are the bushings of the motor, surround this rod to support and buffer the rotation. High-resolution structures of the rod, export apparatus, MS-ring, P-ring, L-ring, flagellar hook and flagellar filament have provided insight into the function of these flagellar components5,6,7,8,9,10,11,12,13. However, past structures for the C-ring are at low resolution (for example, refs. 14,15).

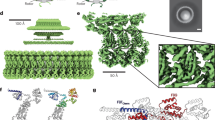
a, Schematic diagram of the flagellar motor showing the L-ring, P-ring, MS-ring and C-ring, as well as the flagellar rod, hook and filament. The switch is housed within the C-ring and is composed of the FliG, FliM and FliN subunits. OM, outer membrane; IM, inner membrane. b, Cropped view of a representative cryoEM micrograph for wild-type MS- and C-rings (1 of 34,381 micrographs) showing the quality of particles used in structure determination. Most particles contain both the MS- and C-rings, although a small number of isolated MS-rings are present. En face views (three are highlighted with black circles) and side views (three are highlighted with white circles) are observed. Tilted views are also observed and give the appearance of a smaller diameter in some cases. Scale bar, 200 Å. The uncropped micrograph is available in the Source data file. Raw micrographs for all structures have been deposited with EMPIAR76 (https://www.ebi.ac.uk/empiar/) and accession codes EMPIAR-11597, EMPIAR-11891 and EMPIAR-11892. c, Surface representation of the C-ring density maps in the CCW pose superimposed on the final model. FliF subunits are shown in blue, FliG subunits are shown in red, FliM subunits are shown in yellow and FliN subunits are shown in shades of pink and purple.
Response regulators affect flagellar rotation and speed. The best studied is the excitatory response regulator CheY (for chemotaxis)16,17, which biases the flagellum toward CW rotation. Others include the fumarate-sensing quinol:fumarate reductase18,19,20, the spermidine-sensing SpeE21 and the cyclic-di-GMP-sensing YcgR22,23. The CW pose18 may also support switch assembly, as well as assembly and disassembly of the entire flagellum24,25.
Key unknowns in chemotaxis are how the motor drives both CCW and CW rotation, how response regulators affect rotation and how torque transfers from the stator to the flagellum. To help inform on these controversies, we determined the structures of the S. enterica serovar typhimurium combined MS- and C-rings in the CCW pose, the C-ring in the CW pose and the CW pose bound to a response regulator. This complements concomitant work from the Lea group showing the isolated C-ring in the CCW pose, the CW pose and the MotA/B interaction with the C-terminus of FliG26.
Results
Structure of the wild-type C-ring
We formed particles from coexpressed FliF, FliG, a region of FliL, FliM, FliN and FliO27. We purified the resultant 6 MDa supercomplex containing both the MS-ring and the C-ring, collected cryogenic electron microscopy (cryoEM) data and determined the structure (Fig. 1b,c, Extended Data Fig. 1a,b, Supplementary Table 1 and Supplementary Video 1). Standard cryoEM workflows could not improve the resolution beyond ~8 Å. We, therefore, used particle subtraction at the level of the micrograph (Extended Data Fig. 1a). This technique improves alignment by obscuring unwanted features in the primary dataset28, with common applications including removing nanodiscs or detergent micelles from membrane protein particles. We removed the MS-ring and determined the structure of the C-ring, where the predominant species was a 34-mer (~50% of the particles). Local resolutions ranged from 2.9 to 6.6 Å, and the average resolution was 4.0 Å (Extended Data Fig. 2a and Supplementary Video 1). Other symmetries included a 33-mer (4.5 Å resolution), 35-mer (4.5 Å resolution) and 36-mer (6.7 Å resolution).
We interpreted the 34-mer maps by docking AlphaFold (v.2.0)29 models of isolated S. enterica domains and manually connecting them (Fig. 2a–f, Extended Data Fig. 2b,c and Supplementary Video 2). This identified that the switch subunits loosely organize into layers. Beginning at the side of the C-ring that faces the MS-ring and the membrane, the layers contain FliF514–560/FliG1–331 in two layers at the top, FliM52–237 (FliMmid) in the middle, and FliM257–330 (FliMC)/FliN45/59/63–137 in a 3:1 ratio at the bottom (Fig. 2e).

In the global views, FliF is blue, FliG is red, FliM is yellow and FliN is pink and purple. In the insets of a–e, each of the subunits is coloured from the N-terminus (blue) to C-terminus (red) to highlight the fold. a, FliFC wraps around FliGD1. b, A FliG protomer folds into five domains: FliGD1 (FliG1–67), FliGD2 (FliG73–99), FliGD3 (FliG107–186), FliGD4 (FliG196–233) and FliGD5 (FliG243–331). c, The FliM subunit, highlighting FliML1 (FliM31–50), FliMmid (FliM51–230), FliML2 (FliM231–256) and FliMC (FliM257–330). d, A 180° rotated view of panel (c). e, Three FliNC subunits are similar but non-equivalent. To highlight the fold, only one protomer (FliN3) is coloured from the N-terminus (blue) to C-terminus (red). The remaining two (FliN1 and FliN2) are coloured pink and purple. f, A side view of a single FliFGMN unit. An ~30 Å cleft between FliFC–FliGD1/D2 and FliGD5 is highlighted. g, A single FliFGMN unit participates in three staves. h, Density for FliGD5 appears to be separated, with the domain having little contact with adjacent subunits. i, Interactions between the PAA motif of FliGD3 and the adjacent FliGL1 linker. j,k, Formation of a curved spiral by the FliMC:3FliNC heterotetramer. j, A schematic that compares the open ring of FliMC:3FliNC in the cryoEM structure to the closed ring of the crystal structure of T. maritima FliNC (1YAB42). This comparison highlights that a pure FliNC superstructure would favour stacked discs in a linear array. FliMC breaks the symmetry, which is necessary to form the helix along the bottom of the C-ring. k, The FliMC:3FliNC forms a spiral that curves along the base of the C-ring to form a closed circle. A single arc is shown.
The density (Extended Data Fig. 3a–o) was consistent with AlphaFold models and X-ray crystal structures of isolated domains of FliG14,30,31,32,33,34,35,36,37,38, FliM21,32,37,39,40,41 and FliN39,42 from homologues (Extended Data Fig. 2c). However, crystal structures of multi-domain FliG31,35,38 required substantial interdomain adjustment to match the cryoEM structure (Extended Data Fig. 4a–f). In addition, past work supports FliG as a three-domain protein38; however, FliG contains five domains when it is assembled into the switch (Fig. 2b). Therefore, the FliG domains are redefined here as FliGD1–FliGD5 (FliG1–67, FliG73–99, FliG107–186, FliG196–233 and FliG243–331; Supplementary Table 2). Interdomain linkers are termed FliGL1–FliGL5.
Mutagenesis is commonly used to validate cryoEM structures. Given the extensive number of published mutants, designing new mutations was unnecessary. From this, we identified many assembly-deficient mutations30,32,43 that affect residues that form strong interactions at subunit interfaces (Extended Data Fig. 4g), which explains their impact on switch assembly.
The CCW pose of the switch
Purified wild-type C-rings exclusively rotate CCW under physiological conditions44, assigning this as the CCW pose. Furthermore, this structure concurs with the CCW pose shown by tomography45,46 (Extended Data Fig. 4h). In this structure (Fig. 2a–f), individual FliG subunits form a V shape. When assembled into a 34-mer, the upper regions form inner and outer rings separated by a 30 Å cleft. The inner ring contains FliFC, FliGD1 and FliGD2, and the outer ring contains FliGD5. At a more detailed level, FliFC forms a curved helix that extends radially from the MS-ring on the membrane side (Fig. 2a). This FliFC interacts intimately with FliGD1 (Fig. 2b) and has strong density for all C-terminal residues of FliFC (Extended Data Fig. 3a). As FliGD1 extends into FliGD2, it forms an armadillo motif, which is an α-helical hairpin that permits rotations. This armadillo motif of FliGD2 completes the fold of the next FliGD1 to form an intercalated structure (Fig. 2g). By contrast, FliGD5 of the outer ring distinctly separates from neighbouring subunits (Fig. 2h).
Below these upper rings, armadillo motifs of FliGD3 and FliGD4 (Fig. 2b,f) intercalate around the ring via domain swaps, as proposed by ref. 38. FliGD4 also forms the base of the cavity between the upper rings, and FliGD3 binds to FliM (Fig. 2f). The FliGL1–FliGL4 linkers between these domains have strong density suggesting rigidity (Extended Data Fig. 3b,c,e,f). FliGL3 is noteworthy because it both stabilizes the position of FliGD2 and makes intimate interactions with a Pro-Ala-Ala (PAA) sequence motif (FliG169–171) in an adjacent FliGD3 protomer (Fig. 2b,i). Deletion of this PAA motif results in flagellar motors that predominantly rotate CW35,47.
FliMmid (Fig. 2c,d,f) and its interface with FliGD3 resemble crystal structures21,32,37,39 except for a new helix containing FliM231––256 (FliML2). This helix domain swaps with the adjacent subunit and extends to the bottom of the structure. Here FliMC and FliNC adopt an organization called a SpoA fold39,42 and form a heterotetrameric building block that resembles a split lock washer (Fig. 2j,k). These FliMC:3FliNC building blocks pack into a spiral with FliMC at the lower edge of the C-ring (Fig. 2c,d,j,k) that is consistent with past biochemical and structural studies38,48.
The CW pose of the switch
A distinct pose of the switch supports CW rotation45,46 and switch assembly18,19. To inform on this pose, we determined the 4.6 Å resolution structure of switch particles containing the extreme CW-biased FliGΔPAA mutation47. Symmetry expansion followed by local refinement gave superior results to particle subtraction (Extended Data Fig. 5a). To build the model (Fig. 3a–f), we docked modules from the assembled CCW pose as rigid bodies into the CW maps. Linkers between domains were then built manually.

a–e, Individual folds of the C-ring subunits in the CW pose: FliF (a), FliG (b), FliM (c), a 180° rotated view of FliM (d), FliN (e). The relative orientation and the colouring are the same as for Fig. 2. f, A side view of the CW pose showing an expanded cleft between FliGD1/D2 and FliGD5. g, Comparison of the CCW pose (transparent) with the CW pose (solid) of a single C-ring subunit. h, A 90° rotation of panel (g) highlights the magnitude of the conformational change. i, Comparison of a top-down view of the CCW and CW poses shows the reversed orientation of the FliFC helix, which changes the connection to the MS-ring. The increased size of the cleft in the upper ring of FliG is also apparent. j, Colouring a single subunit in the context of the C-ring highlights the increased domain swaps in FliG of the CCW pose compared to the CW pose. In the CCW pose, FliFC–FliGD1 is in the inner ring above FliM and crosses staves three times. In the CW pose, FliFC–FliGD1 is in the inner ring behind FliGD5 and crosses staves twice. k, A side view of a single unit aligned to the FliMC:3FliNC spiral highlights the 25° rotation of FliMmid in the CW pose.
The CW pose contains significant domain rearrangement, particularly in FliG. Here FliFC–FliGD1 and FliGD2 of the inner ring and FliGD5 of the outer ring each rotate by ~180° (Fig. 3a,b,g–i). As a part of this, FliGD2 changes the subunit that it binds, altering the domain swaps (Fig. 3j). These changes have multiple impacts. They reverse the orientation FliGD5, which binds the MotA/B stator, and they also reverse FliGD1, which binds to the MS-ring. Finally, these rotations increase the size of the cleft between the inner and outer rings from 30 Å to 40 Å (Figs. 2f and 3f). FliGD3–FliMmid undergoes smaller positional changes, rotating approximately 25° as a unit with a slight adjustment in the binding interface (Fig. 3c,d,k).
The altered domain swaps in the CW pose (Fig. 3j) may have biological implications. First, the reduced number of swaps could facilitate C-ring assembly18,19. The altered domain swaps also suggest how the C-ring supports directional cooperativity49,50, where a change from CCW to CW in one subunit may trigger a similar change in an adjacent subunit. The likely steric clash in a ring of mixed poses would induce one subunit to change the conformation of the adjacent subunit.
Comparison of the CCW and CW poses (Fig. 3g–k) suggests a mechanism for switching in the switchΔPAA mutant (Fig. 4a). Loss of the PAA motif shortens the FliGL3 linker and adjusts the position between FliGD3 and FliGD4. It also moves FliGD2 to eliminate a domain swap. Finally, the PAA motif in FliGD3 normally interacts with the first helix of FliMmid, which is held under tension in the CCW pose51. Removing this interaction allows FliM to rotate. To support this proposal, we mapped directionally biasing mutants52 onto the CCW structure (Fig. 4b). CCW-biasing mutations group to a surface suggested to be a CheY binding site51,53,54. These likely– disrupt CheY binding to reduce CW transitions. Conversely, CW-biasing mutations dominate the ~100 Å pathway that connects the N-terminus of FliMmid to FliGD5 (Fig. 4a,b). These mutations may prevent allosteric conformational changes of FliM and FliG.

a, Allosteric pathway from the FliM N-terminus to the torque helix in FliGD5. Different steps of signal transmission are coloured from blue to red and numbered. The transfer of information starts at (1) the N-terminus of FliM near the FliML1 linker at the FliM-FliN interface. The information passes through the first helix of (2) FliMmid to FliGD3 near the (3) PAA motif, which supports (4) FliGD4. A rotation of (5) the C-terminal FliGD5 changes the orientation of the torque helix. Note that allosteric signal transmission may involve both the pathway that is shown and a concerted signal transmission in adjacent subunits of the ring. A single subunit of the C-ring with a neighbouring FliG (FliGN − 1) is shown. b, Locations of directionally biased mutations in the switch. Red balls highlight CCW-biasing mutations, with the majority of these located in the proposed binding site for CheY. Their mutation could affect CheY binding. Yellow balls mark locations of CW-biasing mutations, which cluster along the pathway in a. Their mutation could release the CCW pose.
The CW pose bound to a regulator
In one of our CW switchΔPAA datasets, we observed density for a binding partner within the cleft in the FliG subunit (Fig. 5a,b and Extended Data Fig. 5b). This identifies one way that a regulatory protein might interact with the CW switch. The bound protein moved both FliFC/FliGD1/D2 of the inner ring and FliGD5 of the outer ring, as a unit, by ~10 Å toward the centre of the ring. This decreased the diameter from 460 Å in the CW pose (470 Å in the CCW pose) to 440 Å in the CW pose with the binding partner.

a, On the left, twelve repeats of the protein-bound CW pose of the switch are shown in grey, with three repeats of the density in the cleft shown in green. For comparison, the right shows the CW pose of the switch not bound to a partner protein. The cleft is still visible but lacks density within it. b, Cross-section of a single subunit with the density for the regulator in green. For comparison, the CW pose with an empty cleft is shown on the right.
The size of the density is consistent with a globular domain of ~120 amino acids. Because it bridges FliFC/FliGD1 and FliGD5, which differ between CCW and CW, this protein should be specific for the CW pose (Fig. 5a,b). Additional density above the cleft resembles an intertwined helical coiled coil and forms a ring. The position of this ring differs from the position of those that appear on the outer perimeter of FliG during assembly and disassembly55.
The local resolution for this density was 9 Å, making it difficult to identify the species from the maps. We used manual docking to evaluate several possibilities: YcgR, CheY-FliM1–16, quinol:fumarate reductase and FliO (5Y6H ref. 56, 4IGA ref. 57, 1KF6 ref. 58, https://alphafold.ebi.ac.uk/entry/A0A5C2LXN8)29. YcgR and CheY could be docked, while quinol:fumarate reductase and FliO fit the density poorly. YcgR22,23 is unlikely because it physiologically stabilizes the CCW pose. While CheY is relevant to the CW pose16,17, past work identifies that CheY binds to the switch at the N-terminus of FliM (FliM1–16)53, in the central domain of FliM (FliM51–228)54 with FliMR94 suggested as key51 and at a hydrophobic patch of a FliN homodimer59 containing FliNV113, FliNV114 and FliNA115. Moreover, past tomography identifies that CheY correlates with the appearance of density on the exterior of the FliM subunit45,46. Thus, we cannot identify the bound species.
Torque transmission
Torque transmits between the MotA/B binding site on the FliGD5 torque helix4,26,60 and FliF. To trace the path of torque transmission, we evaluated the two ends of this pathway. In wild-type C-rings, torque enters the C-ring at the outer FliGD5 (Fig. 6b). In the CW pose, FliGD5 rotates and presents the MotA/B binding site to the inside of the ring, with torque likely transmitted along the same path, albeit in the opposite direction. Tracing the path of torque transmission to the rod requires understanding how the switch connects to the MS-ring1,2,3. To evaluate this, we determined the structure of the MS-ring in the CCW switch (Fig. 6a) by obscuring the C-ring using particle subtraction (Extended Data Fig. 1b). The resultant 3.4 Å resolution structure contained 33-mer MS-rings (FliF50–428) in 58% of 34-mer C-rings (Extended Data Fig. 6a,b). The remaining MS-rings could not be classified into a stoichiometry. MS-rings that could not be classified correlated with grids that had thinner ice, suggesting that the MS-ring preferentially partitions at the air–water interface. However, we cannot exclude the presence of other stoichiometries that we could not classify.

a, A model for the flagellar motor from Gram-negative bacteria was built from our structure and that of the S. enterica flagellar basal body (7CGO ref. 12). The L-ring (light purple) contains FlgH subunits. The P-ring (dark purple) contains the FlgI subunits. In the centre of the LP-ring is a rod (grey). The distal region of the rod (FlgG) connects to the hook and flagellum, while the proximal region of the rod (FliE, FlgB, FlgC and FlgF) connects the LP-ring to the MS-ring. The MS-ring (blue) localizes within the inter-membrane space and contains FliF subunits. Finally, the C-ring (orange), contains FliF C-termini as well as FliG, FliM, and FliN of the switch. b, Pathway of torque transmission through the flagellar motor. The C-ring transmits torque from the MotA/B stator to the MS-ring and flagellar rod. The figure shows a map of the torque transmission pathway highlighted with black arrowheads and coloured from blue (stator) to red (MS-ring). Torque transfer begins with the interaction between the MotA/B stator and the torque helix of FliGD5 of the C-ring. Interactions across the FliG subunit allow the torque to be transmitted to FliGD1, where there is a direct interaction with FliF. This is expected to turn the MS-ring and the flagellar rod within.
While the fold of each FliF subunit is generally consistent with previous reports5,9,10,11, there are two notable differences. The first is the 33-mer stoichiometry. Some past structures show variable stoichiometry5,10. Others suggest that the native stoichiometry is a 34-mer9. A second difference in the MS-ring structure compared to past work was additionally observed regions of N-terminal ring-building motif 1 (RMB1, FliF50–106; Extended Data Fig. 6a). Past MS-ring structures identified either 9 or 11 RBM1s5,9,10,11, but coordinates were not assigned. The present map contained density for 33 RBM1 domains in two positions and allowed coordinates to be assigned to 11 (Extended Data Fig. 6b).
To complete the connections, we modelled FliF1–49 and FliF429–514 with AlphaFold29. The prediction showed high confidence that these were helices (Fig. 6a). Because the particles have a symmetry mismatch between the MS- and C-rings, there could be multiple ways to model the MSC-ring species. A FliF33:FliG34:FliM34:FliN102 assignment is consistent with the symmetric appearance of the C-ring before averaging. By contrast, loss of a FliG subunit, that is, FliF33:FliG33:FliM34:FliN102, does not match our data and suggests that the FliF:FliG ratio need not be 1:1. We propose that these MSC-rings contain 203 subunits: 33 FliF, 34 FliG subunits, 34 FliM subunits and 102 non-equivalent FliN subunits.
A global view of the resultant model shows that the MS- and C-rings stack with a tilt angle of 4° (Fig. 6a). This non-coaxial stacking is similar to what was observed between MS-ring and LP-ring in previous structures6,12 (Fig. 6a). When considering the intact motor (Fig. 6a), it is tempting to align the axis of the C-ring with the LP-ring, which results in a tilted MS-ring between these features. This tilt could explain why the MS-ring looks thicker on edge in low-resolution MSC-ring structures14,15 than in structures of the isolated MS-ring5,9,10,11. A tilt could be expected in any motor with a symmetry mismatch between the MS- and C-rings.
With this tilt, axial rotation of the C-ring and rod during flagellar rotation would make the MS-ring of the motor appear to wobble when viewed edge on (Supplementary Video 3). Symmetry mismatches have other characterized biological effects. They promote a low-energy state and allow efficient rotation61 at high speed, which requires that there be no significant energy minima. Symmetry mismatch between the 33-mer MS-ring and 34-mer C-ring observed in these particles (Fig. 6a and Extended Data Fig. 1) could help support this. This wobble could prevent an energetic minimum during flagellar rotation10 or influence the inherently asymmetric export apparatus62,63 at the interior of the C-ring. Because FliFC is tightly tethered to FliGD1, the ability of the MS- and C-rings to be flexibly attached may be important for both torque transmission and the shift between the CCW to CW poses.
Discussion
In this Article, we report cryoEM structures of three states of the flagellar switch, answering questions about torque transmission, directional control and binding of a response regulator. Torque input (Fig. 6b) involves the MotA/B stator1,2,3 (Extended Data Fig. 7a,b), which uses the transmembrane electrochemical gradient to induce rotation and transmits this to the torque helix on FliGD5 (refs. 4,60,64,65,66). MotA/B and the C-ring then act like interlaced cogwheels to drive flagellar rotation64,65 (Extended Data Fig. 7c). Supporting this model, the interface between MotA subunits64,65 is perfectly positioned to grasp the FliGD5 torque helix (Extended Data Fig. 7a,b). Torque transmits through FliG to FliF of the MS-ring, which connects to the flagellar rod.
FliG has a conformational difference between CCW and CW poses (Figs. 2 and 3) that moves the binding site for MotA/B from facing outward in the CCW pose to facing inward in the CW pose (Fig. 3), which changes the direction of rotation of the C-ring (Supplementary Video 4 and Extended Data Fig. 7c). This model is supported by tomography of the stator bound to the C-ring in the CCW and CW poses46 and is consistent with past proposals for powering rotation in opposite directions64,65. The concomitant ~180° FliFC/FliGD1 rotation optimizes the connection between the C-ring and the MS-ring in these two directions (Fig. 3i).
The CCW and CW poses of the switch also help explain cooperativity49,50, which induces the subunits of the C-ring to preferentially adopt the same pose. Three conformational differences between the CCW and CW would result in steric clash unless the next subunit adjusted. These are the FliFC–FliGD1 rotation, the change in domain swapping of FliGD2 and the ~25° rotation of FliMmid (Fig. 3g,h,k). Biologically, bound signalling proteins influence both the direction and rate of rotation. One of our CW data sets showed density within an ~40 Å cleft between FliG domains, identifying one binding site for regulatory proteins (Fig. 5). This density is consistent with an ~120 amino acid protein that locks FliFC–FliGD1/D2 and FliGD5 into the CW pose. The ~30 Å cleft in the CCW pose may also be large enough to bind to a regulatory protein (Fig. 2f).
The architectures of FliM and FliN also inform on the mechanism of conformational transitions between the CCW and CW poses45,46 (Figs. 2–4). It has previously been unclear how the motor could be conformationally dynamic enough to function and yet stable enough to survive these large structural transitions. Domain swapping with subunit intercalation is a structural feature known to both enhance stability and allow superstructures to dynamically adopt multiple stoichiometries. This has been best studied during protein aggregation that causes disease67. The substantial domain exchange of FliM and FliN within the switch may help to accommodate large molecular reorganization during the transitions between the CCW and CW poses (Figs. 2–4).
Notably, there is currently no consensus over the stoichiometry for the MS-ring5,9,10,12. Some cryoEM studies showed a range of stoichiometries5,10. Other studies only identify a 34-mer9. The variable stoichiometry was interpreted as the MS-ring adapting to load. This largely leveraged parallels to the C-ring’s stoichiometry68 and the number of bound stators69, which can change in response to the strength of the attractant or load.
Studies showing only a 34-mer suggest that other stoichiometries arise from artefacts due to C-terminal proteolysis of FliF or incorrect templating during plasmid expression9. We can exclude C-terminal proteolysis affecting stoichiometry in our structure because we observe density for the full C-terminus of FliF bound to FliG (Figs. 2a and 3a and Extended Data Fig. 3a). In terms of templating, this 33-mer MS-ring and the previously published strict 34-mer9 were similarly expressed in E. coli. This suggests that the E. coli templating machinery can be recruited to assemble the Salmonella MS-ring (~95% identical) and is also unlikely to underlie the stoichiometric difference. Nevertheless, we did not test conditions proposed to affect stoichiometry, which would be required to distinguish between an adaptive and a strict stoichiometry. For example, we did not coexpress the S. enterica templating machinery with the pKLR3 plasmid, and we did not grow cells under conditions with different attractants or different loads. Taken together, the origins of symmetry differences in MS-ring structures remain unclear at this time.
In aggregate, this work reports the high-resolution structure of the most critical piece of the flagellar motor in three states. The structure suggests mechanisms for torque transmission and directional switching during chemotaxis in Salmonella and related bacteria. This structure also allows a large body of data on bacterial chemotaxis to be understood in the context of an architecture.
Methods
Constructs
Plasmid pKLR3 (ref. 27) containing the S. enterica serovar typhimurium fliL, fliF, fliG, fliM, fliN and fliO genes was a generous gift to M.E. from S. Khan.
Protein purification
The S. enterica serovar typhimurium FliFGMN subunits were expressed in E. coli BL21-Gold cells in LB medium supplemented with 0.034 mg ml−1 chloramphenicol. At an OD600 of 0.6, expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside. Following induction, cells were grown at 37 °C for 18 h with shaking, then collected by centrifugation at 6,750 × g at 4 °C.
Following growth, bacterial cells were resuspended in 100 mM Tris–HCl with pH 8.0, 8 mM EDTA and one protease inhibitor tablet for every 50 ml of buffer. Cells were lysed with 1% w/v Triton X100 detergent and 10 mg lysozyme. The cell suspension was stirred at 4 °C for 4 h before adding MgCl2 to a final concentration of 10 mM. For every 50 ml of buffer, 100 U of DNase and 5 mg of RNase were also added. The lysed cells were stirred for 1 h before centrifuging at 18,000 × g for 30 min to remove cell debris. Membranes were separated from this supernatant by centrifugation at 60,000 × g for 1 h. The cell membranes were resuspended in 100 mM HEPES with pH 7.5, 5 mM EDTA, 0.1% v/v Triton X100 for 30 min on ice. Partial purification was achieved via differential membrane extraction. First, the Triton X100 concentration was increased to 10%, and the suspension was mixed gently for 1 h. These partially extracted membranes were centrifuged at 14,000 × g for 30 min, and the supernatant was centrifuged at 60,000 × g for 1 h to collect the membranes. This pellet was resuspended in 100 mM HEPES with pH 7.5, 5 mM EDTA, 0.1% v/v Triton X100 and 0.05% lauryl maltose neopentyl glycol (Anatrace, NG310), then mixed gently for 1 h. The suspension was then spun at 14,000 × g for 30 min, and the supernatant was filtered through a 0.4 µm syringe filter. The filtered sample contained MSC-ring particles and was used for preparing cryoEM grids.
Cryo-EM sample preparation and imaging
A 300 mesh R1.2/1.3 Au Quantifoil grid (Electron Microscopy Sciences) was glow discharged for 15 s. Purified MSC-rings (2 µl of 23 mg ml−1) were added to each grid at 4 °C and 100% humidity. After 15 s of incubation, blotting was performed for 4 s. Grids were plunged into liquid ethane using a Vitrobot Mark IV system (Thermo Fisher). Grids were screened on 200 keV Glacios microscope (Thermo Fisher). Data were collected from the best grids using a 300 keV Titan Krios G4 microscope with a Gatan K3 direct electron detector (Thermo Fisher).
For wild-type MSC-ring, 34,831 movies were motion-corrected using patch motion-based correction in cryoSPARC (v.4.2.1)70. The contrast transfer function (CTF) was estimated using Patch CTF Estimation in cryoSPARC70. Using template picker, we picked 3,906 particles from 3,427 micrographs (10% of the dataset). An ab initio model was created from this and was used as a template to pick particles from the complete dataset.
For the CW MSC-ring, 35,552 movies were first motion-corrected using patch motion-based correction in cryoSPARC70. The CTF was estimated using Patch CTF Estimation in cryoSPARC. Using template picker, we picked 3,906 particles from 3,500 micrographs (10% of the dataset). An ab initio model was created from this and was used as a template to pick particles from the complete dataset.
For the CW MSC-ring with bound partner protein, 26,130 movies were processed using the same steps as for CW C-ring. Using cryoSPARC template picker, we picked 4,546 particles from 3,700 micrographs (14% of the dataset). Using the CW C-ring as an input model, the C-ring was created and was used as a template to pick particles from the complete dataset.
Structure determination
For the wild-type MSC-rings, 295,031 particles were picked, and 50,000 of them were used to build ab initio models without enforcing symmetry. These initial 3D class averages separated into four classes. In the next step, all 295,031 particles were used to perform heterogeneous refinement on the four ab initio classes. Upon completion, only one class contained MSC-rings (21% of particles; Extended Data Fig. 1a). In addition, one class was isolated MS-rings, and two classes were junk classes. Inspection of the density showed the C-ring at low resolution, ~15 Å, and showed clear staves. However, the associated density for the MS-ring was uninterpretable.
Because the lower quality density for the MS-ring may have resulted from a symmetry mismatch between the MS- and C-rings, potentially combined with alignment that was biased toward the larger C-ring, a particle subtraction technique was used to improve the resolution. First, MS-rings were identified from the 3D model. This consensus map was used to build a mask around the MS-ring. The MS-ring was then subtracted at the level of the micrograph (Extended Data Fig. 1a). The MS-ring-subtracted particles were then used to determine high-resolution structures for the C-ring.
Initial heterogeneous refinement resulted in five classes with different symmetries. One class contained 33-fold symmetry (13,081 particles), two classes contained 34-fold symmetry (15,301 and 13,081 particles), one class contained 35-fold symmetry (11,346 particles) and one class contained 36-fold symmetry (6,041 particles). Non-uniform refinement3 was performed on each of the above classes (33-, 34-, 35- and 36-fold symmetry) with the final resolution of the C33 map at 4.5 Å resolution, the C34 map at 4.1 Å resolution, the C35 map at 4.5 Å resolution and the C36 map at 6.7 Å resolution. To improve the resolution of the 34-fold symmetric map even further, particles from all the classes were passed through heterogeneous refinement with C34 symmetry imposed. Following this procedure, the best classes were refined using homogeneous and non-uniform refinement. The final resolution was 4.0 Å.
For the CW MSC-rings, 43,741 particles were picked. Using the ab initio model, these particles were classified into three classes. Upon completion, only one class contained MSC-rings (16% of particles; Extended Data Fig. 5a). In addition, one class was isolated MS-rings, and one class was junk class. Inspection of the density showed the C-ring at low resolution, ~20 Å, and showed clear staves. However, the associated density for the MS-ring was uninterpretable. From the CCW C-ring previous knowledge, we applied C34 symmetry to refine the C-ring. Due to the low number of particles, we were unable to improve the resolution of the map using particle subtraction. Therefore, we expanded the particles by applying C34 symmetry and locally refined a small section by masking three staves of the C-ring. This improved the resolution to 4.6 Å. We used RELION (v.4.0.1) image handler to form a 34-mer C-ring from the refined sectional map71.
For the CW MSC-rings with bound partner protein, 59,404 particles were picked. Using the CW MSC-ring model, these particles were classified into three classes. Upon completion, only one class contained MSC-rings (34% of particles; Extended Data Fig. 5b). In addition, one class was isolated MS-rings, and one class was a junk class. Inspection of the density showed the C-ring at low resolution, ~25 Å, and showed clear staves. However, the associated density for the MS-ring was uninterpretable. We then applied C34 symmetry to refine the C-ring. As we had more particles in this dataset, we both applied particle subtraction to improve the map quality and also applied local refinement on C34 symmetry-expanded particles from three masked staves. This improved the resolution to 5.9 Å. We used RELION image handler to form a 34-mer C-ring from the refined sectional map71.
To evaluate the MS-ring associated with CCW C-rings, the particle subtraction protocol was reversed. The 34-mer class has the most particles and the highest resolution. Thus, 34-mer particles were next extracted from the micrographs, and the C-ring was then subtracted (Extended Data Fig. 1b). Classes were then developed for the MS-ring. This procedure identified that for the 34-mer C-ring, the MS-ring classified exclusively as a 33-mer (Extended Data Fig. 1b) and was associated with a final resolution of 3.4 Å.
Model building and refinement
To assist in model assignment, alphaFold29 was used to develop homology models of appropriate subunits and domains. For FliG domains, FliGD3:FliMmid heterodimers, FliNC homodimers and FliMC:FliNC heterodimers, packing interactions from crystal structures of various unassembled domains from thermophiles were used to create a library of multi-domain models with potential domain exchanges. This library was manually developed using COOT (v.0.9.8.8)72.
The model of FliGD3:FliMmid (developed from PDB entry 4FQ0 (ref. 32))was first docked into the corresponding density in COOT, then optimized in ChimeraX (v.1.7)73. This was followed by docking the models for FliGD4 and FliGD5 into the density at the exterior of the ring (developed from PDB entries 3HJL (ref. 38) and 1LKV (ref. 30)). Next, a model containing the FliFC, FliGN and the domain-swapped armadillo repeat (developed from PDB entry 5WUJ (ref. 36)) was docked into the density on the interior of the ring in COOT72 and optimized in ChimeraX73. Linkers between these domains were built manually in COOT72. The base of the ring used a combination of docked FliNC homodimers and FliMC:FliNC heterodimers (developed from PDB entry 4YXB (ref. 39)), with the linking helix between FliMmid and FliMC built manually in COOT72.
Docking of each of these structures in COOT72 followed by optimization in ChimeraX73 gave an unambiguous match with the density. Refinement was performed by standard methods that alternated rounds of manual model improvement in COOT72 with refinement in PHENIX (v.1.20.1-4487)74. Figures were made using ChimeraX72, and videos were made using Blender v.3.5 (https://www.blender.org/) and Molecular Nodes (v.2.8)75.
Manuscript editing using artificial intelligence
Manuscript length and accessibility were both edited using the formalizer subroutine in goblin.tools.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw, processed, and interpreted data that support the findings of this study are available in public repositories. Raw micrographs have been deposited with EMPIAR76 (https://www.ebi.ac.uk/empiar/), and accession codes are EMPIAR-11597, EMPIAR-11891 and EMPIAR-11892. CryoEM maps have been deposited at the EMDB77 (https://www.ebi.ac.uk/emdb/) with the accession codes EMD-41100, EMD-41101, EMD-41102, EMD-41103, EMD-41104, EMD-43256, EMD-43258, EMD-43327 and EMD-43328. Atomic coordinates of the 34-mer CCW C-ring and the 33-mer MS-ring have been deposited at the Protein Data Bank78 (www.rcsb.org) with the accession codes 8T8O and 8T8P. Atomic coordinates for the single subunit of the isolated CW-locked C-ring are deposited with the accession code 8VIB, the 34-mer isolated CW-locked C-ring are deposited with accession code 8VKQ. Coordinates for a single subunit of the CW-locked C-ring bound to a partner protein have the accession code 8VID, and the 34-mer of the CW-locked C-ring bound to a partner protein have the accession code 8VKR.
Previously reported structures or computational models used to support this work are: Thermotoga maritima FliG (1LKV ref. 30; 5TDY ref. 34), Helicobacter pylori FliG (3USW ref.31, 4FQ0 ref. 32), T. maritima FliN (1YAB ref. 42), S. enterica FliM:FliN fusion (4YXB ref. 39), S. enterica flagellar basal body (7CGO ref. 12), Aquifex aeolicus MotA (8GQY ref. 79), Campylobacter jejunji MotA/B (6YKM ref. 65), Clostridium sporogenes MotA/B (6YSF ref. 64), Bacillus subtilis MotA/B (6YSL ref. 64), E. coli YcgR (5Y6H ref. 56), T. maritima CheY-FliM1–16 (4IGA ref. 57), E. coli quinol:fumarate reductase (1KF6 ref. 58) and FliO (alphafold.ebi.ac.uk/entry/A0A5C2LXN829). Source data are provided with this paper.
Code availability
No custom code was used or developed for the analysis of data reported in this study.
References
Beeby, M., Ferreira, J. L., Tripp, P., Albers, S. V. & Mitchell, D. R. Propulsive nanomachines: the convergent evolution of archaella, flagella and cilia. FEMS Microbiol. Rev. 44, 253–304 (2020).
Mondino, S., San Martin, F. & Buschiazzo, A. 3D cryo-EM imaging of bacterial flagella: novel structural and mechanistic insights into cell motility. J. Biol. Chem. 298, 102105 (2022).
Minamino, T. & Kinoshita, M. Structure, assembly, and function of flagella responsible for bacterial locomotion. EcoSal Plus 11, eesp00112023 (2023).
Zhou, J., Lloyd, S. A. & Blair, D. F. Electrostatic interactions between rotor and stator in the bacterial flagellar motor. Proc. Natl Acad. Sci. USA 95, 6436–6441 (1998).
Johnson, S. et al. Symmetry mismatch in the MS-ring of the bacterial flagellar rotor explains the structural coordination of secretion and rotation. Nat. Microbiol. 5, 966–975 (2020).
Johnson, S. et al. Molecular structure of the intact bacterial flagellar basal body. Nat. Microbiol. 6, 712–721 (2021).
Johnson, S., Kuhlen, L., Deme, J. C., Abrusci, P. & Lea, S. M. The structure of an injectisome export gate demonstrates conservation of architecture in the core export gate between flagellar and virulence type III secretion systems. MBio 10, https://doi.org/10.1128/mBio.00818-19 (2019).
Kato, T., Makino, F., Miyata, T., Horvath, P. & Namba, K. Structure of the native supercoiled flagellar hook as a universal joint. Nat. Commun. 10, 5295 (2019).
Kawamoto, A. et al. Native flagellar MS ring is formed by 34 subunits with 23-fold and 11-fold subsymmetries. Nat. Commun. 12, 4223 (2021).
Singh, P. K., Cecchini, G., Nakagawa, T. & Iverson, T. M. CryoEM structure of a post-assembly MS-ring reveals plasticity in stoichiometry and conformation. PLoS ONE 18, e0285343 (2023).
Takekawa, N. et al. Two distinct conformations in 34 FliF subunits generate three different symmetries within the flagellar MS-ring. MBio 12, https://doi.org/10.1128/mBio.03199-20 (2021).
Tan, J. et al. Structural basis of assembly and torque transmission of the bacterial flagellar motor. Cell 184, 2665–2679 e2619 (2021).
Yamaguchi, T. et al. Structure of the molecular bushing of the bacterial flagellar motor. Nat. Commun. 12, 4469 (2021).
Thomas, D., Morgan, D. G. & DeRosier, D. J. Structures of bacterial flagellar motors from two FliF-FliG gene fusion mutants. J. Bacteriol. 183, 6404–6412 (2001).
Thomas, D. R., Francis, N. R., Xu, C. & DeRosier, D. J. The three-dimensional structure of the flagellar rotor from a clockwise-locked mutant of Salmonella enterica serovar Typhimurium. J. Bacteriol. 188, 7039–7048 (2006).
Hess, J. F., Oosawa, K., Kaplan, N. & Simon, M. I. Phosphorylation of three proteins in the signaling pathway of bacterial chemotaxis. Cell 53, 79–87 (1988).
Welch, M., Oosawa, K., Aizawa, S. & Eisenbach, M. Phosphorylation-dependent binding of a signal molecule to the flagellar switch of bacteria. Proc. Natl Acad. Sci. USA 90, 8787–8791 (1993).
Cohen-Ben-Lulu, G. N. et al. The bacterial flagellar switch complex is getting more complex. EMBO J. 27, 1134–1144 (2008).
Koganitsky, A., Tworowski, D., Dadosh, T., Cecchini, G. & Eisenbach, M. A mechanism of modulating the direction of flagellar rotation in bacteria by fumarate and fumarate reductase. J. Mol. Biol. 431, 3662–3676 (2019).
Zarbiv, G. et al. Energy complexes are apparently associated with the switch-motor complex of bacterial flagella. J. Mol. Biol. 416, 192–207 (2012).
Zhang, H. et al. A putative spermidine synthase interacts with flagellar switch protein FliM and regulates motility in Helicobacter pylori. Mol. Microbiol. 106, 690–703 (2017).
Ko, M. & Park, C. Two novel flagellar components and H-NS are involved in the motor function of Escherichia coli. J. Mol. Biol. 303, 371–382 (2000).
Fang, X. & Gomelsky, M. A post-translational, c-di-GMP-dependent mechanism regulating flagellar motility. Mol. Microbiol. 76, 1295–1305 (2010).
Kaplan, M. et al. In situ imaging of the bacterial flagellar motor disassembly and assembly processes. EMBO J. 38, e100957 (2019).
Kaplan, M. et al. Loss of the bacterial flagellar motor switch complex upon cell lysis. MBio 12, e0029821 (2021).
Johnson, S. et al. Structural basis of directonal switching by the bacterial flagellum. Preprint at Research Square, https://doi.org/10.21203/rs.3.rs-3417165/v1 (2023).
Lux, R., Kar, N. & Khan, S. Overproduced Salmonella typhimurium flagellar motor switch complexes. J. Mol. Biol. 298, 577–583 (2000).
Fernandez-Gimenez, E. et al. A new algorithm for particle weighted subtraction to decrease signals from unwanted components in single particle analysis. J. Struct. Biol. 215, 108024 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Brown, P. N., Hill, C. P. & Blair, D. F. Crystal structure of the middle and C-terminal domains of the flagellar rotor protein FliG. EMBO J. 21, 3225–3234 (2002).
Lam, K. H. et al. Multiple conformations of the FliG C-terminal domain provide insight into flagellar motor switching. Structure 20, 315–325 (2012).
Lam, K. H. et al. Structural basis of FliG-FliM interaction in Helicobacter pylori. Mol. Microbiol. 88, 798–812 (2013).
Lloyd, S. A., Whitby, F. G., Blair, D. F. & Hill, C. P. Structure of the C-terminal domain of FliG, a component of the rotor in the bacterial flagellar motor. Nature 400, 472–475 (1999).
Lynch, M. J. et al. Co-folding of a FliF-FliG split domain forms the basis of the MS:C ring interface within the bacterial flagellar motor. Structure 25, 317–328 (2017).
Minamino, T. et al. Structural insight into the rotational switching mechanism of the bacterial flagellar motor. PLoS Biol. 9, e1000616 (2011).
Xue, C. et al. Crystal structure of the FliF–FliG complex from Helicobacter pylori yields insight into the assembly of the motor MS-C ring in the bacterial flagellum. J. Biol. Chem. 293, 2066–2078 (2018).
Sircar, R. et al. Assembly states of FliM and FliG within the flagellar switch complex. J. Mol. Biol. 427, 867–886 (2015).
Lee, L. K., Ginsburg, M. A., Crovace, C., Donohoe, M. & Stock, D. Structure of the torque ring of the flagellar motor and the molecular basis for rotational switching. Nature 466, 996–1000 (2010).
Notti, R. Q., Bhattacharya, S., Lilic, M. & Stebbins, C. E. A common assembly module in injectisome and flagellar type III secretion sorting platforms. Nat. Commun. 6, 7125 (2015).
Paul, K., Gonzalez-Bonet, G., Bilwes, A. M., Crane, B. R. & Blair, D. Architecture of the flagellar rotor. EMBO J. 30, 2962–2971 (2011).
Vartanian, A. S., Paz, A., Fortgang, E. A., Abramson, J. & Dahlquist, F. W. Structure of flagellar motor proteins in complex allows for insights into motor structure and switching. J. Biol. Chem. 287, 35779–35783 (2012).
Brown, P. N., Mathews, M. A., Joss, L. A., Hill, C. P. & Blair, D. F. Crystal structure of the flagellar rotor protein FliN from Thermotoga maritima. J. Bacteriol. 187, 2890–2902 (2005).
Irikura, V. M., Kihara, M., Yamaguchi, S., Sockett, H. & Macnab, R. M. Salmonella typhimurium fliG and fliN mutations causing defects in assembly, rotation, and switching of the flagellar motor. J. Bacteriol. 175, 802–810 (1993).
Ravid, S., Matsumura, P. & Eisenbach, M. Restoration of flagellar clockwise rotation in bacterial envelopes by insertion of the chemotaxis protein CheY. Proc. Natl Acad. Sci. USA 83, 7157–7161 (1986).
Carroll, B. L. et al. The flagellar motor of Vibrio alginolyticus undergoes major structural remodeling during rotational switching. Elife 9, https://doi.org/10.7554/eLife.61446 (2020).
Chang, Y. et al. Molecular mechanism for rotational switching of the bacterial flagellar motor. Nat. Struct. Mol. Biol. 27, 1041–1047 (2020).
Togashi, F., Yamaguchi, S., Kihara, M., Aizawa, S. I. & Macnab, R. M. An extreme clockwise switch bias mutation in fliG of Salmonella typhimurium and its suppression by slow-motile mutations in motA and motB. J. Bacteriol. 179, 2994–3003 (1997).
McDowell, M. A. et al. Characterisation of Shigella Spa33 and Thermotoga FliM/N reveals a new model for C-ring assembly in T3SS. Mol. Microbiol. 99, 749–766 (2016).
Bren, A. & Eisenbach, M. Changing the direction of flagellar rotation in bacteria by modulating the ratio between the rotational states of the switch protein FliM. J. Mol. Biol. 312, 699–709 (2001).
Bai, F. et al. Conformational spread as a mechanism for cooperativity in the bacterial flagellar switch. Science 327, 685–689 (2010).
Afanzar, O. et al. The switching mechanism of the bacterial rotary motor combines tight regulation with inherent flexibility. EMBO J. 40, e104683 (2021).
Sockett, H., Yamaguchi, S., Kihara, M., Irikura, V. M. & Macnab, R. M. Molecular analysis of the flagellar switch protein FliM of Salmonella typhimurium. J. Bacteriol. 174, 793–806 (1992).
Bren, A. & Eisenbach, M. The N terminus of the flagellar switch protein, FliM, is the binding domain for the chemotactic response regulator, CheY. J. Mol. Biol. 278, 507–514 (1998).
Dyer, C. M., Vartanian, A. S., Zhou, H. & Dahlquist, F. W. A molecular mechanism of bacterial flagellar motor switching. J. Mol. Biol. 388, 71–84 (2009).
Kaplan, M. et al. Novel transient cytoplasmic rings stabilize assembling bacterial flagellar motors. EMBO J. 41, e109523 (2022).
Hou, Y. J. et al. Structural insights into the mechanism of c-di-GMP-bound YcgR regulating flagellar motility in Escherichia coli. J. Biol. Chem. 295, 808–821 (2020).
Ahn, D. R., Song, H., Kim, J., Lee, S. & Park, S. The crystal structure of an activated Thermotoga maritima CheY with N-terminal region of FliM. Int. J. Biol. Macromol. 54, 76–83 (2013).
Iverson, T. M., Luna-Chavez, C., Croal, L. R., Cecchini, G. & Rees, D. C. Crystallographic studies of the Escherichia coli quinol-fumarate reductase with inhibitors bound to the quinol-binding site. J. Biol. Chem. 277, 16124–16130 (2002).
Sarkar, M. K., Paul, K. & Blair, D. Chemotaxis signaling protein CheY binds to the rotor protein FliN to control the direction of flagellar rotation in Escherichia coli. Proc. Natl Acad. Sci. USA 107, 9370–9375 (2010).
Terashima, H., Kojima, S. & Homma, M. Site-directed crosslinking identifies the stator–rotor interaction surfaces in a hybrid bacterial flagellar motor. J. Bacteriol. 203, https://doi.org/10.1128/JB.00016-21 (2021).
Stock, D., Gibbons, C., Arechaga, I., Leslie, A. G. & Walker, J. E. The rotary mechanism of ATP synthase. Curr. Opin. Struct. Biol. 10, 672–679 (2000).
Kuhlen, L. et al. Structure of the core of the type III secretion system export apparatus. Nat. Struct. Mol. Biol. 25, 583–590 (2018).
Sheedlo, M. J. et al. Cryo-EM reveals species-specific components within the Helicobacter pylori Cag type IV secretion system core complex. Elife 9, e.59495 (2020).
Deme, J. C. et al. Structures of the stator complex that drives rotation of the bacterial flagellum. Nat. Microbiol. 5, 1553–1564 (2020).
Santiveri, M. et al. Structure and function of stator units of the bacterial flagellar motor. Cell 183, 244–257 e216 (2020).
Hu, H. et al. Ion selectivity and rotor coupling of the Vibrio flagellar sodium-driven stator unit. Nat. Commun. 14, 4411 (2023).
Garcia-Seisdedos, H., Villegas, J. A. & Levy, E. D. Infinite assembly of folded proteins in evolution, disease, and engineering. Angew. Chem. Int. Ed. Engl. 58, 5514–5531 (2019).
Yuan, J., Branch, R. W., Hosu, B. G. & Berg, H. C. Adaptation at the output of the chemotaxis signalling pathway. Nature 484, 233–236 (2012).
Wadhwa, N., Phillips, R. & Berg, H. C. Torque-dependent remodeling of the bacterial flagellar motor. Proc. Natl Acad. Sci. USA 116, 11764–11769 (2019).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Pettersen, E. F. et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Johnston, B. et al. BradyAJohnston/MolecularNodes: v2.8.1 for Blender 3.5+ (v2.8.1). Zenodo. https://doi.org/10.5281/zenodo.8240420 (2023).
Iudin, A. et al. EMPIAR: the Electron Microscopy Public Image Archive. Nucleic Acids Res. 51, D1503–D1511 (2023).
The wwPDB Consortium. EMDB—the Electron Microscopy Data Bank. Nucleic Acids Res. 52, D456–D465 (2024).
Berman, H. M. et al. The Protein Data Bank. Nucleic Acids Res. 28, 235–242 (2000).
Nishikino, T. et al. Structure of MotA, a flagellar stator protein, from hyperthermophile. Biochem. Biophys. Res. Commun. 631, 78–85 (2022).
Paul, K., Harmon, J. G. & Blair, D. F. Mutational analysis of the flagellar rotor protein FliN: identification of surfaces important for flagellar assembly and switching. J. Bacteriol. 188, 5240–5248 (2006).
Acknowledgements
We thank S. Khan, currently at the Molecular Biology Consortium, Lawrence Berkley National Laboratories and Lahore University of Management Sciences, for the pKLR3 plasmid; T. Nakagawa for technical advice during the early stages of this work; and B. Butler for experimental assistance. We thank W. Chiu and the S2C2 workshop for training in cryoEM. We thank B. DeBuyser for the artificial intelligence-based goblin.tools. This project was funded by National Institutes of Health (NIH) grant GM61606 awarded to T.M.I. and G.C. G.C. is the recipient of a Senior Research Career Scientist award 1K6BX004215 from the Department of Veterans Affairs. M.H.G. is supported by NIH T32 GM007628. Negative stain and cryoEM data were collected at the Center for Structural Biology CryoEM facility at Vanderbilt University. The Glacios cryo-TEM used for screening was acquired by NIH grant S10 OD030292-01.
Author information
Authors and Affiliations
Contributions
P.K.S. expressed and purified protein, determined the structures, analysed the data and wrote the manuscript. P.S. and M.H.G. assisted in the CW structure and data analysis. E.M. and O.A. analysed data. M.E. and G.C. conceived the research, provided scientific advice and contributed to manuscript preparation. T.M.I. conceived the research, supervised the project, assisted in data interpretation, assisted in modelling, performed data analysis and wrote the manuscript with PKS.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Gert Bange and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 CryoEM workflow for the CCW C-ring.
(a) Workflow for the reconstruction of the 34-fold symmetric counterclockwise C-ring at 4.0 Å resolution. Following 2D classification, only particles containing both MS- and C-rings were retained. Symmetry could not be unambiguously classified from the combined MS- and C-rings. To identify the structure of the C-ring, MS-rings were removed through particle subtraction from the micrograph. This was saved as a separate dataset so that the original micrographs were retained. Classification of the separate C-ring revealed rings with 33- to 36-fold symmetry dominated by a prevalent of a 34-mer (~50% of particles). The 34-mer C-ring was subjected to C34 heterogeneous refinement and had a final overall resolution of 4.0 Å after this procedure. (b) To refine the associated MS-ring, the particles containing 34-mer C-rings were re-identified in the original micrographs and the C-ring was subtracted from these 34-mer particles. The subsequent heterogeneous refinement process used five parallel calculations to individually impose multiple C33-C36 symmetries. Among these, only the C33 MS-ring refinement (58% of particles) yielded distinct secondary structure features. The remaining particles (42%) did not classify as any observable symmetry. To ensure that we had not missed a subset of MS-rings with other stoichiometries, we removed the C33 particles from the calculation and separately imposed C34, C35, and C36 symmetry. This did not result in a class with interpretable density. To identify why 42% of the MS-rings that were bound to C-rings could not be classified, we re-evaluated the raw micrographs. We identified that the MS-rings that could not be classified were associated with micrographs that had thinner ice, suggesting a preferential orientation with the MS-ring at the air-water interface. We cannot, however, exclude that other symmetries exist at lower abundance in our samples or in the biological system. Notably, C33 symmetrization revealed the RBM3 and β-collar, but masked the details of RBM1 and RBM2. To achieve high-resolution insights into all domains, we subsequently conducted refinement using C11 symmetrization.
Extended Data Fig. 2 Assignment of the CCW pose of the C-ring.
(a) Surface representation of the C-ring colored by local resolution. The highest resolution (2.9 Å) is in blue, and the lowest resolution (6.6 Å) is in red. (b) AlphaFold models of individual domains were used as starting points in building the structure. The structures are colored by confidence from blue (very high confidence) to orange (very low confidence). (c) Comparison of the final model from the cryoEM structure to isolated domains from crystal structures of homologs. Insets show different regions of the structure. FliGD4-D5 is superposed with the equivalent domains from Thermotoga maritima (rcsb.org/structure/1lkv30). FliGD3 is shown superposed with the equivalent domain from Helicobacter pylori (rcsb.org/structure/3usw31). FliGD1-D2 and FliFC are shown superposed with the equivalent domains from T. maritima (rcsb.org/structure/5tdy34). The FliMmid domain and FliGD3 are superposed with the equivalent domains from H. pylori (rcsb.org/structure/4fq032). FliMC and FliN are superposed with the FliN dimer from T. maritima (rcsb.org/structure/1yab42) and the fused FliMC-FliNC dimer from Salmonella enterica (rcsb.org/structure/4yxb39).
Extended Data Fig. 3 Density for subunits of the C-ring in the CCW pose.
Density for: (a) FliFC; (b)–(g) regions of the FliG subunit; (h)–(l) regions of the FliM subunit (m)–(o) each of the three FliNC domains. The resolution of FliMmid is < 3 Å. In panel (i), the side chains are shown for FliM51-75 to highlight the map quality.
Extended Data Fig. 4 Validation of the assembled C-ring.
(a)–(f) Individual domains of the FliG subunit concur with the domains of this structure. However, the global appearance of FliG differs due to the different interdomain angles. (g) Locations of flagellum-deficient mutations30,31,40,80 in the context of two adjacent protomers of the C-ring. All flagellum-deficient mutations are highlighted with a sphere. Flagellum deficient mutations that map to the interior of folded domains and are likely to prevent flagellar assembly through misfolding of an individual subunit is colored black. Flagellum deficient mutants that interact with adjacent protomers in the ring are the same color as the associated chain. Insets highlight key select intersubunit interactions (yellow dash) that may be disrupted with these mutations. (h) comparison between the CCW pose and a CCW tomogram from Vibrio algintolyticus45.
Extended Data Fig. 5 Workflow for cryoEM structures of CW C-rings.
(a) Workflow for the unbound CW C-ring at 4.6 Å resolution. Representative 3D reconstructions used symmetry expansion followed by local refinement. (b) Workflow for the CW C-ring with a bound partner at 5.9 Å resolution. Representative 3D reconstructions used particle subtraction followed by non-uniform refinement, symmetry expansion, and local refinement. The raw micrographs for CW rings all had thinner ice than the micrographs for CCW rings. Because of this, the MS-ring structure could not be classified in any case, and the structure could be determined without the application of particle subtraction.
Extended Data Fig. 6 Structure of the MS-ring.
(a) The MS-ring is composed entirely of copies of the FliF subunit. Past structures have been determined with different stoichiometries5,6,9,10,12, although there remains debate on whether there is a biologically-relevant exact stoichiometry. Three views of representative cryoEM density (blue mesh) for the 33-mer MS-ring, calculated at 3.4 Å resolution and superposed onto the final model. RBM1 is red, RBM2 is olive and RBM3 is blue. Regions of density corresponding to other positions of RBM1 but where the quality was not sufficient to assign are circled. (b) Model of the MS-ring highlighting the relative positions of the ring-building motifs.
Extended Data Fig. 7 Bidirectional rotation of the C-ring by MotA/B.
MotA subunits are labeled A – E in the panels. (a) Comparison of MotA/B64,65 and MotA79 cryoEM structures identifies varying levels of asymmetry that change the width of the cleft between subunits. The most symmetric structure is that of isolated MotA79. In the context of past biochemistry and structures of MotA/B, the MotA/B stator binds to the torque helix on FliGD5. A compelling model would use a cleft between MotA subunits, a concept with parallels to interlocking cogwheels in macroscopic motors. However, these molecular cogwheels in the chemotaxis machinery undergo shape changes during function, which may benefit from the symmetry mismatch of MotA/B. One possibility is that the open MotA/B clefts64,65 allow rapid binding or release of the torque helix without the need for a rate-limiting induced-fit process. Cleft closure would grasp the FliGD5 torque helix tightly. (b) Complementary electrostatics and sterics of the torque helix of FliGD5 and the MotA/B stator. An electrostatic surface representation of the FliGD5 domain shows that the torque helix is presented as an isolated feature and is negatively charged. (c) A schematic mechanism for bidirectional MotA/B-dependent rotation of the C-ring by moving the MotA/B binding site from the outside to the inside of the ring.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 2.
Supplementary Video 1
Overview of the cryoEM density in the CCW pose. A rocking view of the MS- and C-ring density, displayed as a solid surface. The MS-ring is in blue, and the C-ring is in gold.
Supplementary Video 2
Overview and organization of the C-ring and flagellar switch in the CCW pose. The subunits are shown individually during the rotation of the C-ring to highlight how these interact in the assembled structure.
Supplementary Video 3
A tilted MS-ring and wobble during rotation. The movie shows the MS- and C-rings rotating. This highlights how the symmetry mismatch could result in the wobble of the MS-ring during rotation.
Supplementary Video 4
CCW and CW rotation induced by MotA/B. The movie starts with MotA/B powering the MS- and C-ring in the CCW direction. The MS-ring and MotA/B disappear to highlight how a single FliGMN3 changes from the CCW to CW pose. Finally, the MS-ring and MotA/B reappear to demonstrate CW rotation.
Source data
Source Data Fig. 1
Uncropped micrograph for Fig. 1b.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, P.K., Sharma, P., Afanzar, O. et al. CryoEM structures reveal how the bacterial flagellum rotates and switches direction. Nat Microbiol 9, 1271–1281 (2024). https://doi.org/10.1038/s41564-024-01674-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41564-024-01674-1
- Springer Nature Limited