
Abstract
While first-generation, spike (S)-based COVID-19 vaccines were effective against early SARS-CoV-2 strains, the rapid evolution of novel Omicron subvariants have substantially reduced vaccine efficacy. As such, broadly protective vaccines against SARS-CoV-2 are needed to prevent future viral emergence. In addition, it remains less clear whether peripheral immunization, especially with mRNA vaccines, elicits effective respiratory immunity. Our group has developed a nucleoside-modified mRNA vaccine expressing the nucleocapsid (N) protein of the ancestral SARS-CoV-2 virus and has tested its use in combination with the S-based mRNA vaccine (mRNA-S). In this study, we examined efficacy of mRNA-N alone or in combination with mRNA-S (mRNA-S+N) against more immune evasive Omicron variants in hamsters. Our data show that mRNA-N alone induces a modest but significant protection against BA.5 and that dual mRNA-S+N vaccination confers complete protection against both BA.5 and BQ.1, preventing detection of virus in the hamster lungs. Analysis of respiratory immune response in mice shows that intramuscular mRNA-S+N immunization effectively induces respiratory S- and N-specific T cell responses in the lungs and in bronchoalveolar lavage (BAL), as well as antigen-specific binding IgG in BAL. Together, our data further support mRNA-S+N as a potential pan-COVID-19 vaccine for broad protection against current and emerging SARS-CoV-2 variants.
Similar content being viewed by others


Introduction
Since 2020, tremendous efforts have been devoted to the development of vaccines for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19. Two vaccines based on the mRNA-lipid nanoparticle (LNP) platform were rapidly developed and clinically approved. These first-generation mRNA vaccines targeted the viral spike (S) protein and showed high clinical efficacy against the early strains1,2. However, a number of immune-escaping SARS-CoV-2 variants have emerged since 2020, including Alpha, Beta, Gamma, Delta, and Omicron variants, largely due to mutations in the viral S protein3,4. A number of new Omicron subvariants have also emerged, including BA.2, BA.3, BA.4, BA.5, BQ.1, and others5. Extensive pre-clinical and clinical studies have shown that the first-generation vaccines had reduced efficacy against SARS-CoV-2 variants, especially the Omicron strains6,7. The emergence of variants has posed continuing challenges to host immunity induced by the monovalent or bivalent S-based vaccines. Thus, development of a pan-SARS-CoV-2 vaccine with broad protection against current and future variants is needed.
A broadly protective SARS-CoV-2 vaccine likely requires targeting the conserved viral epitopes, in addition to the S protein. The viral nucleocapsid (N) protein is relatively conserved across SARS-CoV-2 variants8 and is a main target of host immunity in SARS-CoV-2-infected individuals9,10. In our previous study, we generated a nucleoside-modified mRNA vaccine that expresses the SARS-CoV-2 N protein (mRNA-N) and showed that vaccination with dual mRNA-S+N, both based on the prototypical viral sequence, conferred strong protection against both Delta and Omicron BA.1 variants in animal models11. However, the breadth of protection by this mRNA vaccine approach against newer, more immune evasive SARS-CoV-2 variants12,13,14 remains to be determined. In addition, clinically approved mRNA vaccines are administered via the intramuscular (IM) route. Our previous study reported induction of strong systemic immunity by IM mRNA vaccination11. Whether peripheral mRNA vaccination elicits effective antigen-specific respiratory immunity is less clear15,16,17,18. Addressing these questions will not only facilitate the development of a pan-SARS-CoV-2 vaccine, but also have implications for vaccine development against other respiratory infections.
In this study, we further evaluated efficacy of mRNA-N alone or in combination with mRNA-S (mRNA-S+N) against two newer SARS-CoV-2 Omicron variants (BA.5 and BQ.1) in Syrian hamster models. Our results show that mRNA-N alone induces significant albeit incomplete protection against BA.5 in hamsters. Importantly, dual mRNA-S+N vaccination confers complete protection against both BA.5 and BQ.1, preventing detectable viral loads in the hamster lungs. Serum antibody analysis indicates that the complete protection by mRNA-S+N is independent of neutralizing antibodies against these two variants, further supporting a role of T cell immunity in the vaccine-induced protection. In the C57BL/6 mouse model, we show that IM mRNA-S+N vaccination effectively elicits respiratory S- and N-specific T cell responses in the lungs and bronchoalveolar lavage (BAL). The data provides immunological insights for the robust viral control of immune-escaping SARS-CoV-2 variants by peripheral mRNA-S+N vaccination even in the absence of detectable neutralizing antibodies.
Results
mRNA-N alone induces incomplete but significant protection against SARS-CoV-2 Omicron BA.5 variant in hamsters
The coronavirus N protein exhibits higher sequence conservation when compared to the S protein19,20. Our previous study reported the development of a nucleoside-modified mRNA vaccine expressing ancestral SARS-CoV-2 N (mRNA-N)11. Prior to investigating the extent of protection conferred by mRNA-N against newer variants, we conducted an analysis of the sequence diversity of the N protein among various SARS-CoV-2 strains (BA.1, BA.5, and BQ.1 versus Wuhan-Hu-1 strain), compared to the S protein (Table 1). The analysis reveals that compared to Wuhan-Hu-1 strain, the three Omicron variants demonstrate an overall sequence diversity in 53 (out of 1273) residues for the S protein, and 8 (out of 419) residues for the N protein, supporting that N is more conserved among these SARS-CoV-2 strains relative to S (Table 1).
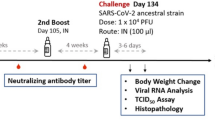
Next, we assessed the efficacy of mRNA-N against Omicron BA.5 in hamsters, which was a dominant circulating variant in human population and showed substantial neutralization escape21,22 when this study was conducted. Two groups of hamsters (n = 10 per group) were IM vaccinated with empty LNP (mock) or mRNA-N (2 μg) at weeks 0 and 3, followed by intranasal (IN) challenge with BA.5 (2 × 104 pfu) at week 5 (Fig. 1A). The mRNA vaccine dose was selected based on our recent study11. Two (n = 5) and four (n = 5) days post infection (DPI), hamsters were euthanized and lung tissues were harvested for viral RNA quantification by RT-qPCR (Fig. 1A). Our primary goal was to evaluate the vaccine efficacy based on reductions in viral RNA copies. Since viral replication kinetics for different SARS-CoV-2 strains in animal models varies, lungs are usually harvested at multiple time points after challenge (e.g., 2 and 4 DPI) for quantification of viral loads11. Hamster body weights were also monitored daily until the terminal harvest (4 PDI) as a secondary measure of vaccine-induced protection. At 2 DPI, lung viral RNA copies were reduced in the mRNA-N-vaccinated group compared to the mock-vaccinated group (60-fold reduction in mean viral RNA copies; mock versus mRNA-N; p < 0.05) (Fig. 1B). At 4 DPI, mRNA-N also induced a significant reduction of lung viral RNA copies (8.3-fold reduction in median copies compared to the mock group; p < 0.01; Fig. 1B). Body weight analysis showed that BA.5 infection caused weight reduction in the mock-vaccinated group by ~3% on 4 DPI (Fig. 1C). However, mRNA-N-vaccinated hamsters were protected from weight loss (4 DPI mRNA-N vs. LNP: p < 0.05) (Fig. 1C). Together, these data indicated that mRNA-N induced a modest but significant protection against SARS-CoV-2 BA.5. This level of protection is consistent with its efficacy against the Delta strain reported in our previous study11.

A Experimental design and timeline (Created with BioRender.com). Two groups of hamsters (n = 10/group) were vaccinated intramuscularly with either empty LNP (mock) or mRNA-N vaccine (2 µg/dose) at weeks 0 and 3. At week 5, hamsters were intranasally challenged with Omicron BA.5 (2 × 104 pfu). B Lungs were harvested at 2 and 4 DPI (days post infection; n = 5 at each time point) for quantification of viral RNA copies by RT-qPCR. C Hamster body weight was monitored from 0 to 4 DPI. In (B) symbols represent individual animals, midlines represent the median, error bars represent the interquartile range (IQR), and the dashed lines represent the lower limit of detection (LOD). The number of animals with viral loads above the LOD is noted. Log10 normalized data was compared by Mann-Whitney test. In (C) symbols represent the mean, error bars represent the standard deviation, and the dashed line highlights 0% weight change. Weight change at individual time point between the two groups was compared by unpaired t-test. *p < 0.05, **p < 0.01.
Intramuscular immunization with mRNA-N alone induces modest respiratory T cell responses in the lung and bronchoalveolar lavage of mice
Prior results are conflicting regarding whether peripheral mRNA immunization (e.g., IM) elicits effective immunity in the airway, in addition to systemic immunity15,16,17,18. Our previous study showed induction of systemic T cell responses and circulating antibodies by mRNA-N in BALB/c mice11. Here, we investigated respiratory immune responses following IM mRNA-N vaccination in C57BL/6 mice. Use of C57BL/6 mice was in part because SARS-CoV-2 N-epitope specific MHC-I tetramer (H2-Db; N219-227: LALLLLDRL)23 is available for the identification of N-specific CD8 T cells in the mouse BAL and lung samples. The N219-227 is one of the dominant CD8 T cell epitopes within the N protein that was bioinformatically predicted8 and then confirmed in humans and mice24,25.
Two groups of C57BL/6 mice (n = 5 per group) were IM vaccinated with either empty LNP (mock) or mRNA-N (1 μg) at week 0 (prime) and week 3 (boost). Two weeks after boost vaccination (week 5), lung and BAL samples were collected for analysis of respiratory immune response. Serum and spleen samples were also collected for analysis of systemic immune response for comparison (Fig. 2A). Single cell suspensions prepared from lung and BAL were stained for CD3, CD4, CD8, CD44, CXCR6, and SARS-CoV-2 N-tetramer, to identify activated T cells (based on CD44)26, T cells with tissue-homing potential (CXCR6)27, and vaccine-elicited, N-specific CD8 T cells (tetramer+). Vaccine-induced binding antibodies (IgG and IgA) were also examined in the BAL.

A Mouse experimental design and timeline (Created with BioRender.com). Two groups of C57BL/6 mice (n = 5/group) were intramuscularly vaccinated with either empty LNP (mock) or mRNA-N vaccine (1 μg) at weeks 0 and 3. Two weeks after booster dose (week 5), immune responses were analyzed. B, C Analysis of tetramer positive, N-epitope specific T cells in the lungs by flow cytometry. B Representative flow cytometry plots for N-tetramer staining of CD8 T cells from LNP and mRNA-N vaccinated mice. C Frequencies of N epitope-specific CD8 T cells in the lungs of LNP and mRNA-N vaccine group. Analysis of total and N epitope-specific T cells in the BAL. Frequencies of activated (CD44+) CD4 and CD8 T cells (D), CXCR6+ CD4 and CD8 T cells (E), and N epitope-specific CD8 T cells (F) in BAL were examined. Data were presented as median and interquartile range, and were compared by Mann-Whiteney test between the two groups. *p < 0.05, **p < 0.01, ***p < 0.001.
Gating strategy to identify T cell populations and N-tetramer+ CD8 T cells in the lungs is shown in Supplementary Fig. 1. Representative flow cytometric plots for N-tetramer+ CD8 T cells are shown in Fig. 2B. Compared to mock control, mRNA-N vaccination elicited comparable levels of total CD4 and CD8 T cells in the lung (Supplementary Fig. 2A), and slightly higher levels of activated CD44+ (Supplementary Fig. 2B) or CXCR6+ (Supplementary Fig. 2C) CD4 and CD8 T cells in the lung. Analysis of N-tetramer straining (Fig. 2B) revealed that compared to mock control, mRNA-N vaccination elicited significant levels of N epitope-specific CD8 T cells in the lungs (p < 0.001) (Fig. 2C). Of interest, this N epitope-specific CD8 T cell response was not evident in the spleens after mRNA-N vaccination (Supplementary Fig. 3).
We next evaluated T cell response in the BAL after mRNA-N vaccination. Gating strategy for flow cytometric analysis of BAL cells is shown in Supplementary Fig. 4. Compared to mock control, mRNA-N vaccination elicited higher levels of CD44+ CD4 T cells and CD44+ CD8 T cells in the BAL, although statistical significance was only achieved for CD4 T cells (p < 0.05 for CD4) (Fig. 2D). A similar trend was observed for CXCR6+ T cells, with mRNA-N eliciting significantly higher levels of CXCR6+ CD4 T cells in the BAL compared to mock control (p = 0.01 for CD4) (Fig. 2E). Analysis of N-tetramer+ cells revealed that mRNA-N vaccination elicited a trend towards higher levels of N epitope-specific CD8 T cells in the BAL compared to mock control (p = 0.055) (Fig. 2F). Analysis of binding antibodies showed that mRNA-N vaccination induced readily detectable N-specific binding IgG in the BAL (Supplementary Fig. 5A). No binding IgA was detected following IM mRNA-N vaccination (Supplementary Fig. 5B), consistent with the previous report15. Together, the data indicate that mRNA-N vaccine elicits a modest respiratory immune response including N-specific CD8 T cells in the mouse lungs and BAL.
mRNA-S+N vaccination induces strong protection against SARS-CoV-2 Omicron BA.5 in hamsters
After demonstrating that mRNA-N alone elicited modest protection against BA.5, we next explored whether dual mRNA-S+N vaccination induces stronger protection against this variant than mRNA-S alone. Three groups of hamsters were vaccinated with empty LNP (mock), mRNA-S (2 μg), or mRNA-S+N (2 μg for each mRNA) at weeks 0 and 3, followed by intranasal challenge at week 5. On 2 (n = 5) and 4 (n = 5) DPI, vaccine-induced protection was analyzed based on viral loads (Fig. 3A). Analysis of viral RNA in the lungs revealed that, compared to mock vaccination, mRNA-S alone induced modest but significant protection, reducing the lung viral RNA copies by 22 and 4 folds at 2 and 4 DPI, respectively (Fig. 3B–C). Relative to mRNA-S alone, mRNA-S+N induced a more robust effect, leading to complete viral control with no detection of the viral RNAs on 4 DPI (mRNA-S vs. mRNA-S+N: p < 0.0001 on 4 DPI) (Fig. 3C). We previously showed that the stronger protection against an Omicron variant (BA.1) by mRNA-S+N relative to mRNA-S alone was not due to the difference in total mRNA or LNP doses11. Lastly, vaccination with mRNA-S alone and mRNA-S+N both protected the hamsters from weight loss, with no significant difference detected between the two vaccine groups (Fig. 3D).

A Hamster experimental design and timeline (Created with BioRender.com). Three groups of hamsters (n = 10/group) were vaccinated intramuscularly with empty LNP (mock), mRNA-S (2 μg), or mRNA-S+N (2 μg for each mRNA) at weeks 0 and 3, followed by intranasal challenge with SARS-CoV-2 Omicron BA.5 (2 × 104 pfu) at week 5. Lungs were harvested at 2 (B) and 4 (C) DPI (n = 5 at each time) for quantification of viral RNA copies by RT-qPCR. (D) Hamster body weights were monitored from 0 to 4 DPI. In (B, C), symbols represent individual animals, midlines represent the median, error bars represent the interquartile range, and the dashed line represents the lower limit of detection (LOD). The number of animals with viral loads above the LOD is noted. Data were compared among the three groups by Kruskal-Wallis test. In (D), symbols represent the mean, error bars represent the standard deviation, and the dashed line highlights 0% weight change. Weight change was compared by 2-way ANOVA followed by Tukey’s multiple comparisons test (two factors; hamsters’ weight and time). *p < 0.05, **p < 0.01, ****p < 0.0001.
mRNA-S+N vaccination induces strong protection against SARS-CoV-2 Omicron BQ.1 in hamsters
To further determine the breadth of protection by mRNA-S+N, we next examined efficacy of mRNA-S and mRNA-S+N against BQ.1 in the hamster model. BQ.1, derived from BA.5, emerged with additional spike mutations that contributed to its strong immune evasion and efficient transmission13,28. Hamsters were vaccinated at weeks 0 and 3 before intranasal challenge with BQ.1 (2 × 104 pfu) at week 5 (Fig. 4A). Lungs were harvested at 2 (n = 5) and 4 (n = 5) DPI with hamster body weights monitored throughout the course of the experiment (Fig. 4A). Compared to BA.5 (Fig. 3) and earlier variants (BA.1 and Delta)11, BQ.1 appears to replicate to lower levels in hamsters and with faster clearance by 4 DPI (Fig. 4B, C). Similarly, body weight data indicated that nonvaccinated (mock) hamsters showed marginal weight loss through 4 DPI (Fig. 4D). This is consistent with the observations that BQ.1 is moderately pathogenic compared to BA.513,29. Vaccination with mRNA-S+N led to complete viral control with no detection of viral RNA in four out of five hamsters at 2 DPI (Fig. 4B) and five out of five hamsters at 4 DPI (Fig. 4C). Both mRNA-S and mRNA-S+N protected hamsters from the mild weight loss resulting from BQ.1 infection with no significant difference detected between the two vaccine groups (Fig. 4D).

A Experimental design and timeline (Created with BioRender.com). Three groups of hamsters (n = 10/group) were vaccinated intramuscularly with empty LNP (mock), mRNA-S (2 μg), or mRNA-S+N (2 μg for each mRNA) at weeks 0 and 3, followed by intranasal challenge with SARS-CoV-2 Omicron BQ.1 (2 × 104 pfu) at week 5. Lungs were harvested at 2 (B) and 4 (C) DPI (n = 5 at each time point) for quantification of viral RNA copies by RT-qPCR. D Hamster body weights were monitored from 0 to 4 DPI. In (B, C), symbols represent individual animals, midlines represent the median, error bars represent the interquartile range, and the dashed line represents the lower limit of detection (LOD). The number of animals with viral loads above the LOD is noted. Log10 normalized data was compared among the three groups by Kruskal-Wallis test. In (D), symbols represent the mean, error bars represent the standard deviation, and the dashed line highlights 0% body weight change. Weight change was compared by 2-way ANOVA followed by Tukey’s multiple comparisons test (two factors; hamsters’ weight and time). *p < 0.01, **p < 0.01, ****p < 0.0001.
mRNA-S+N induces binding antibodies that manifest no detectable neutralizing activities against BA.5 and BQ.1 variants
In the above hamster experiments (Fig. 3, 4), serum samples were collected prior to viral challenge (two weeks after booster) for analysis of vaccine-induced antibody responses (Fig. 5). Binding antibody endpoint titers (EPTs) were determined by ELISA. Compared to the mock control, both mRNA-S alone and mRNA-S+N induced significant levels of S-specific binding antibodies in sera (Fig. 5A–C). Median IgG EPTs for both mRNA-S and mRNA-S+N groups were 104.9 (Fig. 5A). Other than IgG, serum IgA and IgM were also detectable. Median IgA EPTs for mRNA-S and mRNA-S + N were 103.95 and 104.43, respectively (Fig. 5B), and median IgM EPTs for mRNA-S and mRNA-S+N were 102.76 and 103, respectively (Fig. 5C). These serum samples were also examined for neutralization against SARS-CoV-2 WA.1, BA.1, BA.5, and BQ.1 using the plaque-reduction neutralization test (PRNT) with corresponding live virus. While the sera of hamsters immunized with mRNA-S and mRNA-S+N showed strong neutralizing activity against the ancestral WA.1 strain (Fig. 5D), their neutralizing activities against BA.1 were substantially reduced: 4/10 samples had weakly detectable neutralization in the mRNA-S alone group and 5/10 weakly detectable neutralization in the mRNA-S+N group. Notably, neutralizing activities of these sera against BA.5 and BQ.1 variants were completely lost, with all samples showing undetectable neutralization activity for these variants (Fig. 5D). These data are consistent with the observations that BA.5 and BQ.1 manifest strong immune escape from the ancestral S-based vaccine induced neutralization. The data also show that the strong viral control by mRNA-S+N in the hamster lungs is occurring even in the absence of detectable neutralizing antibodies, indicating a role of cellular immunity in mRNA-S+N induced protection against variants as described previously11.

S-specific binding IgG (A), IgA (B), and IgM (C) endpoint titers (EPTs) in the hamster sera at week 5 after immunization (n = 4 for LNP; n = 10 for mRNA-S or mRNA-S+N). Log10 normalized EPTs for each group is indicated. D Neutralization of the hamster sera (n = 10 for all three groups) against SARS-CoV-2 WA.1, BA.1, BA.5, and BQ.1 as measured by PRNT50. Data are presented as median with interquartile range. Dotted lines in each plot indicates LOD for each assay. Number of animals in each group with neutralizing titers above the LOD is noted in (D). Data were compared among the three groups by Kruskal-Wallis test. *p < 0.05, **p < 0.01, ****p < 0.0001.
mRNA-S+N vaccination induces strong respiratory T cell responses in the lungs and bronchoalveolar lavage
After showing that mRNA-S+N vaccine induced complete viral control in the hamster lungs in the absence of detectable neutralizing antibodies, we next examined respiratory immune responses induced by mRNA-S+N as compared to mRNA-S alone. For this, we utilized the C57BL/6 mouse model and the available S- (S539–546: VNFNFNGL) and N- (N219–227: LLLDRLNQL) specific MHC-I tetramers. Similar to N219–227 (Fig. 2), S539-546 (VNFNFNGL) is a highly dominant CD8 T cell epitope within the S protein that was predicted based on bioinformatic analysis and later validated in the context of both infection and vaccination30,31,32. Multiple vaccine studies have confirmed a protective role of the CD8 T cell response induced against this particular S epitope33,34.
Three groups of C57BL/6 mice (n = 5 per group) were vaccinated (IM) with empty LNP (mock), mRNA-S alone (1 μg), or mRNA-S+N (1 μg per mRNA) at week 0 and 3, followed by analysis of vaccine-induced immune responses in the lungs and BAL (Fig. 6A). T cell responses in the spleen were also examined for comparison. The gating strategies to identify T cell populations and tetramer+ CD8 T cells are shown in Supplementary Fig. 1, Supplementary Fig. 4, and the main figures.

A Mouse experimental design and timeline (Created with BioRender.com). Three groups of C57BL/6 mice (n = 5/group) were intramuscularly vaccinated with either empty LNP (mock), mRNA-S vaccine (1 μg), or mRNA-S+N vaccine (1 μg for each mRNA) at week 0 and 3. Two weeks after final dose (week 5), mice were euthanized and immune analyses were performed. Analysis of activated T cells in the lungs (B), BAL (C), and spleens (D) of mice. Expression of CD44 on CD4 and CD8 T cells was examined by flow cytometry and shown as percent CD44+ of the parental population. Expression of CXCR6 on CD4 and CD8 T cells in the lungs (E), BAL (F), and spleens (G) was examined by flow cytometry and shown as percent CXCR6+ of the parental population. H Representative flow cytometry plots for S- and N-tetramer staining of CD8 T cells from LNP, mRNA-S, and mRNA-S+N vaccinated mice. Tetramer+ (S- and N-epitope specific CD8 T cells) in the lungs (I), BAL (J), and spleens (K) of mice. Data are presented as median and interquartile range. Kruskal-Wallis test was used for statistical comparison among the three groups. *p < 0.05, **p < 0.01.
In the lungs, compared to mock control, both mRNA-S and mRNA-S+N elicited slightly higher levels of total activated (CD44+) CD4 T cells, although only the mRNA-S group reached statistical significance (p < 0.05 for mock versus mRNA-S) (Fig. 6B, left). A significant increase in the frequency of total activated CD8 T cells in the lungs of mRNA-S+N-vaccinated mice (median: 15%) was observed when compared to the mock group (median: 6.53%) (p < 0.01) (Fig. 6B, right). In the BAL, a stronger effect was observed. Compared to mock control, mRNA-S+N vaccine elicited substantially higher levels of activated CD4 (Mock versus mRNA-S+N: 20.1%, 88.5%; p < 0.01) and CD8 (Mock versus mRNA-S+N: 14.6%, 92.9%; p < 0.01) T cells (Fig. 6C). Compared to mRNA-S+N, mRNA-S alone induced weaker CD4 and CD8 T cell activation in the BAL and no statistical significance was detected between the two vaccine groups (Fig. 6C). In contrast, in the spleen, we only noted a modest increase in CD4 and CD8 T cell activation in the mRNA-S+N group compared to mock control (Fig. 6D). In all these compartments, no significant difference in the frequency of total CD4 and CD8 T cells was observed among the groups (Fig. S6).
We next examined frequencies of T cells positive for CXCR6 in the lungs, BAL, and spleen (Fig. 6E–G). A pattern similar to the activated T cells was revealed. Compared to the mock control, mRNA-S+N elicited significantly higher levels of CXCR6+ CD8 T cells in the lungs (1.47% versus 6.99%; p < 0.01) (Fig. 6E) as well as in the BAL (6.08% versus 51.6%, p < 0.01) (Fig. 6F). In the BAL, mRNA-S+N also induced a markedly higher level of CXCR6+ CD4 T cells when compared to the mock control (7.87% versus 66.9%, p < 0.01) (Fig. 6F). These data indicate lung-homing potential of the induced T cells by mRNA-S+N. In contrast, mRNA-S alone only elicited a trend towards increase in CXCR6+ CD4 and CXCR6+ CD8 T cells in the BAL when compared to the mock control (Fig. 6F). As anticipated, lower levels of CXCR6+ T cells were observed in the spleens when compared to those in the BAL and lung following mRNA-S or mRNA-S+N vaccination (Fig. 6G).
Antigen-specific CD8 T cells in these compartments were measured (Fig. 6H–K). Representative flow cytometry plots for S- and N-tetramer staining showed that mRNA-S alone elicited S-specific, but not N-specific, CD8 T cells, while mRNA-S+N vaccine elicited both S- and N-specific CD8 T cells, supporting the specificity of the tetramer staining (Fig. 6H). We observed that, compared to mRNA-S alone, mRNA-S+N vaccine elicited higher levels of S-specific CD8 T cells in the lungs (p < 0.05) (Fig. 6I). A more profound effect was observed in the BAL, where mRNA-S+N induced much higher levels of S-specific CD8 T cells (~36%) than mRNA-S (~14%) (Fig. 6J). Of note, while mRNA-S+N also elicited detectable N-specific CD8 T cells in the lungs (Fig. 6I), their levels in the BAL were low (Fig. 6J). Lastly, only low levels of S-epitope specific CD8 T cells were detected in the spleen after mRNA-S+N vaccination (Fig. 6K). Together, these data indicate that mRNA-S+N elicits strong respiratory T cell responses, especially S-specific CD8 T cells, after IM mRNA immunization. The data also reveal that the presence of N for co-immunization has some synergistic effect and augments S-specific T cell response in the respiratory tract, a finding also observed for systemic immunity in our previous study11.
Vaccine-induced binding antibodies in the BAL were also examined. Compared to mock control, both vaccines induced significant levels of S-specific binding IgG (Fig. S7). As expected, only mRNA-S+N vaccination resulted in the production of both S- and N-specific binding IgG in the BAL (Fig. S7), whereas mRNA-S alone only elicited S-specific binding IgG. Unlike IgG, there was a lack of detectable antigen-specific IgA in the BAL, consistent with the result of mRNA-N vaccination (Supplementary Fig. 5) as well as with a previous study reporting limited mucosal IgA production following IM mRNA immunization15.
Discussion
The emergence of novel SARS-CoV-2 variants has greatly reduced effectiveness of the first-generation COVID-19 vaccines35. Our previous study reported the efficacy of the dual mRNA-S+N vaccine against Delta and Omicron BA.1 strain in animal models11. The present study demonstrates that mRNA-S+N, based on the ancestral viral sequence, induces robust protection against the more immune evasive Omicron subvariants, BA.5 and BQ.1, in the absence of detectable neutralizing antibodies. Additionally, in contrast to our previous study which examined vaccine-induced systemic immunity11, this study investigates mRNA vaccine-induced respiratory immunity and shows that peripheral immunization with mRNA-S+N effectively elicits respiratory S- and N-specific CD8 T cell responses in the animal lungs and BAL at the time examination (2 weeks after booster). The data together support mRNA-S+N as a promising pan-SARS-CoV-2 vaccine approach and provide additional immunological insights in mechanisms of protection conferred by mRNA-S+N against immune-escaping variants.
There are ongoing efforts to develop a next-generation, broadly protective SARS-CoV-2 vaccine against current and emerging variants. Targeting conserved regions of the virus, e.g., N protein, in addition to the S protein, is considered an attractive strategy for pan-COVID-19 vaccine development36,37. In support, our analysis reveals that N protein has higher sequence conservation among the different SARS-CoV-2 variants (BA.1, BA.5, BQ.1., and Wuhan-Hu-1) as compared to the S protein (Table 1). N is a structural protein of SARS-CoV-2 with abundant expression in infected cells and plays a role in the coronavirus life cycle, including viral genome packaging and immune regulation38. Notably, the N-induced memory T cell response is cross-reactive39. In this study, we further demonstrate that mRNA-N alone induces incomplete but significant control of Omicron BA.5 infection in hamster lungs. The data are consistent with our previous report showing a modest protective effect of this vaccine alone against mouse-adapted SARS-CoV-2 and Delta variant11.
Our study also determines the efficacy of dual mRNA-S+N vaccine against newly emerged immune-escaping Omicron subvariants BA.5 and BQ.1. Compared to the early variants, BA.5 possesses additional changes (69-70del, L452R, and F486V) and a reversion (R493Q) in the S protein (Table 1), which contribute to its enhanced fusogenicity and infectivity22. Studies in rodents showed that BA.5 has superior viral fitness to that BA.240. In addition, when the present study was conducted, the BQ.1 variant which bears additional mutations in its S protein (i.e., N460K, R346T, and K444T) (Table 1) had emerged as a dominant circulating strain with enhanced transmissibility and immune evasion35,41,42. In line with these observations, our data showed that while the sera of mRNA-S or mRNA-S+N-immunized hamsters manifested strong neutralization of the prototypical WA.1 strain, and even the Delta strain11, their neutralizing activities were greatly reduced against Omicron BA.1 (only a few animals in each vaccine group showed weak neutralization) and were completely lost against BA.5 and BQ.1 (Fig. 5D)43,44. However, even in the absence of detectable neutralizing antibodies, we still observed complete viral control in hamster lungs by mRNA-S+N against both BA.5 and BQ.1. Indeed, our previous study using in vivo CD8 cell depletion did support a role of T cell immunity in protection11. Despite these data, immune mechanisms other than T cell immunity, such as other effector functions of antibodies induced by S and N23,45,46, should not be excluded and warrant further investigation. Nevertheless, our data confirmed the breadth of protection conferred by mRNA-S+N, irrespective of S-based mutations, and support the idea that vaccine approaches inducing robust, cross-protective cellular immunity should be pursued47,48.
Respiratory immunity is important for protection against SARS-CoV-2 infection. Studies on induction of respiratory immunity by peripheral mRNA vaccination have reported inconsistent results. Some studies reported minimal T cell and antibody responses in the airway mucosa after peripheral vaccination15,16, while others showed detectable mucosal IgA along with CD4 and CD8 T cells in vaccinated individuals17,18. In our study, we analyzed total and antigen-specific T cells in the lungs and BAL of the immunized mice. We demonstrated that both S and N epitope-specific CD8 T cells were detectable following IM mRNA-S+N vaccination in the lungs and/or BAL. This induction of a robust respiratory T cell response correlates with the strong control of BA.5 and BQ.1 by mRNA-S+N in the absence of neutralizing antibodies. Thus, our data favor that peripheral mRNA vaccination could induce T cell responses in the airway, although it remains unclear if these T cell responses are durable, since we only examined the response at the peak immunogenicity following vaccination (2 weeks after booster). Our analysis of antibodies showed that only binding IgG was detectable in the BAL. Lack of detection of mucosal IgA following IM mRNA vaccination in our study is consistent with previous reports15,16 and supports the need for a mucosal booster to further improve respiratory immunity after peripheral mRNA vaccination.
The present study utilized MHC-I tetramer staining to identify antigen-specific CD8 T cells in the mouse lungs and BAL. Compared to intracellular cytokine staining (ICS), tetramer staining requires fewer cells and no peptide restimulation, thus making it less challenging to measure antigen-specific T cells in samples of low cell numbers (e.g., BAL), although tetramer staining only identifies T cells specific to single epitope. In our study, two MHC-I epitopes (N219-227: LLLDRLNQL; S539-546: VNFNFNGL) were used to detect N- and S-specific CD8 T cells in mouse BAL/lungs. Both are dominant CD8 T cell epitopes within the SARS-CoV-2 N and S protein, respectively. The two epitopes were initially predicted by bioinformatical analysis8 and subsequently validated in humans and mice24,25,30,31,32. In particular, the protective role of CD8 T cell responses against the single S539-546 epitope was confirmed in multiple SARS-CoV-2 vaccine studies33,34. Consistent with these reports, our study shows that IM mRNA-S+N vaccination elicits both S539-546 and N219-227 epitope-specific CD8 T cell populations in the mouse airway (Fig. 6). It is interesting to note that the magnitude of S539-546-specific CD8 T cell response is markedly higher than N219-227-specific CD8 T cell response in the mouse BAL/lungs, although the dose of mRNA-S and mRNA-N used in the dual immunization regimen (mRNA-S+N) is identical. This is likely due to multiple reasons. First, the co-immunization of N augments S-specific immunity, but not vice versa. This pattern is particularly evident in the CD8 T cell responses, as seen here and in our previous study11. The underlying mechanisms are not yet clear. Second, the S539-546 epitope may be more dominant within the S protein than the N219-227 epitope within the N protein following mRNA vaccination. Also, it is possible that CD8 T cells specific to other N epitopes are also induced by mRNA vaccines but not detectable using the N219-227 tetramer staining.
Other than CD8 epitopes, a number of CD4 T cell epitopes within the SARS-CoV-2 S and N proteins were also identified8,49, despite their protective roles in vaccination are less clear. Our study examined respiratory CD4 T cells with activation (CD44) or lung-tissue residency (CXCR6) phenotype and showed that IM mRNA-S+N vaccination elicits high levels of activated and CXCR6+ CD4 T cells, especially in the BAL (Fig. 6C, F). The present study did not measure respiratory antigen-specific CD4 T cells, largely due to the current lack of tetramers for these CD4 epitopes. However, our previous study used ICS assay and showed the induction of robust systemic antigen-specific CD4 T cells by mRNA-S+N vaccine in BALB/c mice11. We thus speculate that peripheral mRNA vaccination should also elicit antigen-specific CD4 T cells in BAL/lungs. Nevertheless, a more comprehensive characterization of the respiratory CD4 and CD8 T cell responses specific to the full S and N protein in the BAL/lungs following mRNA vaccination is needed in future studies.
The present study has several limitations. First, in the respiratory immune analysis, single cell suspensions were prepared from the whole lung tissue without discriminating immune cells from circulating blood, raising the concern that the total and antigen-specific T cells identified in the lungs could also contain contaminated cells from the blood. However, our analysis of T cells in the BAL supports that mRNA vaccination induces respiratory T cell responses in the airway. Second, while we show the induction of antigen-specific T cells in the lung and BAL at peak immunogenicity, whether they are long-lasting, tissue-resident T cells and can confer durable protection remain unclear. Lastly, with the vast majority of the human population having received the S-based vaccines or being naturally infected with SARS-CoV-2, mRNA-N as a booster component for inducing a broad protection in the context of pre-existing immunity should be investigated, which could expand the potential utility of this vaccine candidate.
Materials and methods
Study design
The aim of the present study was to investigate efficacy of mRNA-N and mRNA-S+N against newer SARS-CoV-2 Omicron variants and the respiratory immunity induced by these mRNA vaccines in animal models. Evaluation of vaccine efficacy against Omicron BA.5 and BQ.1 challenges were conducted in Syrian hamsters, and analysis of vaccine-induced respiratory immunity was conducted in C57BL/6 mice. The animal study protocols were approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch (Protocol numbers: 1703020, 2009087). All animal experiments were carried out following the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. During the studies, all animals were monitored by animal resources center or laboratory staff daily. The number of animals utilized per group was determined by a power analysis for a medium effect size prior to the onset of the study and all data points were utilized for analysis. All animals were allowed a minimum of 3 days to acclimate to their environment before study onset and anesthetized with 1–5% isoflurane via vaporizer prior to each procedure apart from weights. Mice were humanely euthanized with CO2 asphyxiation followed by cervical dislocation. Hamsters were humanely euthanized with CO2 asphyxiation followed by bilateral thoracotomy. The study design was not blinded to researchers or staff at the animal facility, and animals were randomly assigned to each group.
mRNA synthesis and LNP formulation
mRNA synthesis and LNP formulations were prepared as published previously11. Briefly, mRNA-N (full-length) and mRNA-S (prefusion-stabilized S protein with two proline mutations; mRNA-S-2P) were synthesized by using the T7 RNA polymerase (MegaScript, Thermo Fisher Scientific) in vitro transcription kit. The sequences are based on the ancestral SARS-CoV-2 Wuhan-Hu-1 strain (GenBank MN908947.3). Uridine triphosphate was replaced with one-methylpseudouridine (m1)-5′-triphosphate. For improved protein expression, polyadenylated tails were added to the ends of modified mRNAs. ScriptCap m7G capping system and ScriptCap 2′-O-methyltransferase kit were used to cap in vitro transcribed mRNAs, which were then underwent cellulose-based purification. As previously reported, the mRNAs were formulated into LNPs using an ethanolic lipid mixture of ionizable cationic lipid and an aqueous buffer system. mRNA-LNPs were prepared in accordance with RNA concentrations (1 µg/µl) and stored at −80 °C prior to animal immunizations. Protein expression in cells after mRNA-N and mRNA-S LNP treatment was confirmed in our previous publication11.
Animal immunizations and SARS-CoV-2 challenge
mRNA vaccine-induced immune response was evaluated in mice. 6-week-old female C57BL/6 mice purchased from the Jackson Laboratory (strain no. 000664) were randomly assigned into four groups (n = 5 per group) that were respectively immunized IM (thigh muscles of the hind limb) with 50 µl of either empty LNP (mock), mRNA-S (1 μg), mRNA-N (1 μg), or mRNA-S+N (1 μg per mRNA) at week 0 (prime) and week 3 (boost). 2 weeks after booster vaccination (week 5), all mice were euthanized (CO2 and physical dislocation) for terminal blood collection and tissue harvests. Lung and BAL samples were collected for analysis of vaccine-induced respiratory immune responses. In some experiments, sera and spleen samples were also collected to analyze vaccine-induced systemic immune response for comparison.
mRNA vaccine efficacy was evaluated in hamsters. 3-week-old male golden Syrian hamsters purchased from Envigo (strain HsdHan: AURA, catalog no. 8901M) were randomly assigned into four groups (n = 10 per group). They were respectively vaccinated IM (thigh muscles of the hind limb) with 100 µL of either LNP (mock), mRNA-S (2 µg), mRNA-N (2 µg), or combined mRNA-S+N (2 µg per mRNA) at weeks 0 and 3. Two weeks after booster (week 5) and prior to viral challenge, all hamsters were subject to serum collection under anesthesia. The sera were used to measure vaccine-induced antibodies. Hamsters were then transferred to ABSL-3 facility and intranasally challenged with either SARS-CoV-2 Omicron BA.5 (2 × 104 pfu in 100 µl/hamster) or BQ.1 (2 × 104 pfu in 100 µl/hamster) under anesthesia. At 2- and 4 DPI, five hamsters of each group (for each time point) were euthanized and the lung tissues were collected for evaluating vaccine-induced protection. Hamster body weights were also monitored daily from the day of viral challenge to 4 DPI.
Spike-specific binding antibodies in hamster sera
Vaccine-induced S-specific binding antibody titers (IgG, IgA, and IgM) in the hamster sera were determined by ELISA. Plates (Greiner Bio-One) were coated with recombinant S protein (1 µg/ml; 40589-V08B1, Sino Biological) overnight at 4°C. Plates were washed three times and then blocked with blocking buffer [8% fetal bovine serum (FBS) in Dulbecco’s PBS (DPBS)] for 1 h at 37 °C, followed by washing and incubation at 37 °C for 2 h with serially diluted serum samples (initial dilution 1:100, followed by 3-fold serial dilution) in blocking buffer at 50 µl per well. Plates were washed again and incubated with HRP-conjugated anti-hamster IgG (1:1000; Southern Biotech; 6060-05), anti-hamster IgA (1:1000; Brookwood Biomedical; sab3003a), or anti-hamster IgM (1:500; Brookwood Biomedical; sab3003m) secondary antibodies for 1 h (IgG) or overnight (IgA and IgM) at 37 °C. After incubation, plates were washed and developed using TMB 1-Component Peroxidase Substrate (Thermo Fisher Scientific), followed by termination of reaction using the 2 N HCl solution. Plates were read at 450 nm wavelength within 15 min by using a Microplate Reader (BioTek). Binding antibody EPTs were calculated.
Spike- and nucleocapsid-specific binding antibodies in mouse bronchoalveolar lavage
S- and N-specific binding IgG and IgA in mouse BAL were measured by ELISA as described above except the BAL samples underwent 1:1 dilution.
Flow cytometry analysis
Single cell suspensions prepared from lung, BAL, and spleen were directly stained for viability (65-0866-14, Invitrogen; Fixable Viability Dye eFluor 506) and surface markers including CD3- BUV805 (741982, BD Biosciences; clone 17A2), CD4-APC/Fire 570 (100460, BioLegend; clone GK1.5), CD8-BV570 (100740, Biolegend; clone 53-6.7), CD44-PerCP (103036, Biolegend; clone IM7), CXCR6-BV711 (151111, Biolegend; clone SA051D1). To detect S- and N-specific CD8 T cells, samples were incubated with S539–546 MHC-I tetramer-Alexa488 (H-2Kb, 1:500 dilution) and N219–227 MHC-I tetramer-BV421 (H2-Db, 1:500 dilution) (NIH Tetramer Core) for 30 min at room temperature. Cells were washed and then acquired on a flow cytometer FACSymphony A5 (BD Biosciences). Data were analyzed using FlowJo (Flowjo LLC).
Quantification of viral RNA by RT-qPCR
Hamster lungs were collected in maintenance medium (2% FBS in DMEM with 1% pen-strep) and subjected to homogenization for 1 min. Debris was pelleted by centrifugation for 5 min at 16,000 g. Following homogenization, the supernatants of tissue homogenates were combined with a fivefold volume of TRIzol LS Reagent (Thermo Fisher Scientific). Extraction of viral RNAs was performed in accordance with the instructions provided by the manufacturer. The final RNA solutions were stored at −80 °C until use for RT-qPCR. Viral RNA copies were determined in the lungs by using the one-step RT-qPCR kit (Bio-Rad, 1725151) on CFX Connect Real-Time PCR Detection System (Bio-Rad). SARS-CoV-2 E gene primers (forward, 5′-GGAAGAGACAGGTACGTTAATA-3′; reverse, 5′-AGCAGTACGCACACAATCGAA-3′) were used. The PCR reaction was composed of primers (10 μM), RNA sample (2 μl), iTaq universal SYBR Green 1-step reaction mix (2×; 10 μl), iScript reverse transcriptase (0.25 μl), and molecular grade water, for a total volume of 20 μl. The PCR cycling conditions were as follows: 95 °C for a duration of 3 min, followed by 45 cycles of 95 °C for 5 s and 60 °C for 30 s. A standard curve was included in each RT-qPCR analysis, utilizing an RNA standard that was synthesized in vitro. This RNA standard consisted of 3839 base pairs and encompassed genomic nucleotide locations 26,044 to 29,883 of the SARS-CoV-2 genome. By using the standard curve we determined the absolute number of viral RNA copies present in the lung tissue11.
Virus neutralization analysis
Serum neutralizing activity was examined using PRNT assay11. The experiments were carried out on Vero E6 cells (ATCC, CRL-1586) with the SARS-CoV-2 wild-type or Omicron variants. Briefly, serum samples were heat-inactivated (at 56 °C for 30 min) and serially diluted twice (1:10 initial dilution and then twofold serial dilutions), followed by incubation for 1 h at 37 °C with wild-type SARS-CoV-2 (USA-WA1/2020), BA.5, or BQ.1, respectively. The above mixtures were added to monolayers of Vero E6 cells in 6-well plates for incubation at 37 °C for 1 h. After incubation, a 2 ml of semisolid overlay medium (minimum essential medium containing 1.6% agarose, 2% FBS, and 1% penicillin-streptomycin) was added to the cells, and then incubated for 48 h at 37 °C. After incubation, monolayer cells were stained with 0.03% liquid neutral red for 3–6 h. A manual counting method was used to count the number of plaques and PRTN50 was calculated.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 10.1.1 software. Data were presented as median ± IQR or mean ± standard deviation, as denoted in the figure legends. Statistical comparison among groups was performed using Mann-Whitney test, Kruskal-Wallis test, or two-way ANOVA with Tukey’s multiple comparison test where appropriate as denoted in the figure legends. Two-tailed p values were denoted, and p < 0.05 were considered significant. All analyses were conducted assuming a 95% confidence interval.
Data availability
The datasets generated and analyzed in this study are presented in the manuscript and the supplementary files. Original raw data are also available from the corresponding authors upon request.
References
Baden, L. R. et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 384, 403–416 (2020).
Polack, F. P. et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 383, 2603–2615 (2020).
Harvey, W. T. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424 (2021).
Viana, R. et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 603, 679–686 (2022).
Chatterjee, S., Bhattacharya, M., Nag, S., Dhama, K. & Chakraborty, C. A detailed overview of SARS-CoV-2 omicron: its sub-variants, mutations and pathophysiology, clinical characteristics, immunological landscape, immune escape, and therapies. Viruses 15, 167 (2023).
Garcia-Beltran, W. F. et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 184, 2372–83.e9 (2021).
Lau, J. J. et al. Real-world COVID-19 vaccine effectiveness against the Omicron BA.2 variant in a SARS-CoV-2 infection-naive population. Nat. Med 29, 348–357 (2023).
Grifoni, A. et al. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 27, 671–680.e2 (2020).
Grifoni, A. et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 181, 1489–501.e15 (2020).
Peng, Y. et al. Broad and strong memory CD4(.) and CD8(.) T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 21, 1336–1345 (2020).
Hajnik, R. L. et al. Dual spike and nucleocapsid mRNA vaccination confer protection against SARS-CoV-2 Omicron and Delta variants in preclinical models. Sci. Transl. Med. 14, eabq1945 (2022).
Saksena, N. K. et al. SARS-CoV-2 variants, its recombinants and epigenomic exploitation of host defenses. Biochim. Biophys. Acta Mol. Basis Dis. 1869, 166836 (2023).
Ito, J. et al. Convergent evolution of SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant. Nat. Commun. 14, 2671 (2023).
Case, J. B. et al. Characterization of the SARS-CoV-2 BA.5.5 and BQ.1.1 Omicron variants in mice and hamsters. J. Virol. 97, e0062823 (2023).
Tang, J. et al. Respiratory mucosal immunity against SARS-CoV-2 following mRNA vaccination. Sci. Immunol. 7, eadd4853 (2022).
Lim, J. M. E. et al. SARS-CoV-2 breakthrough infection in vaccinees induces virus-specific nasal-resident CD8+ and CD4+ T cells of broad specificity. J. Exp. Med. 219, e20220780 (2022).
S, Guerrieri, S, Lazzarin, C, Zanetta, A, Nozzolillo, M, Filippi & L, Moiola Serological response to SARS-CoV-2 vaccination in multiple sclerosis patients treated with fingolimod or ocrelizumab: an initial real-life experience. J. Neurol. 269, 39–43 (2022).
Ssemaganda, A. et al. Expansion of cytotoxic tissue-resident CD8(+) T cells and CCR6(+)CD161(+) CD4(+) T cells in the nasal mucosa following mRNA COVID-19 vaccination. Nat. Commun. 13, 3357 (2022).
Yoshimoto, F. K. The proteins of severe acute respiratory syndrome coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J. 39, 198–216 (2020).
Wu, A. et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27, 325–328 (2020).
Miller et al. Substantial neutralization escape by SARS-CoV-2 Omicron variants BQ.1.1 and XBB.1. N. Engl. J. Med. 388, 662–664 (2023).
Tegally, H. et al. Emergence of SARS-CoV-2 Omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 28, 1785–1790 (2022).
Dangi, T. Improved control of SARS-CoV-2 by treatment with a nucleocapsid-specific monoclonal antibody. J. Clin. Investig. 132, e162282 (2022).
Saini, S. K. et al. SARS-CoV-2 genome-wide T cell epitope mapping reveals immunodominance and substantial CD8(+) T cell activation in COVID-19 patients. Sci. Immunol. 6, eabf7550 (2021).
Joag, V. et al. Cutting edge: mouse SARS-CoV-2 epitope reveals infection and vaccine-elicited CD8 T cell responses. J. Immunol. 206, 931–935 (2021).
Ponta, H., Sherman, L. & Herrlich, P. A. CD44: from adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 4, 33–45 (2003).
Wein, A. N. et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med 216, 2748–2762 (2019).
Scarpa, F. et al. Genetic and structural data on the SARS-CoV-2 Omicron BQ.1 variant reveal its low potential for epidemiological expansion. Int. J. Mol. Sci. 23, 15264 (2022).
Case J. B., et al. Characterization of the SARS-CoV-2 BA.5.5 and BQ.1.1 Omicron variants in mice and hamsters. J. Virol. 97, e0062823 (2023).
Li et al. Mechanisms of innate and adaptive immunity to the Pfizer-BioNTech BNT162b2 vaccine. Nat. Immunol. 23, 543–555 (2022).
Kingstad-Bakke, B. et al. Vaccine-induced systemic and mucosal T cell immunity to SARS-CoV-2 viral variants. Proc. Natl Acad. Sci. USA 119, e2118312119 (2022).
Poluektov, Y., George, M., Daftarian, P. & Delcommenne, M. C. Assessment of SARS-CoV-2 specific CD4(+) and CD8 (+) T cell responses using MHC class I and II tetramers. Vaccine 39, 2110–2116 (2021).
Pardieck, I. N. et al. A third vaccination with a single T cell epitope confers protection in a murine model of SARS-CoV-2 infection. Nat. Commun. 13, 3966 (2022).
Montoya B., et al. mRNA-LNP vaccine-induced CD8(+) T cells protect mice from lethal SARS-CoV-2 infection in the absence of specific antibodies. Mol. Ther. 32, 1790–1804 (2024).
Carabelli, A. M. et al. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nat. Rev. Microbiol. 21, 162–177 (2023).
Dutta, N. K., Mazumdar, K. & Gordy, J. T. The nucleocapsid protein of SARS-CoV-2: a target for vaccine development. J. Virol. 94, e00647–20 (2020).
Wu, W., Cheng, Y., Zhou, H., Sun, C. & Zhang, S. The SARS-CoV-2 nucleocapsid protein: its role in the viral life cycle, structure and functions, and use as a potential target in the development of vaccines and diagnostics. Virol. J. 20, 6 (2023).
Bai, Z., Cao, Y., Liu, W. & Li, J. The SARS-CoV-2 nucleocapsid protein and its role in viral structure, biological functions, and a potential target for drug or vaccine mitigation. Viruses 13, 1115 (2021).
Le Bert, N. et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 584, 457–462 (2020).
Uraki, R. et al. Characterization of SARS-CoV-2 Omicron BA.4 and BA.5 isolates in rodents. Nature 612, 540–545 (2022).
Li, D. et al. Neutralization of BQ.1, BQ.1.1, and XBB with RBD-Dimer Vaccines. N. Engl. J. Med. 388, 1142–1145 (2023).
Jiang, X. L. et al. Omicron BQ.1 and BQ.1.1 escape neutralisation by omicron subvariant breakthrough infection. Lancet Infect. Dis. 23, 28–30 (2023).
Zou, J. et al. Neutralization of BA.4-BA.5, BA.4.6, BA.2.75.2, BQ.1.1, and XBB.1 with bivalent vaccine. N. Engl. J. Med. 388, 854–857 (2023).
Hachmann, N. P. et al. Neutralization escape by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4, and BA.5. N. Engl. J. Med. 387, 86–88 (2022).
Mackin, S. R. et al. Fc-gammaR-dependent antibody effector functions are required for vaccine-mediated protection against antigen-shifted variants of SARS-CoV-2. Nat. Microbiol. 8, 569–580 (2023).
Zhang, A. et al. Beyond neutralization: Fc-dependent antibody effector functions in SARS-CoV-2 infection. Nat. Rev. Immunol. 23, 381–396 (2023).
Wherry, E. J. & Barouch, D. H. T cell immunity to COVID-19 vaccines. Science 377, 821–822 (2022).
Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 23, 186–193 (2022).
Tye, E. X. C. et al. Mutations in SARS-CoV-2 spike protein impair epitope-specific CD4(+) T cell recognition. Nat. Immunol. 23, 1726–1734 (2022).
Acknowledgements
Virus stocks were obtained from the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA). We thank Meredith Weglarz and the flow cytometry and cell sorting (FCCS) core at UTMB for assistance in flow cytometry. The research was supported by a UTMB COVID-19 pilot grant (to H.H.) and NIH grant R24AI120942 (to S.C.W.). H.H. was supported by NIH grants AI157852 and AI181134. D.W. was supported by NIH grant P01-AI158571. R.L.H. was supported by UTMB Mclaughlin Fellowship. The funders played no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.R.B., H.H., R.L.H., and K.S.P., designed experiments; R.L.H. and S.R.B. performed animal immunization experiments with assistance from Y.L. and N.C.H.; S.R.B., J.A.P., K.S.P., R.A.R. and J.H.W., performed animal challenge experiments. R.L.H. and S.R.B. performed ELISA, flow cytometry, RT-qPCR, and data analysis. J.A.P., J.H.W., and G.H.R. performed viral neutralizing experiments. D.H.W. performed data analysis; M-G.A., D.W. provided experimental materials; H.H., K.S.P., S.C.W. supervised experiments and the study. S.R.B., H.H., and R.L.H. wrote manuscript draft with editing from all authors.
Corresponding authors
Ethics declarations
Competing interests
H.H. and D.W. are inventors on a patent (PCT) titled COVID-19 mRNA vaccine (WO2023056045A1). All other authors declare no competing interests where relevant.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hajnik, R.L., Plante, J.A., Reddy Bonam, S. et al. Broad protection and respiratory immunity of dual mRNA vaccination against SARS-CoV-2 variants. npj Vaccines 9, 160 (2024). https://doi.org/10.1038/s41541-024-00957-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-024-00957-2
- Springer Nature Limited