
Abstract
Variational quantum eigensolvers (VQEs) represent a powerful class of hybrid quantum-classical algorithms for computing molecular energies. Various numerical issues exist for these methods, however, including barren plateaus and large numbers of local minima. In this work, we consider the Adaptive, Problem-Tailored Variational Quantum Eiegensolver (ADAPT-VQE) ansätze, and examine how they are impacted by these local minima. We find that while ADAPT-VQE does not remove local minima, the gradient-informed, one-operator-at-a-time circuit construction accomplishes two things: First, it provides an initialization strategy that can yield solutions with over an order of magnitude smaller error compared to random initialization, and which is applicable in situations where chemical intuition cannot help with initialization, i.e., when Hartree-Fock is a poor approximation to the ground state. Second, even if an ADAPT-VQE iteration converges to a local trap at one step, it can still “burrow” toward the exact solution by adding more operators, which preferentially deepens the occupied trap. This same mechanism helps highlight a surprising feature of ADAPT-VQE: It should not suffer optimization problems due to barren plateaus and random initialization. Even if such barren plateaus appear in the parameter landscape, our analysis suggests that ADAPT-VQE avoids such regions by design.
Similar content being viewed by others
Introduction
Quantum computers have long been viewed as a promising technology for quantum simulation1. However, the limited capabilities of Noisy, Intermediate-Scale Quantum (NISQ) devices restrict the types of algorithms that can be implemented at present2. While quantum phase estimation (QPE) provides a route to efficient molecular simulation3, the presence of both noise and errors on NISQ devices make near-term implementation of large-scale phase estimation intractable.
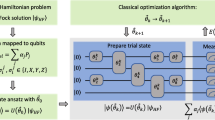
In response to the intractability of QPE, the variational quantum eigensolver (VQE) was introduced by Peruzzo et al.4 as a hybrid quantum-classical approach to finding approximate eigenvalues of a Hamiltonian, \({{{\mathcal{H}}}}\). In VQE, a quantum processor is used to apply a parameterized unitary transformation expressed as a quantum circuit (or even a direct pulse5,6,7), \({{{\mathcal{U}}}}\left({{{\boldsymbol{\theta }}}}\right)\), to some easily prepared reference state, \(\left\vert 0\right\rangle\)4,8,9,10,11. The target Hamiltonian is then measured with the prepared state to obtain the energy as a function of circuit parameters:
Using such quantum resources to prepare states and measure observables, a VQE will classically optimize θ in order to minimize \(E\left({{{\boldsymbol{\theta }}}}\right)\). The quality of the optimal energy for a given VQE is naturally dependent on the quality of the parameterization \({{{\mathcal{U}}}}\left({{{\boldsymbol{\theta }}}}\right)\), but because unitary operators are norm-preserving, the energy in Eq. (1) is variationally bounded from below by the ground-state energy of \({{{\mathcal{H}}}}\). The main advantage of VQEs is relatively low circuit depth4, avoiding the long, coherent evolutions of QPE12. This makes VQEs more appealing in the absence of fault-tolerant quantum computers. The circuit depth of a VQE is defined by the choice of \({{{\mathcal{U}}}}\), so that there is generally a trade-off between accuracy and circuit depth.
An outstanding challenge with many VQE ansätze is that the cost function, Eq. (1), creates a rough parameter landscape full of local minima, complicating the parameter optimization. Bittel and Kliesch have identified situations where there are so many far-from-optimal local minima that VQEs must be NP-hard in general13. The problem of local minima can be ameliorated through overparametrization in both quantum optimal control5,14 and classical neural network settings15,16. This idea of overparametrization avoiding local minima has since been applied to VQEs: Rivera-Dean et al. used this philosophy by employing a neural network to distort their cost function landscape mid-VQE17. This enabled them, in some cases, to escape from local minima. (The neural network temporarily adds additional “weight” parameters to the optimization. Even when the neural network is then reset to the identity, a better set of parameters θ was sometimes found for the undistorted cost function.) Alternative strategies for avoiding local minima include collectively optimizing an ansatz for several Hamiltonians at the same time with a “snake” algorithm18 and a “sweeping” approach to energy minimization called Unitary Block Optimization19.
A recent theoretical analysis by Larocca et al. suggests that quantum neural networks (of which VQEs are a special case) undergo a sort of phase transition where local minima cease to be a problem20. This transition tends to occur when the number of parameters surpasses the dimension of the associated ansatz’s dynamical Lie algebra, or DLA. The DLA for an ansatz of the form \(\left\vert {{\Psi }}\right\rangle ={e}^{{\theta }_{1}{A}_{1}}{e}^{{\theta }_{2}{A}_{2}}\ldots {e}^{{\theta }_{M}{A}_{M}}\left\vert {\phi }_{0}\right\rangle\) is defined as the span of the set of repeated commutators of \(\{{\hat{A}}_{i}\}\). As the authors point out, their results imply that this desirable overparametrization is likely to be unachievable for ansätze due to the exponential scaling of the DLA dimension with ansatz length. Perhaps even more alarmingly, Wierichs et al. were able to identify situations where adding additional parameters actually hurts the performance of gradient descent methods 21.
In addition to the problems with local traps, it has recently been recognized that VQEs might also become impossible to optimize (even to a local mininum) as the system size increases. For sufficiently flexible or expressive VQE ansätze (formal arguments have largely been restricted to 2-design structures), it has been found that the energy landscape flattens (as quantified by the variance in the parameter gradients) exponentially fast as the system size increases22. The exponential growth of these flat landscapes (so-called “barren plateaus”), means that only a vanishingly small region of parameter space exists which has gradients large enough to measure with high enough precision to perform gradient descent. This region of concentrated cost has been termed a “narrow gorge”23. As a result, initializing the optimization from a random point in parameter space is bound to land in a barren plateau, meaning that the number of circuit executions (shots) needed to resolve the search direction increases exponentially with the number of qubits, preventing any opportunity for quantum advantage. While intelligent heuristics for parameter initializations might help protect an optimization from getting stuck in a barren plateau (e.g., starting from a Hartree-Fock solution in molecular VQEs), the success is largely determined on a case-by-case basis 22.
In this work, we present arguments and numerical simulations that indicate that our recently introduced adaptive variational algorithm, ADAPT-VQE24, is expected to be effectively immune to local minima and barren plateaus in the parameter landscape, at least in the noise-free case. Both issues are avoided because the algorithm systematically “burrows” a deep well in the landscape until the global minimum is reached. In other words, ADAPT-VQE dynamically modifies its parameter landscape in such a way that problematic regions are never explored. This phenomenon can be understood directly from the gradient criterion used to iteratively update the wavefunction ansatz. We illustrate this behavior with simulations of several different molecules. In Supplementary Note 2, we also show that the smoothness of the landscape can be controlled by intentionally overparameterizing the ansatz. In Supplementary Note 6, we show how the fidelity (overlap with the target state) is affected by the number of parameters.
Methods
ADAPT-VQE
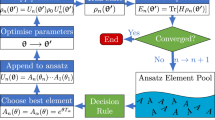
In recent work, we developed a dynamic framework for constructing ansätze that have much faster energy convergence with respect to circuit depth. This approach, referred to as ADAPT-VQE24,25, uses measurements of the molecular energy gradient to dynamically grow an ansatz, operator by operator, creating a highly compact ansatz that quickly converges to the exact solution. Defining a pool of anti-Hermitian operators, \({{{\mathcal{A}}}}=\{{A}_{i}\}\), we outline the steps in Algorithm 1.
Algorithm 1
ADAPT-VQE Algorithm

At each ADAPT-VQE iteration, the gradient, \(\frac{\partial E}{\partial {\theta }_{i}}\), is measured with respect to all operators in the pool. The operator with the largest gradient magnitude is then added to the ansatz with the associated parameter initialized to zero. The other parameters in the ansatz are initialized using the optimal values from the previous step (we refer to this as parameter “recycling”). At this point, an ordinary VQE is performed using some classical optimization algorithm. In this work, we exclusively use the Broyden-Fletcher-Goldfarb-Shanno (BFGS) method26, a quasi-Newton strategy, because we are explicitly seeking information about local minima, and because we are not including any noise models in our simulations. In all cases, a gradient norm of 1 × 10−8 was pursued, but not necessarily achieved, by the solver. In cases where the solver could not achieve this accuracy, its output was still used. Because we initialize the new parameter added during each ADAPT-VQE iteration to zero, the new trial circuit is equivalent to the previous one during the first VQE iteration. Consequently, the energy can only improve during this VQE, i.e., the energy decreases monotonically. Parameters are added one-by-one in this fashion until some convergence criteria are achieved. Reasonable choices include the norm (either l2 or l∞) of the vector of gradients, g, or the number of operators in the ansatz.
All simulations were conducted using a locally developed code which can be found on GitHub at https://github.com/hrgrimsl/adapt. OpenFermion27 was used to construct matrix representations of operators under the Jordan-Wigner transformation and PySCF28 was used to obtain molecular integrals. Because our focus in this work is to first understand the noise-free parameter landscapes associated with ADAPT-VQE, all simulations are performed without any noise models. Future work will explore how the presence of noise affects the landscapes. For all the ADAPT-VQE calculations in this work, the unitary coupled cluster with singles and doubles (UCCSD) operator pool is used24, without spin-complemented or spin-adapted operators. While many different pools can be used for ADAPT-VQE calculations, in this paper we focus primarily on the original fermionic pool due to its robustness in that it seems to consistently converge to an exact eigenstate and has a connection with the stationary conditions of the Anti-Hermitian Contracted Schrödinger equation29. Details of this pool are provided in Supplementary Note 1.
Results
Prevalence and distribution of local minima
In this section, we numerically explore the parameter landscapes of several example systems using ADAPT-VQE. Our aim is to characterize the way in which the number and distribution of local minima change as ADAPT-VQE gradually increases the length of the ansatz (and thus the depth of the circuit). For each molecule and bond distance considered, we first run ADAPT-VQE normally, where the initial parameter values used in the VQE at each iteration of the algorithm are chosen to be the “recycled” parameters, i.e., the optimal values obtained from the previous iteration. This yields an ansatz that reproduces the target ground state with high accuracy.
After using ADAPT-VQE to define the ansatz, we then use this ansatz to search for local minima by repeatedly reinitializing each VQE with randomly chosen parameters, and reoptimizing. (Each parameter was randomly initialized on an interval of length 2π in order to coincide with the period of \({e}^{{A}_{i}{\theta }_{i}}\) for the chosen pool.) In this work, we performed 1000 such random initializations for each ansatz considered unless otherwise specified. The numbers of samples were chosen due to computational considerations, and tests were performed to verify that increasing the number of random initializations does not change the results qualitatively. For each layer of the ansatz and each random initialization, we record the minimum energy obtained by the VQE subroutine. These values correspond to the energies of local minima in the landscape associated with each ansatz.
In addition to these random initializations, we also include both the “recycled” parameters from the previous VQE (the default initialization in ADAPT-VQE24) and the 0 parameter vector associated with the Hartree-Fock (HF) reference. All 1002 initializations of a given ansatz are then optimized with BFGS, and the resulting energy errors are shown with rainbow-colored bars in each figure. The colors indicate relative energy ordering at a given ansatz, such that red corresponds to the highest energy and violet to the lowest energy. The recycled initialization’s outcome is of particular interest since this is the default, deterministic initialization for ADAPT-VQE, and the approach used when growing the ansätze used in the data. These conventions will be used throughout this work.
We consider linear H4 (8 qubits) at 1 and 3 Å and linear H6 (12 qubits) at 1, 2, and 3 Å as toy models exhibiting varying degrees of electron correlation (and entanglement in the target wavefunction). While not interesting as chemistry agents, the fictitious molecules H4 and H6 provide an excellent testbed for quantifying the effect of strong correlation. Such ‘molecules’ are often used as surrogates for real strongly correlated systems such as ones involving transition metals, which are too large to simulate classically. In addition, we study LiH (12 qubits) at 1.62 Å and BeH2 (14 qubits) at 1.33 Å as examples of real molecules at equilibrium geometries. These geometries were obtained through optimization at the B3LYP30/6-31G*31,32,33,34 level of theory in PySCF28, and are included as a separate file. All ADAPT-VQE calculations were performed in the STO-3G35,36 basis. No symmetries were used to reduce the number of qubits. In cases where the exact solution was not obtained, the number of ADAPT-VQE iterations was determined by computational considerations.
H4 molecule
In Fig. 1 we show the energies (relative to the global minimum obtained from a full configuration interaction (FCI) calculation) of the various local minima as a function of ansatz length (as defined by the ADAPT-VQE algorithm). After a short period without local minima, the random initializations begin to diverge to an increasing number of distinct local minima as the number of parameters increases. In contrast to the random initializations, both the HF and the recycled initializations converge to the same minimum for H4 at 1 Å, which is consistently better than the average random initialization. This is our first indication that good initializations can reliably avoid high-energy traps. Interestingly, even though ADAPT-VQE doesn’t always find the lowest energy trap, it does eventually converge. Additionally, we observe that there are still many local minima even after these “chemically informed” guesses are able to reach the exact ground state. In Supplementary Note 2, we consider the prospect of removing local minima through systematic overparameterization for H4 at 1 Å. While we are successful in removing local minima using our “ADAPTN” approach, deeper circuits are actually required to achieve the overparameterization than to simply add operators until ADAPT-VQE reaches the ground state in spite of local minima.

The x-axis corresponds to the number of ADAPT-VQE iterations, i.e. the number of operators in the ansatz at a given step. The y-axis corresponds to the error from the exact FCI energy. The red curve corresponds to the energy obtained through BFGS minimization using an HF guess, i.e. one where all parameters are zero. The green curve corresponds to the energy obtained through BFGS minimization using the standard ADAPT-VQE in which optimal parameter values in one iteration are recycled as initial guesses in the next iteration, and with the new parameter initialized to zero. The colored dots correspond to all the energies obtained through BFGS optimizations, with red being the highest energy and violet the lowest.
In Fig. 2, we see that for the more strongly correlated 3 Å bond distance, the HF and recycled initializations differ. The recycled initialization is able to reach the ground state with fewer parameters than the HF initialization, though this behavior is not consistently observed in other systems. Again, we see ADAPT-VQE converging to the exact solution far faster than a typical (yellow–green) random initialization.

The axes and colors are as in Fig. 1.
H6 molecule
In Fig. 3, we begin to see the true power of an intelligent guess by simulating H6 at 1 Å. As the ansatz grows longer, a massive gap opens up between the random guesses and the HF/recycled ones. This gap implies that in practice, it is very difficult to do better than simply recycling the previous parameters in ADAPT. This gap is further numerical evidence of a “narrow gorge”, in which the exact solution is hypothesized to exist23. Although such a landscape is often associated with optimization difficulties, here we see that ADAPT-VQE is able to stay very close to the narrow gorge, avoiding such issues. We emphasize that this feature is not only a result of good initialization37, but rather a cooperative effect between initialization and the gradient-guided ansatz construction. In Supplementary Note 4, we demonstrate this explicitly by performing simulations using the recycled initialization, but on randomized (not gradient-guided) ansätze. We finally notice a sharp increase in the median around 140 parameters. This indicates that as the number of parameters increases, so too does the number of local traps. Furthermore, these new traps are preferentially high in energy, thus moving the median solution to higher energies. This further implies that as the system grows in size, the overwhelming number of solutions will be high in energy, making random sampling of VQE initializations intractable.

The axes and colors are as in Fig. 1. Plots a, b, and c correspond to bond lengths 1, 2, and 3 Å respectively.
In Fig. 3, the same gap appears for H6 at 2 Å that appeared at 1 Å. As the ansatz grows in depth (i.e., around 50 parameters), we notice an earlier rise in the median energy of the traps found.
In Fig. 3, for H6 at 3 Å, the energy distribution of the local traps significantly increases at the beginning, but chokes up around 100 parameters where the large gap is seen again. The HF and recycled initializations are still far better than random ones. We see the sharp increase in the median again here.
LiH molecule
In Fig. 4 we see similar behavior for LiH to that of H6 at 1 Å. While the solution gap is less pronounced, both HF and the recycled initialization are always significantly better than nearly every random initialization.

The axes and colors are as in Fig. 1.
BeH2 molecule
We observe similar behavior once again in Fig. 5 for BeH2, with the exception that a large gap is observed.

The axes and colors are as in Fig. 1.
In all cases, we observe that for more than a few parameters, local minima emerge, and for large numbers of parameters, these minima often dominate the energy landscape. In many cases initializing all parameters to 0 (HF) is a reasonable choice that leads to low energy minima.
Trap “Burrowing”
The problem of local minima seems to be partially mitigated by ADAPT-VQE itself. Even in cases where the recycled initialization converges to a high-energy trap, ADAPT-VQE progresses by adding an operator which is chosen to preferentially deepen the current trap (via the gradient criterion). As such, over a sequence of ADAPT-VQE iterations, the current trap becomes increasingly deep relative to the other parameter traps, such that a gap can open up between the current minimum (which approaches the global minimum) and all other local minima. Thus ADAPT-VQE appears to “burrow” into the parameter landscape, creating a single deep well as opposed to stabilizing all local minima (i.e., reaching overparameterization). This burrowing effect is depicted graphically in Fig. 6.

Schematic cartoon of how the parameter landscapes change as parameters are added using (a) ADAPT-VQE, and (b) a “controllable” or overparameterized ansatz which has a greater number of parameters than the rank of the DLA. Here the y-axis is meant to convey error, and the x-axis is meant to convey a generalized coordinate in parameter space.
Insensitivity to barren plateaus
In the previous section, we demonstrated that while the parameter landscapes exhibit a large number of local traps that are high in energy, ADAPT-VQE is robust due to the fact that any local minimum in early stages of the algorithm can often be deepened into a global minimum at later stages. This same mechanism implies a similar robustness to the presence of barren plateaus. As mentioned above, the barren plateau phenomenon has been recently recognized as a serious obstacle to the use of VQEs in practical settings. The problem arises from the observation that highly expressive ansätze (more specifically, circuits which form a 2-design), which are attractive from an accuracy perspective, exhibit an exponentially decreasing gradient variance with increasing system size. This means that the vast majority of parameter space becomes essentially flat. In the course of optimizing the parameters of such an expressive ansatz, a randomly chosen initialization will (with overwhelming probability) correspond to a point in parameter space where the gradient of the cost function is so small that an exponentially large number of measurements are needed to resolve a meaningful search direction in the presence of noise. As a result, the ability to optimize or train such expressive circuits is suspect at best. While a physically inspired parameter initialization can be effective (e.g., HF initialization), difficult cases (like those exhibiting strong correlation) may prevent efficient initialization.
Unlike the non-adaptive situation in which a static ansatz is first defined and then optimized, ADAPT-VQE slowly brings a given stationary point (initially the reference state) to the exact solution, via this burrowing mechanism. As such, each VQE subroutine performed along the way is “warm-started”, in that one already has a decent initialization coming from the previous optimization. Using this recycled initialization, we have a clear characterization of the parameter landscape about the initial point: all previous parameters are optimized, and thus have zero gradients, and the newly added operator has a large gradient by design, since we specifically add the operator with the largest gradient. This means that each VQE subroutine in the ADAPT-VQE algorithm is initialized with a single parameter which is guaranteed to be greater than ϵ (the ADAPT-VQE convergence threshold). Based on this argument, we do not expect difficulty due to barren plateaus when training ADAPT-VQE ansätze as system sizes are scaled up. We emphasize that this argument does not suggest that the ansätze constructed by ADAPT-VQE are free from barren plateaus, only that our algorithm remains localized to a region in parameter space with significant gradients.
We note that our analysis focuses exclusively on barren plateaus that arise from highly expressive circuits. ADAPT-VQE may still suffer from noise-induced barren plateaus (NIBP’s)38, which present problems for any VQE ansatz that scales polynomially in depth with system size, since they are a direct consequence of decoherence. Due to the problem-tailored nature of ADAPT-VQE and the computational difficulty of simulating increasingly large system sizes classically, we do not yet know how ADAPT-VQE ansätze scale with system size. Extrapolations from small system simulations will likely provide an overly pessimistic estimation due to the fact that correlation length will not simultaneously increase (at least for gapped systems). For a constant accuracy threshold, we expect the ansatz length to scale at least linearly (and thus ultimately suffer from NIBP’s), though a detailed study of this is not yet available. However, even if we assume that ADAPT-VQE might have an exponential scaling asymptotically, the problems of interest to chemistry are far from the asymptotic limit (around 100 logical qubits), and it is possible that a quantum advantage could still be demonstrated on finite problem instances. As such, further investigation into ADAPT-VQE’s performance in the presence of noise in general is indeed warranted.
“Gradient troughs”
Although barren plateaus seem to pose no threat to the ability to scale up ADAPT-VQE based on the arguments in the previous section, there is still a related issue that might prevent ADAPT-VQE from converging to accurate solutions. As described above, at each ADAPT-VQE step, the ansatz is extended using the operator with the largest gradient:
The ansatz is then repeatedly extended until the largest gradient in the operator pool is smaller than some threshold, ϵ. (In the first paper the convergence criterion was taken to be the norm of the gradients in the pool, rather than the maximum.) Noise on a NISQ device, however, defines some lowest possible threshold, \({\epsilon }_{\min }\), that can be resolved using a given shot allowance. In our earlier work24, we sometimes observed non-monotonic convergence of the gradients as a function of ansatz length (although the energy convergence is guaranteed to be monotonic), such that as the ansatz is extended, the pool gradients might first decrease, then increase again before finally converging. This “gradient trough”, therefore presents a challenge in the presence of noise. If a gradient trough appears and drops below the NISQ resolvable threshold, \({\epsilon }_{\min }\), then the ADAPT-VQE algorithm may halt prematurely.
How do these gradient troughs grow with system size? If we were to find that they grow exponentially fast, meaning that the largest gradient in the operator pool is exponentially suppressed as the number of qubits increases, then this would suggest concern for the scalability of ADAPT-VQE. However, this does not need to be the case. Choosing a local orbital basis one can imagine trivial situations where the gradients not only avoid exponential suppression, but any suppression at all. (One is always free to rotate occupied or virtual orbitals without changing the associated Slater determinant, due to orbital subspace rotational invariance.) Consider the nth iteration of an ADAPT-VQE calculation of a molecular wavefunction, \(\left\vert {\psi }_{n}\right\rangle\). If one were to double the number of qubits by adding another molecule (at infinite distance so as to remove interactions between the systems), the total wavefunction at iteration 2n would have a product form, \(\left\vert {\psi }_{2n}^{{{{\rm{AB}}}}}\right\rangle =\left\vert {\psi }_{n}^{{{{\rm{A}}}}}\right\rangle \left\vert {\psi }_{n}^{{{{\rm{B}}}}}\right\rangle\). Any pool operator \({\hat{O}}_{i}\) that is local to either subsystem has the exact same gradient in the supersystem, \(\left\vert {\psi }_{2n}^{{{{\rm{AB}}}}}\right\rangle\), as it does in the subsystem, \(\left\vert {\psi }_{n}^{{{{\rm{A}}}}}\right\rangle\). For example, consider an operator, \({\hat{O}}_{i}^{{{{\rm{A}}}}}\), local to subsystem A:
The additive separability of non-interacting subsystems is referred to as “size-consistency” in the chemistry literature. However, in addition to additive separability of the energy, size-extensive wavefunctions (like UCCSD) also demonstrate “size-intensivity” for intensive properties (e.g., density, optical gaps, etc). As shown in Eq. (3), the gradient with respect to a local rotation is not affected by the presence of an additional non-interacting system, thus demonstrating size-intensivity.
In the limit of a large system, any further additions to the system size will necessarily be too far away from a given subsystem to interact. Based on this argument, we don’t expect gradient troughs to deepen asymptotically with system size. However, more work is needed to characterize the behavior of gradient troughs as the system size increases in the presence of interactions.
Effect of low-lying FCI eigenstates
In order to understand the nature of the “gradient troughs” discussed in Sec. III C, and shown in Figs. 7 and 8, we superimposed the low-lying FCI energies with the ADAPT-VQE energies computed. The FCI spectrum is plotted as a set of blue horizontal lines. We only plot H4 and H6, as the other systems studied have no nearby excited states, nor do they exhibit any gradient troughs. In the region of the gradient trough, the energy also becomes very flat, (i.e., consider operators 9-16 in Fig. 7 and operators 50–100 in Fig. 8).

The x-axis corresponds to the ADAPT-VQE iteration. The green curve depicts the gradient associated with the operator to be added at each step, while the red curve depicts the energy at each ADAPT-VQE step. The blue lines depict the excited FCI eigenstates which are lower than the HF energy.

Overlay of the ADAPT-VQE error vs. iteration (red line, right axis) with the FCI excited states (blue horizontal lines, right axis) for H6. Plots a, b, c, and d correspond to bond lengths 2, 3, 4, and 5 Å respectively. The x-axis corresponds to the ADAPT-VQE iteration. The largest pool gradient associated with the operator to be added at each step is shown in green (left axis).
By plotting the exact eigenstates on top of these curves, one readily sees that the gradient troughs occur when ADAPT-VQE falls inside of a nearly degenerate manifold of FCI excited states. Should the ADAPT-VQE threshold be chosen loose enough (or if there is too much device noise to measure the gradient below this value) that the algorithm is aborted in this region, then ADAPT-VQE will be unable to advance further toward the ground state, remaining stuck as an approximation to an excited state (or in general some arbitrary superposition of the nearly degenerate eigenstates). This appearance of gradient troughs was first noticed in the paper that introduced ADAPT-VQE24, however the origin of the onset and the interpretation was not clear at that time.
As a consequence, although ADAPT-VQE isn’t expected to suffer from the more general problem of barren plateaus, more work is needed to understand how to escape any gradient troughs to ensure smooth convergence to the exact solution, particularly when noise is included. This remains an outstanding problem associated with ADAPT-VQE, warranting more research.
Discussion
Underparameterized ansätze are difficult to optimize due to large numbers of local minima, while highly expressive ansätze are difficult to optimize due to barren plateaus. In this paper, we find that ADAPT-VQE does not necessarily suffer from these challenges. We have studied the parameter landscapes arising from various ADAPT-VQE generated ansätze and have arrived at the following conclusions:
-
1.
Chemically informed initialization helps avoid traps: ADAPT-VQE’s process of re-using parameters at each step focuses the search space on a local region, keeping the algorithm relatively easy to train despite the rough overall landscape. The parameter vector from the previous iteration tends to be a relatively good initial guess for the following ADAPT-VQE iteration. This means that by simply “recycling” the parameters from one ADAPT-VQE iteration to the next, the vast majority of parameter traps are entirely avoided. Similarly, it seems that the chemical intuition granted by the HF state avoids most traps.
-
2.
Trap burrowing corrects local minima: Even if the early iterations get stuck in a trap, the adaptive construction iteratively extends the ansatz in a direction that is guaranteed to improve the cost function near the current stationary point. By continuously focusing on a local point in parameter space, ADAPT-VQE can “burrow” into a given local minimum, even if the vast majority of traps remain high in energy.
-
3.
Barren plateau avoidance: The nature of the ADAPT-VQE algorithm suggests that barren plateaus should not prove problematic in the parameter optimization step. This originates from the fact that ADAPT-VQE specifically adds a large gradient operator, generating a steep landscape, such that a search direction is resolvable without an exponential number of shots.
-
4.
Gradient troughs: ADAPT-VQE can still exhibit numerical challenges. An exponentially vanishing pool operator gradient could potentially arise, resulting in ADAPT-VQE becoming stuck during the operator addition step (in contrast to the parameter optimization step). Numerical evidence suggests that these gradient troughs appear when the ADAPT-VQE energy starts to converge near one or more excited states. Heuristics for diagnosing and addressing such issues will be the focus of future work.
Despite the presence of local minima and the possibility of barren plateaus in standard ADAPT-VQE ansatze, we conclude that ADAPT-VQE can be optimized reasonably well through parameter recycling. Consequently, in addition to being parameter- and gate- efficient, ADAPT-VQE appears to be relatively immune to the problems of both local minima and barren plateaus in VQEs.
Data availability
All data were generated with code available at https://github.com/hrgrimsl/adapt. Data is available upon request.
Code availability
Code developed for this project is open-source and available at https://github.com/hrgrimsl/adapt.
Change history
15 March 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41534-023-00694-9
References
Feynman, R. P. Simulating physics with computers. Int. J. Theor. Phys. 21, 467–488 (1982).
Preskill, J. Quantum computing in the NISQ era and beyond. Quantum 2, 79 (2018).
Aspuru-Guzik, A., Dutoi, A. D., Love, P. J. & Head-Gordon, M. Simulated quantum computation of molecular energies. Science 309, 1704–1707 (2005).
Peruzzo, A. et al. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun. 5, 4213 (2014).
Asthana, A. et al. Minimizing state preparation times in pulse-level variational molecular simulations. Preprint at http://arxiv.org/abs/2203.06818 (2022).
Meitei, O. R. et al. Gate-free state preparation for fast variational quantum eigensolver simulations. npj Quantum Inf. 7, 155 (2021).
Magann, A. B. et al. From pulses to circuits and back again: a quantum optimal control perspective on variational quantum algorithms. PRX Quantum 2, 010101 (2021).
Cao, Y. et al. Quantum chemistry in the age of quantum computing. Chem. Rev. 119, 10856–10915 (2019).
Cerezo, M. et al. Variational quantum algorithms. Nat. Rev. Phys. 3, 625–644 (2021).
Tilly, J. et al. The variational quantum eigensolver: a review of methods and best practices. Phys. Rep. 986, 1–128 (2022).
Fedorov, D. A., Peng, B., Govind, N. & Alexeev, Y. VQE method: a short survey and recent developments. Mater. Theory 6, 2 (2022).
Kitaev, A. Y. Quantum measurements and the Abelian Stabilizer Problem. Preprint at http://arxiv.org/abs/quant-ph/9511026 (1995).
Bittel, L. & Kliesch, M. Training variational quantum algorithms is NP-hard. Phys. Rev. Lett. 127, 120502 (2021).
Riviello, G. et al. Searching for quantum optimal controls under severe constraints. Phys. Rev. A 91, 043401 (2015).
Lopez-Paz, D. & Sagun, L. Easing Non-Convex Optimization with Neural Networks (ICLR, 2018).
Du, S. S. & Zhai, X. Gradient Descent Provably Optimizes Over-parameterized Neural Networks (ICLR, 2019).
Rivera-Dean, J., Huembeli, P., Acín, A. & Bowles, J. Avoiding local minima in variational quantum algorithms with Neural Networks. Preprint at http://arxiv.org/abs/2104.02955 (2021).
Zhang, D.-B. & Yin, T. Collective optimization for variational quantum eigensolvers. Phys. Rev. A 101, 032311 (2020).
Slattery, L., Villalonga, B. & Clark, B. K. Unitary block optimization for variational quantum algorithms. Phys. Rev. Res. 4, 023072 (2022).
Larocca, M., Ju, N., García-Martín, D., Coles, P. J. & Cerezo, M. Theory of overparametrization in quantum neural networks. Preprint at http://arxiv.org/abs/2109.11676 (2021).
Wierichs, D., Gogolin, C. & Kastoryano, M. Avoiding local minima in variational quantum eigensolvers with the natural gradient optimizer. Phys. Rev. Res. 2, 043246 (2020).
McClean, J. R., Boixo, S., Smelyanskiy, V. N., Babbush, R. & Neven, H. Barren plateaus in quantum neural network training landscapes. Nat. Commun. 9, 4812 (2018).
Arrasmith, A., Holmes, Z., Cerezo, M. & Coles, P. J. Equivalence of quantum barren plateaus to cost concentration and narrow gorges. Quantum Sci. Technol. 7, 045015 (2022).
Grimsley, H. R., Economou, S. E., Barnes, E. & Mayhall, N. J. An adaptive variational algorithm for exact molecular simulations on a quantum computer. Nat. Commun. 10, 3007 (2019).
Tang, H. L. et al. Qubit-ADAPT-VQE: An adaptive algorithm for constructing hardware-efficient ansätze on a quantum processor. PRX Quantum 2, 020310 (2021).
Fletcher, R. Practical Methods of Optimization 2nd edn (Wiley, Chichester, 2000).
McClean, J. R. et al. OpenFermion: the electronic structure package for quantum computers. Quantum Sci. Technol. 5, 034014 (2020).
Sun, Q. et al. PySCF: the python-based simulations of chemistry framework. Wiley Interdiscip. Rev. Comput. Mol. Sci. 8, e1340 (2018).
Mazziotti, D. A. Anti-hermitian contracted schrödinger equation: direct determination of the two-electron reduced density matrices of many-electron molecules. Phys. Rev. Lett. 97, 143002 (2006).
Becke, A. D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Dill, J. D. & Pople, J. A. Self consistent molecular orbital methods. XV. Extended Gaussian type basis sets for lithium, beryllium, and boron. J. Chem. Phys. 62, 2921–2923 (1975).
Ditchfield, R., Hehre, W. J. & Pople, J. A. Self consistent molecular orbital methods. IX. An extended Gaussian type basis for molecular orbital studies of organic molecules. J. Chem. Phys. 54, 724–728 (1971).
Hariharan, P. C. & Pople, J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 28, 213–222 (1973).
Hehre, W. J., Ditchfield, R. & Pople, J. A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56, 2257–2261 (1972).
Hehre, W. J., Stewart, R. F. & Pople, J. A. Self consistent molecular orbital methods. I. Use of Gaussian expansions of slater type atomic orbitals. J. Chem. Phys. 51, 2657–2664 (1969).
Collins, J. B., von R. Schleyer, P., Binkley, J. S. & Pople, J. A. Self consistent molecular orbital methods. XVII. Geometries and binding energies of second row molecules. A comparison of three basis sets. J. Chem. Phys. 64, 5142–5151 (1976).
Skolik, A., McClean, J. R., Mohseni, M., van der Smagt, P. & Leib, M. Layerwise learning for quantum neural networks. Quantum Mach. Intell. 3, 5 (2021).
Wang, S. et al. Noise-induced barren plateaus in variational quantum algorithms. Nat. Commun. 12, 6961 (2021).
Acknowledgements
N.J.M., S.E.E., and E.B. are grateful for financial support provided by the U.S. Department of Energy. N.J.M. and E.B. acknowledge Award No. DE-SC0019199. S.E.E. acknowledges the DOE Office of Science, National Quantum Information Science Research Centers, Co-design Center for Quantum Advantage (C2QA), Contract No. DE-SC0012704. H.R.G. acknowledges support provided by the Institute for Critical Technology and Applied Science at Virginia Tech. The authors thank the Advanced Research Computing at Virginia Tech for the computational infrastructure.
Author information
Authors and Affiliations
Contributions
H.R.G. wrote the code used in this work and ran all simulations. All authors contributed to the design of simulations, theoretical developments, and writing in the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Grimsley, H.R., Barron, G.S., Barnes, E. et al. Adaptive, problem-tailored variational quantum eigensolver mitigates rough parameter landscapes and barren plateaus. npj Quantum Inf 9, 19 (2023). https://doi.org/10.1038/s41534-023-00681-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41534-023-00681-0
- Springer Nature Limited
This article is cited by
-
Theoretical guarantees for permutation-equivariant quantum neural networks
npj Quantum Information (2024)
-
Quantifying the effect of gate errors on variational quantum eigensolvers for quantum chemistry
npj Quantum Information (2024)
-
Variational quantum algorithms: fundamental concepts, applications and challenges
Quantum Information Processing (2024)
-
Overlap-ADAPT-VQE: practical quantum chemistry on quantum computers via overlap-guided compact Ansätze
Communications Physics (2023)
-
Using Differential Evolution to avoid local minima in Variational Quantum Algorithms
Scientific Reports (2023)