
Abstract
Alternative UO2 nuclear fuels, incorporating Cr as a dopant, are currently in use in light–water reactors. Dissolution experiments using Cr-doped UO2, performed as a function of Cr content in a simplified groundwater solution and under oxic conditions, established that the addition of Cr to the UO2 matrix systematically reduced the normalised dissolution rate of U at 25 and 40 °C. This effect was most notable under dilute solution conditions, and is the result of galvanic coupling between Cr and U, resulting from the presence of Cr2+ in the UO2 matrix, as corroborated by activation energy determination. Under conditions of solution saturation, where schoepite ((UO2)8O2(OH)12·(H2O)12) and Na2U2O7·6H2O were identified as secondary phases, the rate of U dissolution was invariant with Cr content. Moreover, at 60 °C, the trend was reversed and the rate of U dissolution increased with increasing Cr content. Under these conditions, other factors, including U solubility or bicarbonate-surface interactions, exert a stronger influence on the U dissolution kinetics than Cr. Increased grain size, a feature of Cr-doped UO2 fuel, was also found to reduce the normalised dissolution rate of U. In establishing the mechanisms by which Cr dopants influence UO2 fuel dissolution, it can be concluded that, overall, Cr-doped UO2 nuclear fuel possesses similar dissolution kinetics to undoped UO2 fuel, giving confidence for its eventual disposal in a geological facility.
Similar content being viewed by others

Introduction
The current option for the final disposal of spent UO2 nuclear fuel is a burial in an engineered geological disposal facility. This will consist of a multi-barrier containment system, designed for the retention of radionuclides and mitigation against radioactive elements reaching the biosphere. While engineered to promote minimal degradation, there is expected to be a point in time at which groundwater will reach spent fuel and facilitate dissolution processes.
The dissolution of spent UO2 fuel has been well established through many years of investigation of dissolution under anoxic, reducing and oxic conditions, e.g. refs. 1,2,3. For the latter, dissolution rates are quoted to range from between 1 and 7 mg m−2 d−1, with the highest rates obtained in the presence of carbonate ions due to the propensity of U6+ to complex with CO32− to form soluble species3,4. However, the development and use of alternative UO2 fuels, adapted by doping with elements such as Cr2O3 to enhance in-reactor performance5,6,7, necessitates further investigation to understand whether dissolution rates are influenced by doping.
Although complex in final composition and microstructure post-fission4, dissolution of the UO2 matrix is understood to be the rate-determining step of spent fuel degradation, which proceeds via the oxidation of U4+O2 to soluble U6+O22+ species. This mechanism is governed by the availability of oxygen to diffuse through the lattice, enhanced by the presence of oxygen vacancies (Ov)8. In the new alernative nuclear fuel, Cr-UO2, it has been shown that Cr doping of UO2 results in the substitution of Cr2+ onto U4+ sites in the lattice, with the concurrent formation of Ov and U5+ to form a compound with a stoichiometry of \(\left( {\left( {U_{1 - x}^{4 + }U_x^{5 + }} \right)_{1 - y}Cr_y^{2 + }} \right)O_{2 - \frac{y}{2}}\) 9. As such, the presence of Ov and U5+ defect species should tend to increase the oxidative dissolution of doped UO2, which may reduce durability in a geological disposal facility when compared with conventional, undoped, UO2 fuel types.
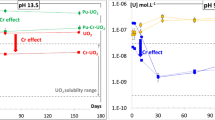
However, a recent study focused on Cr-doped UO2 dissolution behaviour indicated the opposite behaviour, that the presence of Cr decreased the dissolution rate of U relative to pure UO210. The authors postulated that the high pH conditions of their study (cementitious water, pH 13.5 and bicarbonate water, pH 9) caused the surface of Cr-UO2 to be more resistant to oxidation than UO2, or it otherwise prevented the release of U6+ species10. A slower rate of U dissolution compared with undoped UO2 was also reported for the dissolution of commercially fabricated Cr/Al-doped UO2 under oxidative conditions (H2O2), although it was stated that the behaviour may be due to differences in the surface area between samples, rather than the presence of Cr11. These authors were unable to establish a mechanism to satisfactorily explain why the addition of Cr influenced the UO2 behaviour.
It may be possible to partly explain these observations by reference to other doped systems, for example, a reduction in U dissolution rate compared to pure UO2 was also observed for trivalent Gd3+-doped UO212,13,14,15. At room temperature and in bicarbonate solution (pH 7– 8.5), the dissolution rate of U was an order of magnitude lower for Gd-doped UO2, while a drop of almost two orders of magnitude was observed at 50 and 75 °C, thought to confirm a 'matrix stabilisation effect' of Gd in UO215. It was postulated that stabilisation was conferred by a decrease in the degree of UO2 oxidation (U4+O2 to U4+/5+4O9) due to the substitution of U4+ cations for Gd3+ and potential formation of MO8-type defect clusters11,14. The same behaviour was observed for Dy3+-doped UO215,16. MO8-type defect clusters have not been reported for Cr-doped UO2, therefore, another mechanism must exist.
The addition of Cr to UO2 fuel is known to result in an enlarged grain size when compared to standard UO25,6,7. Grain boundaries have been shown to influence the dissolution behaviour of UO2 and spent fuel analogue materials including CeO2 and ThO2 and Ln-doped CeO217,18,19,20,21,22,23,24. Assessment of the transformation of grain boundaries during dissolution found that these features contribute significantly to the release of U (or U analogue element). It was hypothesised that the concentration of Ov defects at grain boundaries creates high-energy reactive surface sites for dissolution to initiate18. This suggests that a decrease in the quantity of grain boundaries, which is associated with a larger grain size, as observed for Cr-doped UO2, could reduce the number of energetically reactive sites and, thus, reduce dissolution rates.
To establish the influence of Cr as a dopant on the dissolution kinetics of UO2, and to underpin the mechanisms that govern U release from Cr-doped UO2, semi-static oxic dissolution experiments in a simplified bicarbonate groundwater solution were performed. Two possible mechanisms were evaluated: (1) Cr-induced changes to the crystal chemistry and (2) microstructure. For (1), materials were prepared using a range of Cr-dopant concentrations, above and below the solubility limit of Cr in UO2, and the dissolution kinetics of U were determined as a function of Cr content and dissolution temperature. These materials were prepared such that there was a fixed grain size, to enable isolation of the chemical effects of Cr on the dissolution rate from those of the microstructure. For (2), materials were prepared at different sintering temperatures, producing pellets with varying grain size, to quantify the extent to which microstructure influences the dissolution of Cr-doped UO2.
Results and discussion
Effect of Cr concentration on the dissolution of UO2
Pellets sintered at 1700 °C were used to study the effect of Cr content and dissolution temperature on the dissolution rate of U, independently from microstructure effects.
The microstructure and defect chemistry of the Cr-doped UO2 materials utilised in the dissolution experiments was detailed fully by Smith et al. 9. Briefly, for all concentrations of Cr-doped UO2, the sintered pellets presented a homogenous distribution of Cr2+ within the UO2 matrix, charge compensated by Ov defects. Above the solubility limit of Cr within UO2 (700–1200 ppm)25,26,27,28, precipitates of Cr were observed to reside within grain boundaries, hypothesised to be Cr3+2O39,29. When sintered at 1700 °C, the presence of these precipitates in Cr-doped UO2 resulted in a reduction in grain size when compared to undoped UO2 (Fig. 1a), due to the grain boundary pinning effect of precipitates, which inhibit diffusion and grain growth30.

a Grain size as a function of Cr content; b the measured Cr content of Cr-doped UO2 sintered at different temperatures (1400–1700 °C); and c Archimedes density. Error bars represent one standard deviation of triplicate measurements.
The intentional absence of large grains (>30 μm), as observed in industrially synthesised Cr-doped UO26, is a direct result of sintering in a fully reducing atmosphere; it is well understood that a controlled oxygen potential of the sintering atmosphere can promote grain growth in Cr-doped UO2 materials26. Indeed, the grain size did not vary significantly with Cr content when sintered at 1700 °C (Fig. 1a). Complete digest of the pellets sintered at 1700 °C showed that the Cr content was below the expected, nominal, Cr concentration (Fig. 1b), which is attributed to volatilisation at the high sintering temperature9,31,32.
The normalised mass loss of U, NL(U) (g m−2), at 25, 40 and 60 °C as a function of time (Fig. 2a–c) exhibited several distinct regimes of normalised dissolution rate, RL(U) (g m−2 d−1), as shown in Figs. 3, 4. The interpretation of these regimes was guided predominantly by the concentration, in mol L−1, of U leached from undoped UO2, which is reported to reach a solubility limit with respect to schoepite, of around 10−5 mol L−1, in saline, carbonate-containing solutions under oxic conditions33. The presence (or absence) of secondary phases was also used to support the identification of the regimes, as discussed later. The common regimes for all temperatures were interpreted as being: (1) RL,i, reflecting an initial dissolution rate where solution conditions were dilute and saturation effects were not observed, i.e. where U concentrations were ~10−7 mol L−1 (Fig. 3a, b); (2) RL,t, describing a transitional stage of dissolution where the effects of solution saturation commenced and the dissolution rate of U began to decrease, i.e. where U concentrations were ~10−6 mol L−1 (Fig. 3c, d); and (3) RL,ss where the thermodynamic, solubility-related effects of elements in solution were observed (steady state), i.e. where U concentrations were ~10−5 mol L−1 and a plateau, or drop, in NL(U) was observed (Fig. 4a).

Data derived from dissolution experiments performed at a 25 °C, b 40 °C and c 60 °C, using materials sintered at 1700 °C. Error bars represent one standard deviation of triplicate measurements.

Data derived from dissolution experiments performed at a, b 25 °C, c, d 40 °C and e, f 60 °C, using materials sintered at 1700 °C. Error bars represent one standard deviation of triplicate measurements.

Showing a the steady state (RL,ss) and b the grain boundary-influenced (RL,gb) regimes. Data were acquired using materials sintered at 1700 °C. Error bars represent one standard deviation of triplicate measurements.
For Cr-doped materials dissolved at 25 °C only, an additional regime, RL,gb, where the normalised dissolution rate of U increased at later time points (Figs. 2a, 4b) was observed. Dissolution of UO2 in acidic media has attributed this apparent increase in dissolution to an increase in the surface area associated with the dissolution of grain boundaries23,34,35,36. Table 1 details the time period (days) of each regime for materials dissolved at each temperature, Table 2 gives the absolute dissolution rate values in g m−2 d−1 and Supplementary Table 1 in mol L−1 d−1.
25 °C experiment
The normalised dissolution rate of U decreased with increasing Cr content in UO2 dissolved at 25 °C, in both the RL,i and RL,t regimes (Fig. 3a, b). For example, RL,i = (2.12 ± 0.17) × 10−3 g m−2 d−1 for undoped UO2 and (1.31 ± 0.13) × 10−3 g m−2 d–1 for 1490 ppm Cr-doped UO2. The corresponding dissolution rates in the RL,t regime were (1.10 ± 0.09) × 10−3 g m−2 d−1 and (0.39 ± 0.04) × 10−3 g m−2 d−1 for undoped and Cr-doped UO2, respectively (Table 2). From these results, it is apparent that the presence of Cr in UO2, in solid solution with the UO2 matrix and/or at present as precipitates at grain boundaries, influences the dissolution behaviour of U.
Since UO2 dissolution is dependent on the oxidation of U4+ to soluble U6+ species, especially under the oxic conditions of the present study, it is hypothesised that preferential oxidation of Cr2+/Cr3+ allows U to maintain a reduced state through the action of a galvanic coupling effect, according to the simplified form of the redox couples in Eqs. 1 and 2.
Clearly, the Cr3+-induced reduction of U6+ to U4+ is thermodynamically unfavourable (Eq. 2), meaning that the presence of Cr2+ species in the UO2 matrix is responsible for the decreased U dissolution rate. Increased Cr2+ content as a function of Cr doping would explain why the galvanic coupling effect is most pronounced in the more highly Cr-doped UO2, in the RL,i and RL,t regimes.
During the latter stages of the dissolution experiment, in the solubility-controlled RL,ss regime, a steady state was reached for U leached from Cr-doped UO2 at 25 °C (Fig. 2a). Importantly, in this steady state regime, there appeared to be no significant difference in the normalised dissolution rate of U as a function of Cr content (Fig. 4a). However, the absence of steady-state conditions for the undoped UO2 signifies an effect of Cr on the thermodynamics of U dissolution.
Since no Cr was measured in solution, it is assumed that sufficient Cr2+ remained within the doped UO2 to promote the galvanic coupling effect during the earlier regimes of dissolution; however, this process clearly no longer controlled the dissolution rate for the RL,ss and RL,gb regimes (Fig. 4a, b, respectively). It is possible that, at later time points, the Cr2+ involved in this mechanism was fully oxidised and, therefore, had no further potential for galvanic coupling. Alternatively, solution saturation of U may have hindered further dissolution. The measured molar concentration of U in the RL,ss regime was ~0.6–1.8 × 10−6 mol L−1 (Supporting Information, Fig. 1), which is below, but close to, the solubility limit of schoepite, reported to be around 10−5 mol L−1 under similar conditions33,37. As such, the solution was close to saturation with respect to U-containing secondary phases, reducing the thermodynamic driver for further dissolution of U. It was not possible to identify any secondary phases precipitated at the surface of the 25 °C pellets, even after 761 days of dissolution; therefore, the former explanation, that Cr2+ was fully oxidised, is tentatively favoured.
Beyond 616 days of dissolution at 25 °C, the role of grain boundary dissolution increased the U dissolution rate from ~10−4 g m−2 d−1 in the RL,ss regime to ~10−3 g m−2 d−1 in the RL,gb regime (Fig. 4b and Table 2).
40 °C experiment
The normalised dissolution rate of U, at 40 °C, in the RL,i (Fig. 3c) regime also decreased as a function of Cr content, from (4.15 ± 0.42) × 10−3 g m−2 d−1 to (2.56 ± 0.26) × 10−3 g m−2 d−1 for undoped UO2 and the highest Cr-doped UO2 material, respectively. Similar behaviour was observed in the RL,t regime (Fig. 3d), with rates of (2.45 ± 0.24) × 10−3 g m−2 d−1 for undoped UO2 and (1.44 ± 0.14) × 10−3 g m−2 d−1 for the highest concentration of Cr-dopant. It follows that the galvanic coupling effect of Cr2+ in UO2, as described for the 25 °C experiment, also occurs at 40 °C.
A steady-state regime was reached for all experiments at 40 °C after 442 days (Table 2), where the RL,ss was (0.03 ± 0.01) × 10−3 g m−2 d−1 for UO2 and (0.74 ± 0.10) × 10−3 g m−2 d−1 for 147 ppm Cr. The molarity values of U (3.7 × 10−6 mol L−1 and 3.4 × 10−6 mol L−1, respectively) were close to the solubility limit of U in saline, bicarbonate solution under oxic conditions, meaning that the solution was close to saturation with respect to U-containing secondary phases. The absolute normalised dissolution rate of U for UO2 doped with higher concentrations of Cr were negative, concurrent with the precipitation of U-bearing secondary phases, identified via SEM imaging of the material surfaces post-dissolution (Fig. 5).

Acquired from a sample of 1490 ppm Cr-doped UO2 (prepared at 1700 °C) and dissolved at 40 °C in bicarbonate solution for 525 days.
60 °C experiment
At 60 °C, the undoped UO2 exhibited a lower NL(U) than the Cr-doped UO2 (Fig. 2c) and the reverse trend in normalised dissolution rate of U was found for RL,i (Fig. 3e) and RL,t (Fig. 3f) when compared with 25 and 40 °C, i.e. the dissolution of U increased with increasing Cr content. It has previously been shown that bicarbonate-promoted dissolution of UO2, where bicarbonate ligands bind to the initially oxidised UO2 surface, is strongly dependent on temperature, with dissolution rates increasing in the range of 10 to 60 °C38. This is because the activation energy of UO2 surface oxidation is greater than that of the surface attachment of the bicarbonate ligand38. Therefore, given the higher temperature of this particular experiment, we postulate that the role of bicarbonate in the dissolution process is more significant than the galvanic coupling effect of Cr2+.
Calculations of the apparent activation energy (Ea) (see method section for details), determined for undoped and Cr-doped UO2 at 25, 40 and 60 °C using the RL,i dissolution rates are shown in Fig. 6a. In agreement with published data for UO2 dissolution in bicarbonate solution, the undoped UO2 gave an Ea value of 49.40 ± 0.04 kJ mol−1, consistent with a dissolution mechanism that proceeds via surface-controlled reactions38. As the Cr content is increased, the activation energy significantly increased, reaching (78.90 ± 0.01) kJ mol−1 for the highest concentration of Cr-dopant. This suggests that, in the initial stage of dissolution (i.e. without the effects of solution saturation), Cr influences surface-controlled dissolution reactions, consistent with the hypothesised galvanic coupling effect, even in bicarbonate solutions.

a Showing activation energies derived from rates in the initial dissolution rate regime (RL,i) and b During the steady-state dissolution regime (RL,ss). Error bars (smaller than data points but shown within points) represent one standard deviation of triplicate measurements.
The steady-state regime was reached after 100 days of dissolution at 60°C for undoped UO2 and after 79–86 days for Cr-doped UO2 (Table 1). The absolute normalised dissolution rates of U were negative (Table 2), indicating the formation of U-bearing secondary phases, which were more prominent at 60 than 40 °C and, as such, identifiable by XRD (Fig. 7). XRD diffraction patterns were consistent between duplicate pellets; therefore, only one representative pattern of each is shown.

a XRD pattern of Cr-doped UO2 surfaces dissolved at 60 °C in bicarbonate solution after 226 days of dissolution in comparison to diffraction patterns of UO2, Na2U2O7.6H2O, and ((UO2)8O2(OH)12(H2O)12) and b XRD pattern of isolated precipitate phase removed from the dissolved surface. Point (a) refers to the unpublished peak at low angles of 2θ, and points (b) represent absent reflections due to the preferred orientation of platy phases at the Cr-doped UO2 surface.
Two distinct precipitate morphologies were identified: a plate-like phase (Figs. 8a, 9), present on all dissolved materials (doped and undoped UO2); and an angular chip-shaped phase (Fig. 8b), visible only at the surface of the highest concentrations of Cr dopant. EDX point analysis of each precipitate confirmed that both phases comprised U, O and Na (Figs. 8a, 9). A darker phase was also identified in some areas, containing Na and Cl (Supporting Data, Fig. 2a; Point 1). This phase appeared not to contribute to the plate-like or chip-like morphologies in EDX elemental mapping (Supporting Information, Fig. 2b) and was likely remnant NaCl from the solution.

a 741 ppm Cr-doped UO2 plate-like precipitate, identified as Na2U2O7·6H2O and b 1490 ppm Cr-doped UO2 chip-like precipitate, identified as schoepite ((UO2)8O2(OH)12·(H2O)12).

SEM image of plate-like precipitate and corresponding EDX point spectrum representative of Points 1, 2 and 3.
XRD data were compared with UO239, and analysis of the plate-like precipitate matched with most of the reflections of hydrated Na-diuranate40, Na2U2O7·6H2O (Fig. 7b). This identification is tentatively corroborated by the EDX measured stoichiometry U:Na ratio of 1:1 (Table 3, taken from the precipitate layer with greatest apparent thickness). An additional reflection at ~6.7° 2θ (Fig. 7a, b; Point a), not previously published in the literature due to the restricted angular range used, corresponds to a d spacing of ~13.1 Å, which is in agreement with the postulated structure of Na2U2O7·6H2O40. Absent peaks at higher angles of 2θ (Fig. 7b; Points labelled b) can be attributed to preferred orientation arising from the flat, plate-like morphology. Reflections consistent with schoepite ((UO2)8O2(OH)12·(H2O)12) were also present in UO2 doped with the highest concentration of Cr (1490 ppm Cr, Fig. 7a)41.
The apparent activation energy in the RL,ss regime (Fig. 6b), where solution saturation influences the dissolution behaviour, was shown to remain within the same range as the Ea for RL,i regime (49.60 ± 0.01 to 79.70 ± 0.01 kJ mol−1); however, there was no clear trend between Ea and Cr content. This supports the hypothesis that at later stages of dissolution, when the solubility limit of U in bicarbonate solution has been reached, the effect of Cr is negligible. Interestingly, the Ea remains unchanged for UO2 doped with the highest concentration of Cr, which gave an Ea of RL,i = 74.63 ± 0.01 kJ mol−1 and RL,ss = 79.68 ± 0.02 kJ mol−1. To fully understand the mechanistic behaviour, a wider range of Cr-doped UO2 concentrations should be investigated in future.
Effect of grain size on the dissolution of undoped and Cr-doped UO2
To investigate the role of grain size on dissolution, undoped and Cr-doped UO2, prepared in a reducing atmosphere, were subject to heat treatment at different sintering temperatures of 1400, 1500, 1600, and 1700 °C. A clear increase in grain size with increasing sintering temperature was observed, from ~2–14 μm (Fig. 1a).
Complete digestion of these sintered materials showed that the Cr content was below the expected, nominal, Cr concentration (Fig. 1b) due to volatilisation31,32. The extent of volatilisation was much greater for materials sintered at temperatures of ≤1600 °C when compared with those at 1700 °C. It is understood that the solubility of Cr increases with increasing temperature26; therefore, at lower sintering temperatures, where less Cr is incorporated in the UO2 lattice, a greater loss due to volatilisation was observed. The greater extent of volatilisation of Cr at the lower sintering temperatures is corroborated by the measured densification rate, determined via Archimedes density measurements (Fig. 1c). The densification rate was highest for materials sintered at 1700 °C (~98%), as expected, but only reached ~91–96% in materials sintered ≤1600 °C. Since Cr incorporation into the lattice is attributed to increased densification of Cr-doped UO232, it follows that when there is less Cr in the lattice, at lower sintering temperatures, a reduced densification rate would be observed. As such, neither the Cr content (Fig. 1b), nor the density (Fig. 1c), were consistent between pellets, i.e. grain size was not the only variable during the study.
These materials were subject to dissolution in simulant groundwater solution (1 mM NaHCO3 + 19 mM NaCl) at 25 °C for 100 days (assumed to be the RL,i regime, based on data in Table 2). For materials sintered at 1700 °C, which display a high rate of densification (~97%, Fig. 1c), there is an obvious correlation between average grain size and the normalised dissolution rate of U (Fig. 10a, b). This correlation is also evident for materials sintered at lower temperatures (Fig. 10a). This result agrees with the hypothesis that a reduction in grain size, and therefore the quantity of grain boundaries, results in fewer energetically reactive surface sites for initiation of dissolution17,18. By observation of the gradient of RL(U) within each set of materials sintered at the different temperatures, it is clear as the sintering temperature is increased, the change in grain size has less of an effect on the normalised dissolution rate, i.e., the gradient becomes more shallow with increasing temperatures, most probably due to the level of densification attained.

Data were derived from experiments performed at 25 °C in a bicarbonate solution for 100 days. a Showing the relationship between sintering temperature, average grain size and RL(U) and b The relationship between Cr content, average grain size and RL(U). Error bars represent one standard deviation of triplicate measurements.
When the measured Cr content of each material is considered (Fig. 10b) at sintering temperatures of ≤1600 °C, there is no relationship between the dissolution rate and the Cr content. We postulate that variability in density between pellets may give rise to this behaviour. Further study, using Cr-doped UO2 materials prepared under conditions designed to stimulate enhanced grain growth while ensuring constant density and Cr content, is required to fully assess the effects of each variable in complete isolation.
Methods
Cr-doped and undoped UO2 preparation
A wet chemical, nitrate precipitation method was used to synthesise both Cr-doped and undoped UO2 materials at the HADES National Nuclear User Facility42. Nominal concentrations of Cr dopant were chosen, above and below the proposed solubility limit of Cr in UO2 (700–1200 ppm)26,27,28,29. Uranium (VI) nitrate hexahydrate in solution (UO2(NO3)2·6H2O, The British Drug House (BDH). B.D.H Laboratory Chemicals Division, >98%, 0.3 mol L−1) was mixed with various amounts of chromium (III) nitrate nonahydrate in solution (Cr(NO3)3·9H2O, Sigma Aldrich, 99.99%, 1.6 mol L−1) and precipitated at room temperature using concentrated ammonium hydroxide solution (NH4OH, Sigma Aldrich, 28–30% NH3 in H2O 5 mol L−1). The pH was monitored to reach between pH 8–10 for the successful co-precipitation of U and Cr, confirmed by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES, Thermo Fisher iCAP Duo6300) of the supernatants, where 99.9% precipitation for each element was achieved. The yellow precipitate was washed in deionised water, vacuum filtered and dried overnight at 90 °C to eliminate any remaining hydroxide. The resultant precursor powder was converted to oxide via thermal treatment at 750 ˚C for 4 h under a reducing (95% N2(g)–5% H2(g)) atmosphere, followed by dry milling at 35 Hz for 15 min to increase homogeneity and powder reactivity. Pellets of 6 mm were uniaxially pressed at 2.5 tonnes using a stainless steel die, and sintered for 8 h in a reducing (95% N2(g)–5% H2(g)) atmosphere. A range of sintering temperatures, of 1400, 1500, 1600 and 1700 °C, were used to control the microstructure (grain size). All surfaces were ground using SiC paper, polished to 1 μm using diamond suspension and thermally etched at ~85–90% sintering temperature to ensure an equal surface finish for all materials and to reveal the grain structure for characterisation.
Cr-doped and undoped UO2 characterisation
Confirmation of the UO2 single phase was carried out via X-ray diffraction (XRD) using a Bruker D2 Phaser diffractometer utilising a Cu Kα source from 10° to 100° 2θ with a step size of 0.02° and a step time of 2 s. Geometric densities of sintered pellets were calculated using the average geometry, measured using calibrated digital callipers, and average mass, measured using a calibrated five-point balance, to determine the geometric specific surface area of each pellet. Archimedes density was also determined for each individual pellet prior to dissolution, presented as an average of 10 measurements. The microstructures of the Cr-doped and undoped UO2 were characterised by scanning electron microscopy (SEM) using a Hitachi TM3030 SEM operating with an accelerating voltage of 15 kV. Images were taken at 500x magnification across five points of each pellet and the average grain area of ~500 grains was measured using ImageJ.
The total Cr content within the sintered materials was assessed by a complete digest in concentrated nitric acid (2 M HNO3). Pellets were crushed using a pestle and mortar and ~20 mg of powder was completely dissolved, under temperature and stirring, in 5 mL HNO3. The solutions were then measured for Cr concentration by Inductively Coupled Plasma Mass Spectroscopy (ICP-MS, Thermo Fisher single quadrupole iCAP RQ). Grain size and density were quantified as a function of both Cr content and sintering temperature.
Oxic dissolution experiments
Long-term durability experiments were conducted in the PLEIADES National Nuclear User Facility, using thermally etched, sintered pellets in duplicate, which were submerged in 50 mL of simulant groundwater solution (19 mM NaCl + 1 mM NaHCO3 bicarbonate solution) in a PTFE container. No atmospheric control was applied throughout the experiment, which is important to note since the dissolution medium was in equilibrium with CO2 in the air. The pH was measured to be in the range of 7.8 to 8.2 (±0.2 pH units) for all experiments at all times.
To understand the role of Cr content on dissolution behaviour, experiments were performed in ovens at 25, 40 and 60 °C (±2 °C) on UO2 with increasing Cr content, sintered at 1700 °C. To establish the role of grain size on Cr-doped UO2 dissolution, further experiments were performed at 25 °C on a selection of Cr-concentrations sintered at 1400, 1500, 1600 and 1700 °C. Both experiments are discussed and compared to assess the influence of Cr content, temperature and microstructure on dissolution behaviour.
At specific time points, an aliquot of 2 mL of the dissolution medium was removed, filtered (0.22 µm) and diluted by a factor of 10 in 1% ultra-pure conc. HNO3 for analysis by ICP-MS. A fresh solution of the same volume was replaced to maintain the surface area to volume (SA/V) ratio of approximately 1.3 m−1. Aliquots of solution were taken on days 1, 3, 7, 14, 21, then weekly up to day 98 and then bi-weekly for the duration of each experiment, which varied depending on dissolution temperature. It should be noted that Covid-19-induced laboratory closures severely restricted the sampling points for several experiments, over a period of ~6 months. Each long-term experiment was terminated after a constant concentration of U was measured, within error, for four consecutive time points.
The concentration of U (C(U), ng L−1) was converted to the mass of the element in solution for each time point (m(U)(t), mg) using the volume of each aliquot (V, 2 mL) via Eq. 3. The concentration of Cr was below limits of detection (2.5 ng L−1 for KED and 28 ng L−1 for STD analysis modes) for the duration of all experiments and, therefore, could not be measured.
The cumulative mass loss over time (mloss(U)(t), mg) of each pellet, and the initial pellet mass (minitial, mg) was used to determine the percentage of mass remaining for each pellet via Eq. 4, where f(U) is the mass fraction of U in each pellet. The normalised mass loss NL(U) (g m−2) was calculated from Eq. 5 using the specific geometric surface area (SSA, m2 g−1) of each pellet, while the normalised rate of U dissolution (RL(U), g m−2 d−1) was determined via the gradient of NL(U) as a function of time.
The dependence of the dissolution rate on temperature was assessed by determination of the activation energy (Ea) according to the Arrhenius law, Eq. 6, where RL(U) was taken in the initial regime of dissolution (RLi, Table 1) as well as when the effects of solution saturation were observed in the steady state of U dissolution (RLss, Table 1). The gradient of ln(RL(U)), for the intended regime, over the reciprocal absolute temperature was taken to determine the Ea (kJ mol−1) for undoped and Cr-doped UO2, as a function of Cr content.
Secondary phase characterisation
The presence of secondary phases, formed during dissolution, was determined via SEM/EDX using a Hitachi TM3030 SEM coupled with Bruker Quantax EDX system. XRD of the pellet surface, post-dissolution, was performed as described above, between 5° and 100° 2θ with a step size of 0.02° and a step time of 2 s. To improve diffraction pattern analysis, the precipitates were gently removed from the surfaces of pellets and analysed. XRD patterns were indexed using PDF SIEV + software. Geochemical modelling using PHREEQC-3 and the LLNL database was used to identify potential species that reached saturation limits in the dissolution.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Badley, M. & Shoesmith, D. W. The Corrosion/Dissolution of Used Nuclear Fuel in a Deep Geologic Repository. Report No. NWMO-TR-2022-09 (Nuclear Waste Management Organisation (NWMO), Toronto, Canada, 2022).
Oversby, V. M. Uranium Dioxide, SIMFUEL and Spent Fuel Dissolution Rates – A Review of Published Data. Report No. TR-99-22 (Svensk Karnbranslehantering AB (SKB), Stockholm, Sweden, 1999).
Shoesmith, D. W. Fuel corrosion processes under waste disposal conditions. J. Nucl. Mater. 282, 1–31 (2000).
Bruno, J. & Ewing, R. C. Spent nuclear fuel. Elements 2, 343–349 (2006).
Guillet, J. L. & Guerin, Y. Nuclear Fuels. Report No. ISBN 978-2-281-11345-7 (Commissariat à l’énergie atomique (CEA), Gif-sur-Yvette Cedex, France, 2009).
Massih, A. R. An Evaluation of High-Temperature Creep of Zirconium Alloys: Data versus Models. Swedish Radiation Safety Authority Report. Report No. 2014:20 ISSN 2000-0456 (Strålsäkerhetsmyndigheten (SSM), Stockholm (Sweden), 2014).
Assmann, H., Dörr, W. & Peehs, M. Control of UO2 microstructure by oxidative sintering,. J. Nucl. Mater. 140, 1–6 (1986).
He, H., Qin, Z. & Shoesmith, D. W. Characterising the relationship between hyperstoichiometry, defect structure and local corrosion kinetics of uranium dioxide. Electrochim. Acta 56, 53–60 (2010).
Smith, H. et al. Cr2+ solid solution in UO2 evidenced by advanced spectroscopy. Comm. Chem. 5, 163 (2022).
Cachoir, C., Mennecart, T. & Lemmens, K. Evolution of the uranium concentration in dissolution experiments with Cr(Pu)-doped UO2 in reducing conditions at SCK CEN. MRS Adv. 6, 84–89 (2021).
Nilsson, K., Roth, O. & Jonsson, M. Oxidative dissolution of ADOPT compared to standard UO2 fuel. J. Nucl. Mater. 488, 123–128 (2017).
Casella, A., Hanson, B. & Miller, W. The effect of fuel chemistry on UO2 dissolution. J. Nucl. Mater. 476, 45–55 (2016).
Kim, J.-G. et al. Effect of a trivalent dopant, Gd3+, on the oxidation of uranium dioxide. J. Nucl. Mater. 297, 327–331 (2001).
Liu, N. et al. Influence of Gd doping on the structure and electrochemical behaviour of UO2. Electrochim. Acta 247, 496–504 (2017).
Razdan, M. & Shoesmith, D. W. Influence of trivalent-dopants on the structural and electrochemical properties of uranium dioxide (UO2). J. Electrochem. Soc. 161, H105–H113 (2014).
Liu, N., He, H., Noël, J. J. & Shoesmith, D. W. The electrochemical study of Dy2O3 doped UO2 in slightly alkaline sodium carbonate/bicarbonate and phosphate solutions. Electrochim. Acta 235, 654–663 (2017).
Corkhill, C. L. et al. Contribution of energetically reactive surface features to the dissolution of CeO2 and ThO2 analogues for spent nuclear fuel microstructures. Appl. Mater. Interfaces 6, 12279–12289 (2014).
Corkhill, C. L. et al. Role of microstructure and surface defects on the dissolution kinetics of CeO2, a UO2 fuel analogue. Appl. Mater. Interfaces 8, 10562–19571 (2016).
Claparede, L. et al. Influence of crystallisation state and microstructure on the chemical durability of cerium-neodymium mixed oxides. Inorg. Chem. 50, 9059–9072 (2011).
Horlait, D. et al. Environmental SEM monitoring of Ce1−xLnxO2−x/2 mixed-oxide microstructural evolution during dissolution. J. Mater. Chem. A 2, 5193–5203 (2014).
Szenknect, S. et al. Kinetics of structural and microstructural changes at the solid/solution interface during dissolution of cerium(IV)-neodymium(III) oxides. J. Phys. Chem. C. 116, 12027–12037 (2012).
Myllykylä, E. et al. Solution composition and particle size effects on the dissolution and solubility of a ThO2 microstructural analogue for UO2 matrix of nuclear fuel. Radiochim. Acta 103, 565–576 (2015).
Cordara, T. et al. Microstructural evolution of UO2 pellets containing metallic particles of Ru, Rh and Pd during dissolution in nitric acid solution: 3D-ESEM monitoring. Hydrometallurgy 188, 182–193 (2019).
Claparede, L. et al. Dissolution of Th1‑xUxO2: effects of chemical composition and microstructure. J. Nucl. Mater. 457, 304–316 (2015).
Kashibe, S. & Une, K. Effect of additives (Cr2O3, Al2O3, SiO2, MgO) on diffusional release of 133Xe from UO2 fuels. J. Nucl. Mater. 254, 234–242 (1998).
Bourgeois, L., Dehaudt, P., Lemaignan, C. & Hammou, A. Factors governing microstructure development of Cr2O3-doped UO2 during sintering. J. Nucl. Mater. 297, 313–326 (2001).
Milena-Pérez, A. et al. Raman spectroscopy coupled to principle component analysis for studying UO2 nuclear fuels with different grain sizes due to the chromia addition. J. Nucl. Mater. 543, 152581 (2021).
Cardinaels, T. et al. Chromia doped UO2 fuel: investigation of the lattice parameter. J. Nucl. Mater. 424, 252–260 (2012).
Kuri, G. et al. Local atomic structure of chromium bearing precipitates in chromia doped uranium dioxide investigate by combined micro-beam X-ray diffraction and absorption spectroscopy. J. Nucl. Mater. 449, 158–167 (2014).
Riglet-Martial, C. et al. Thermodynamics of chromium in UO2 fuel: a solubility model. J. Nucl. Mater. 447, 63–72 (2014).
Finkeldei, S. C. et al. Synthesis and Characterization of UO2 Feedstocks Containing Controlled Dopants. Report No. M3FT-19OR02021075 (Oak Ridge National Laboratory, Oak Ridge Tennessee (USA), 2019).
Peres, V. et al. High temperature chromium volatilisation from Cr2O3 powder and Cr2O3-doped pellets in reducing atmospheres. J. Nucl. Mater. 423, 93–101 (2012).
Ollila, K. & Ahonen, L. Solubilities of Uranium for TILA-99 Report No. 98-13 Svensk Kärnbränslehantering AB, Stockhom (Sweden), (1998).
Cordara, T. et al. Kinetics of dissolution of UO2 in nitric acid solutions: a multi-parametric study of the non-catalysed reaction. J. Nucl. Mater. 496, 251–264 (2017).
Bertolotto, S. et al. Effect of surface orientation on dissolution rate and surface dynamics of UO2 single crystals in nitric acid. Corros. Sci. 176, 109020 (2020).
Podor, R. L. et al. 3D-SEM height map series to monitor materials corrosion and dissolution. Mater. Charact. 150, 220–228 (2019).
Ollila, K. Dissolution of Unirradiated UO2 Fuel in Synthetic Groundwater – Progress Report ’97. Report No. 98-06 (Svensk Kärnbränslehantering AB, Stockholm (Sweden), 1998).
De Pablo, J. et al. The oxidative dissolution mechanism of uranium dioxide. I. The effect of temperature in hydrogen carbonate medium. Geochim. Cosmochim. Acta 63, 3097–3103 (1999).
Desgranges, G. et al. Neutron diffraction study of the in situ oxidation of UO2. Inorg. Chem. 48, 7585–7592 (2009).
Chernorukov, N. G., Nipruk, O. V. & Kostrova, E. L. Synthesis and study of sodium uranate Na2U2O7·6H2O and of products of its dehydration and thermal decomposition. Radiochemistry 58, 124–127 (2016).
Finch, R. J., Cooper, M. A., Hawthorne, F. C. & Ewing, R. C. Distinguishing among schoepite, [(UO2)8O2(OH)12](H2O)12, and related minerals by X-ray powder diffraction. Can. Mineral., 34, 1071–1088 (1996).
Hyatt, N. C. et al. The HADES facility for high activity decommissioning engineering and science: part of the UK National Nuclear user facility. IOP Conf. Ser. Mat. Sci. 818, 012022 (2020).
Acknowledgements
This work was funded by the European Commission Horizon 2020 Research and Training Programme, DISCO, of the European Atomic Energy Community (EURATOM), under grant agreement number 755443. We also wish to acknowledge funding from the UK Engineering and Physical Science Research Council (EPSRC) under grant numbers EP/N017374/1 and EP/S020659/1. This research utilised the HADES/MIDAS facility at the University of Sheffield, established with financial support from EPSRC and BEIS, under grant EP/T011424/1, and also the PLEIADES National Nuclear User Facility, established with funding from EPSRC under grant number EP/V035215/1, and the Henry Royce Institute for Advanced Materials Nuclear ICP-MS facility, under grant numbers EP/P02470X/1 and EP/P025285/1. We would like to thank Dr. Chris Dixon Wilkins at the University of Sheffield for support with XRD pattern indexing, Dr. Colleen Mann for support with ICP-MS analysis and all the members of the DISCO project for continued support and enjoyable, lively discussion.
Author information
Authors and Affiliations
Contributions
H.S.—data collection, formal analysis and original draft preparation. T.C.—sample preparation and experimental setup. C.G.—data collection. S.E.P.—data collection. C.L.C.—funding acquisition, supervision, data collection, formal analysis, reviewing and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Smith, H., Cordara, T., Gausse, C. et al. Oxidative dissolution of Cr-doped UO2 nuclear fuel. npj Mater Degrad 7, 25 (2023). https://doi.org/10.1038/s41529-023-00347-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-023-00347-4
- Springer Nature Limited
This article is cited by
-
Fabrication, defect chemistry and microstructure of Mn-doped UO2
Scientific Reports (2024)