
Abstract
The development of machine learning models has led to an abundance of datasets containing quantum mechanical (QM) calculations for molecular and material systems. However, traditional training methods for machine learning models are unable to leverage the plethora of data available as they require that each dataset be generated using the same QM method. Taking machine learning interatomic potentials (MLIPs) as an example, we show that meta-learning techniques, a recent advancement from the machine learning community, can be used to fit multiple levels of QM theory in the same training process. Meta-learning changes the training procedure to learn a representation that can be easily re-trained to new tasks with small amounts of data. We then demonstrate that meta-learning enables simultaneously training to multiple large organic molecule datasets. As a proof of concept, we examine the performance of a MLIP refit to a small drug-like molecule and show that pre-training potentials to multiple levels of theory with meta-learning improves performance. This difference in performance can be seen both in the reduced error and in the improved smoothness of the potential energy surface produced. We therefore show that meta-learning can utilize existing datasets with inconsistent QM levels of theory to produce models that are better at specializing to new datasets. This opens new routes for creating pre-trained, foundation models for interatomic potentials.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Introduction
Machine learning is fundamentally changing and expanding our capabilities for modeling chemical and materials systems1,2,3,4,5,6,7,8. A growing array of properties have been successfully predicted with machine learning models from materials’ band gaps and formation energies to molecular energies and bond orders9,10,11,12. The development of machine learning models for various applications has involved the creation of a large number of datasets containing quantum-mechanical calculations at different fidelities (levels of theory)13,14,15,16,17,18. However, incorporating this multi-fidelity information into machine learning models remains challenging. In this work, we show that multiple datasets can be used to fit a machine learning model, even if the datasets were calculated with many varying QM levels of theory. To overcome this challenge, we incorporate meta-learning techniques into the training process and subsequently demonstrate improvements in accuracy for multiple applications. The aim of meta-learning is to use a wide collection of data to train a machine learning model that can be easily re-trained to specialized tasks. Here, we demonstrate the applicability of the meta-learning method to MLIPs.
In the landscape of broader efforts to incorporate machine learning and molecular and material modeling, a particular attention has been paid to MLIPs10,19,20,21,22,23,24. Accurate atomistic simulations rely on interatomic potentials that closely recreate the interactions present between atoms and molecules25,26. Recreating these interactions involves a trade-off between accuracy and computational cost, with quantum mechanical techniques offering highly accurate simulations whilst classical force fields are fast and capable of modeling much larger systems over long timescales27,28,29. Within the last decade, MLIPs have increasingly been seen as a method that could provide a model that is both fast and accurate10,19,20. However, the development of MLIPs that are transferable to unseen organic molecules requires datasets that cover a large fraction of chemical space. This requirement has lead to the production of numerous datasets13,14,15,16,17,18. These datasets contain the quantum mechanical (QM) energies and forces of millions of structures spanning large regions of chemical space. However, the QM methods used to calculate the energies and forces vary considerably. As different QM methods result in different potential energy surfaces, this inconsistency in QM techniques limits the extent that datasets can used together to fit potentials.
Numerous organic molecule datasets have been created for training MLIPs13,14,15,16,17,18. However, a consensus on the best QM techniques to employ to create these datasets has never been reached as a compromise between accuracy and computational cost must always be considered when performing QM calculations. This lack of consensus has led to a variety of different software, methods, basis sets and exchange-correlation functionals being used. For example, the QM7-x and ANI-1x datasets both contain energies and forces for millions of small organic molecules. However, QM7-x was calculated using the PBE0 exchange-correlation functional with many body dispersion whilst ANI-1x was calculated with the ωB97x functional and 6-31G* basis set14,16 and does not include dispersion effects. Therefore, these two datasets describe similar, but slightly different potential energy surfaces. If both datasets were joined together to train a potential then problems would likely arise as contradictory information is present. For example, identical structures at different levels of theory can have different energy and forces. Whilst datasets from different sources have been fit together without further refinement30, this approach does not account for differences in the interactions described. Techniques exist in the machine learning literature to address the difference in the potential energy surface.
Previous work on fitting MLIPs to multiple datasets is limited. In Ref. 31, a transferable molecular potential was first trained to ~ 5 million density functional theory (DFT) training points before being refit, with frozen parameters, to 0.5 million CCSD(T)* energies. This technique, known as transfer learning has been used in several works32,33,34,35,36. The advantage of using transfer learning for training MLIPs is that it requires fewer calculations at a higher, and more expensive, level of theory. However, approach to transfer learning, freezing neural network (NN) parameters, is limited to just two datasets. If we want to use multiple existing datasets, and expand the size and variety of training data, then new methods must be found.
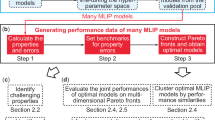
Fortunately, this problem is being explored in a branch of machine learning research known as meta-learning37,38,39,40. Meta-learning seeks to build a model that, although not specialized to any particular task, can be quickly re-trained to many new tasks – where a task is a specific learning problem (Fig. 1). Furthermore, this retraining can be effective even if the amount of new data is limited37,39.
For transferable MLIPs, the concepts of tasks naturally lends itself to quantum mechanical datasets calculated with different methods. By using meta-learning techniques, we will show how information from multiple levels of theory can be incorporated together. We begin by investigating training data with multiple levels of theory for an individual aspirin molecule and for the QM9 dataset (which contains over 100,000 molecules in their equilibrium configuration). With these systems, the problems associated with naively combining datasets together are seen and the benefits of meta-learning are clearly observed in the test set errors. We then move on to combining several large molecule datasets to pre-train an MLIP. Subsets, chosen using active learning, of six existing datasets (ANI-1x, GEOM, QMugs, QM7-x, Transition-1x and the QM9 dataset from Ref. 13) were used to fit an adaptable potential using meta-learning – see Fig. 2 for a visualization of the space the datasets cover13,14,15,16,17,18. Figure 2 demonstrates the increase in chemical space possible when multiple datasets are combined together. The benefits of pre-training are then shown by retraining to the 3BPA molecule and testing various properties. These tests show that pre-training models using meta-learning produces a more accurate and smoother potential. The benefits of pre-training include enhanced accuracy and generalization capabilities in modeling interatomic potentials (Fig. 2).

This work uses Reptile to build a potential that incorporates information from multiple molecular datasets, calculated at different levels of theory. This meta-learned potential adapts well to new tasks, and outperforms potentials that were trained only to the data for a single task.

A diverse collection of datasets, with varying levels of theory, molecule sizes, and energies, will be incorporated into a single meta-learned potential. The distributions of the number of atoms and energy of the structures contained in the datasets used for training a potential in this work are shown. The structures included contain only C,H,N,O. Energies are made comparable using linear scaling as detailed in Multiple Organic Molecules section.
Training machine learning models to large amounts of data before re-training to a specific task is related to the concept of foundation models41. This concept has been used to create large language models, ie. GPT-4, which have been pre-trained to extremely large datasets before being fine-tuned to specific tasks, i.e., ChatGPT which is fine-tuned for conversational usage42. Creating foundation models allows a wide range of information to be encoded before specialisation. With meta-learning techniques, we can now pre-train interatomic potentials to numerous large datasets and this is a step towards foundation models for MLIPs – MLIPs that could be quickly re-trained to diverse molecular systems.
The number of QM datasets has grown rapidly over the last few years. However, a major bottleneck in exploiting this information has been the absence of methods that can effectively combine all of this information. In this work, we have overcome this limitation by exploiting techniques which enable the incorporation of datasets with different fidelities. Whilst we focus on MLIPs, these techniques are applicable to the wide range of predictive models that exist for material and molecular property prediction. By showing how meta-learning can be applied, we aim to encourage researchers to fully utilize the vast amount of existing data that the scientific community has already collected.
Results
A simple case study on aspirin
As the initial test case we investigate the performance of meta-learning on a dataset containing a single aspirin molecule. Aspirin structures were produced by molecules dynamic simulations at 300K, 600K and 900K. The QM energies and forces were then calculated at three different levels of theory: two distinct DFT functionals, and Hartree-Fock. This created three different datasets, with each temperature corresponding to a different level of theory. These three datasets were used to pre-train a molecular potential to the energy and forces of 1,200 structures. The pre-trained potential was then refit to a new dataset of 400 MD configuration at the MP2 level of theory from the 300K simulation.
The change in the RMSE error for the forces is shown with the value of k used in the meta-learning algorithm in Fig. 3. The k parameter controls the number of steps taken towards each dataset. As k is increased the speed of the algorithm also increases and this is an additional consideration in choosing the optimal value. In the limit of k → ∞ the algorithm would correspond to iterative training to each dataset and then transfer learning to a new task. However, while this may work for small problems, this approach is impractical for large datasets.

The error as a function of the value of k used in the meta-learning algorithm for an aspirin molecule. The potential is first pre-trained to multiple levels of theory before retraining to 400 structures at MP2 level of theory. When no pre-training is used the root mean squared error (RMSE) is 5.35 ± 0.41 kcal mol−1 Å−1.
Figure 3 shows that as the k parameter is increased the error in the test set decreases with the minimum error at around k = 400. There is therefore an improvement in test set error in comparison to both no pre-training (5.35 ± 0.41 kcal mol−1 Å−1) and k = 1 (3.38 ± 0.16 kcal mol−1Å−1). Note that k = 1 effectively corresponds to simultaneous training to all tasks. Therefore, when we attempt to combine multiple datasets at different levels of theory an improvement in performance can be seen when meta-learning is incorporated into the training process.
Meta-learning many levels of theory using QM9
Next, we move onto the QM9 dataset that contains multiple different small organic molecules in their equilibrium structures. The QM9 dataset has been calculated at 228 different levels of theory and therefore provides an ideal dataset for analysing meta-learning techniques. We can use this dataset to test whether meta-learning can develop a potential which can be refit to a new level of theory encountered for the QM9 dataset with less data. In order to do this, a subset of the QM9 dataset was used to train a potential to 10,000 molecules, 50 different exchange-correlation functionals and three different basis set. The potential was then refit to a new exchange-correlation functional, that had not been previously encountered, and the performance of this new model was assessed and compared to no pre-training and k = 1 meta-learning.
The test set error for the meta-learning potential refit to a new level of theory in the QM9 dataset is shown in Fig. 4. Pre-training the potential greatly improves the test set error for this case. In Supplementary Fig. 9 a comparison between meta-learning and k = 1 is shown and we see that k = 1 does not perform as well as k = 10. This is because it does not account for the discrepancy in the interaction present. These results show that even when the number of levels of theory is relatively large, at 150, and multiple molecules are present that meta-learning improves test set error over k = 1.

The error as a function of the number of structures averaged across 5 different DFT functions and 3 basis sets, for different fitting procedures for the QM9 dataset. The meta-learning algorithm fits the different levels of theory together with k = 10 used. The standard deviation of the error bars corresponds to the variation across different, randomly selected, specialization tasks. Freezing the parameters before retraining was also attempted for 400 structures, however, no improvement in accuracy was observed.
Making the most of scarce data at CCSD(T) level
We will now move to the datasets used to train transferable interatomic potentials. As a starting example, we will look at pre-training to the multiple levels of theory (ωB97x/ 6-31G* and ωB97x/ def2-TZVPP) contained in the ANI-1x dataset14. We will then retrain to the ANI-1ccx dataset14. Figure 5 shows the distribution in error when pre-training to multiple levels of theory with meta-learning and k = 1. The RMSE is 3.30 ± 0.10 kcal mol−1 and 2.39 ± 0.00 kcal mol−1 for k = 1 and meta-learning respectively. Therefore, we can again see that meta-learning with a higher k values improves results compared to k = 1. The comparative results for direct training to ωB97x/ 6-31G* and ωB97x/ def2-TZVPP and then transfer learning to CCSD(T) is 2.20 ± 0.01 kcal mol−1 and 2.09 ± 0.02 kcal mol−1 respectively. Therefore, in this case fitting to multiple datasets does not improve results over fitting to just one. This is in part because both datasets contain the same structures and cover the same chemical and configurational space. The potential trained to multiple organic datasets was also refit to the CCSD(T) dataset and the benefits of meta-learning over k = 1 were also seen with errors of 2.89± and 3.32±, respectively. However, this is notably higher than training to the ANI-1x dataset alone. The CCSD(T) dataset is a subset of the ANI-1x dataset and contains identical structures. For these cases, adding additional data in other areas of chemical space may not improve results.

The error distribution for the CCSD(T) specialization task after pre-training to ANI-1x with meta-learning (k = 50) using and k = 1. The RMSE is 3.47 kcal mol−1 and 2.39 kcal mol−1.
Training to multiple transferable organic molecule datasets
Numerous datasets have been created that contain quantum mechanical calculations for organic molecules. However, as these datasets use different levels of theory and software, combining the information from different datasets requires advanced training techniques. By using meta-learning, a pre-trained model was created that uses information from seven different datasets. This is the first instance, to our knowledge, of combining information from multiple organic molecule datasets in this manner.
We have already seen that meta-learning can improve results compared to k = 1 when multiple datasets are used. We will now use the pre-trained model to explore the benefits of pre-training with meta-learning in comparison to no pre-training, and k = 1 when retraining to a single molecular system. The pre-trained model was re-trained to the 3BPA dataset taken from Ref. 36 and various properties explored43. The bond dissociation calculated using DFT is 119.6 kcal mol−1 and 111.2 kcal mol−1 with the bond dissociation energies estimator from Ref. 44.
The first properties we will analyze are the energy and force RMSE errors. The force errors for a dataset taken from MD at 1200K is shown in Fig. 6 with the energy and force learning curves for datasets at 300K, 600K and 1200K given in Supplementary Fig. 4. From these graphs, the improved performance of pre-training using the meta-learning approach (with three passes through the dataset) to both k = 1 and no pre-training can be seen for energies and forces. Therefore, just by adapting the training scheme, with no change in the model architecture or the dataset itself, consistent improvements in accuracy can be seen with meta-learning. The importance of the training method used has previously been seen in Ref. 45. Here we see how it can improve performance for fitting multiple datasets together. In comparison to when the ANI-1x model is used for pre-training, meta-learning performs slightly better at force errors but slightly worse for energy predictions. This implies that the gradients modeled with meta-learning can be readily adapted to new data sources. By contrast, shifting the potential energy surface to recreate energies may be more complex or data intensive. Given that the ANI-1x model is fit to the same level of theory as the 3BPA dataset, the performance of the meta-learning potential is encouraging.

The (a) 3BPA molecule, b the force error versus the number of structures used for the final training and c hydrogen bond dissociation for 3BPA with the frequency of C–H bonds in the 3BPA training set. The results shown in (b) demonstrate the difference in performance with varying pre-training approaches – with meta-learning producing the lowest energy results. The bond dissociation calculated using DFT is 119.6 kcal mol−1 and 111.2 kcal mol−1 with the bond dissociation energies estimator from Ref. 44. All other atoms are fixed with the removal of the hydrogen atom. The frequency of the C–H bond is for the 3BPA dataset used for re-training and the maximum length of the bond is 1.27Å.
However, it is known that RMSE errors alone are not enough to verify the performance of a potential36,46. We will therefore examine additional properties. The 3BPA molecule has three central dihedral angles which are illustrated in Fig. 6. The energy scans along these dihedral angles are shown in Fig. 7 with the model refit to the energies and forces of just 62 3BPA conformations. When no pre-training is used, the surface at β = 120 significantly over-estimates the high energy point and lacks smoothness. A similar shape is seen for the k = 1 potential. However, when meta-learning is used for pre-training the surface remains noticeably smoother with significantly less over prediction. When k = 1 is used, multiple different potential energy surfaces are combined together in a nonphysical way which destroys the smoothness of the underlying potential. The error in the gradient of the 2D energy surface is shown in Fig. 7b and emphasizes this difference in smoothness. When meta-learning is used, the contradiction in the potential energy surface described is corrected resulting in a smoother model. When no pre-training or k = 1 is used, an additional problem can occur with the high energy regions at α = 0 failing to be recreated for the β = 180 and β = 150 scan respectively. In contrast, both the meta-learning pre-training model correctly recreate this behavior. The results for ANI-1x pre-training are given in Supplementary Fig. 6.

a Torsional energy scans (of α and γ) for potentials with different pre-training approaches. The β angle is set to three different values. The potentials are re-trained to 62 structures from the 3BPA dataset. An ensemble of 8 models is used. b The gradient of the torsional energy surface with respect to α. The units are kcal mol−1/degrees.
One advantage of pre-training with multiple datasets over ANI-1x or QM7-x, is that reactive systems can be added that are not contained in ANI-1x. To test if this information has been effectively passed to the meta-learning potential, hydrogen bond dissociation for the 3BPA molecules was performed. There is no reactive information contained within the 3BPA training set and so this test relies entirely on the information contained in the pre-training.
Figure 6 shows the change in energy as a hydrogen molecule is removed from the 3BPA. The potential pre-trained with meta-learning recreates the smooth dissociation curve expected. In contrast, when no pre-training, k = 1 or ANI-1x is used the curve lacks smoothness and has an additional barrier present. In Supplementary Fig. 7, the bond dissociation energy when just 31 structures are used for retraining. Even in this low data limit the smooth dissociation curves for the meta-learning potential remain. To demonstrate that this is not unique to 3BPA, the hydrogen bond dissociation for ethanol is shown in Supplementary Fig. 8. Again, k = 1 fails to recreate the smooth curve expected whilst the meta-learning potential captures the correct shape.
We have therefore shown how meta-learning can be used to combine multiple datasets and the resulting improvements in the error, torsion energy scans and bond dissociation. Joint-fitting can improve on no-pre-training. However, not accounting for the difference in QM level of theory causes a reduction in performance that can be seen in the test set errors, smoothness of the potential and performance in extrapolation regions.
Discussion
The quantum mechanical properties of millions of molecular species and many materials systems have already been calculated and composed into extended datasets13,14,15,16,17,18. However, the varying levels of theory used to perform the QM calculations has previously prevented different datasets being used together to make machine learning models, for example for MLIPs. In this work, we have shown that meta-learning techniques can be used to jointly fit multiple datasets and demonstrated the improvement in performance that results from including a diverse selection of datasets.
We show the wide applicability of meta-learning by creating MLIPs for a variety of systems, from a single aspirin molecule to the ANI-1ccx dataset. By pre-training a model to multiple large organic molecule datasets we show that these datasets (QM7-x, QMugs, ANI-1x, Transition-1x and GEOM) can be combined together to pre-train a model. The benefits of using a pre-trained model are then shown for the 3BPA molecule, with a more accurate and smoother potential produced. Meta-learning greatly expands the variety of fitting data available for MLIPs and establishes the possibility of creating readily pre-trained, foundation models for MLIPs.
When datasets are built with different levels of quantum mechanical theory, disparities may exist between the potential energy surfaces described. The meta-learning approach enables training to broader, possibly contradictory datasets with the goal of building models that can be rapidly fine-tuned to new tasks, e.g., to higher levels of theory, or to specific problems such as studying a single molecule. By allowing parameters to differ in the optimization process, the potential energy surfaces for different levels of theory are allowed to vary. However, as a limited number of optimization steps are executed the parameter space is restricted and fundamental properties that are shared across datasets remain. As the NN parameters can vary, the contradictory information across datasets no longer causes problems associated with inconsistent data sources. Comparisons may also be drawn between meta-learning and regularization techniques. As meta-learning limits the optimization steps, the parameters are restrained to a region of parameter space. Therefore, meta-learning effectively acts as a form of regularization even though it is not explicitly described in the loss function.
Pre-training machine learning models has been extensively discussed in the machine learning literature in recent years47,48,49. Whilst pre-training has been carried out for MLIPs, its use has been limited to training from one dataset to another31,32,36. With techniques such as meta-learning, this pre-training does not need to be limited to one specific dataset but can include large numbers of existing datasets. In this work, we added only a single reactive dataset to pre-train a model. However, many different reactive datasets exist and combining this large amount of information could help build a general transferable potentials for reactions in both condensed and gas phase without the need for millions of new QM calculations. Additionally, datasets have been created for many different combinations of elements. Meta-learning techniques could help build more transferable MLIPs over a wider range of elements with fewer calculations required.
However, combining multiple datasets together and training with meta-learning will not always improve results. This was seen with the CCSD(T) results where fitting straight from ANI-1x to CCSD(T) resulted in the lowest error. Therefore, adding more data when there is a specific application in mind is not always the best approach, particularly if the additional data is far from the final application. For specific applications, transfer learning from one dataset to another may yield the best training and test set errors. However, if multiple data sets need to be incorporate together, or a general model is desired which can be specialized to multiple different tasks, meta-learning methods are preferable.
With the techniques described in this work, multiple datasets can be fit at once. However, this advancement has exposed a more practical problem with the datasets currently published. There is not a standard format for storing information. Manual manipulation of datasets to a standard format is extremely time-consuming. The need for uniformity in the structure of datasets produced is therefore becoming increasingly important.
The growth of available datasets containing quantum mechanical information for molecular and material structures has given researchers unprecedented levels of QM information. However, combining data from multiple data-sources is a major challenge. We have shown how meta-learning can be used to combine information from multiple datasets generated with varying levels of theory. This advancement changes the way that existing datasets should be viewed, and opens up new avenues for MLIP fitting. Beyond this, the results suggest that meta-learning can be seen as a general approach for combining training datasets for the broad array of chemical and materials processes where data science models can benefit.
Methods
Meta-learning algorithm
Meta-learning is an area of machine learning concerned with improving the learning process to produce models that can easily adapt to new problems37,38,39,40. A key component of meta-learning is the concept of different ‘tasks’. Tasks are datasets with similar properties but slight differences. For example, if we were interested in animal classification of a cat and a dog, a similar task might be to classify a lion and a bear. The task is not the same but we would expect fundamental similarities in the model needed to perform the classification. By using a meta-learning algorithm to learn multiple different tasks, less data will be required when a new learning problem is introduced.
The objective of meta-learning algorithms is to train a model that can generalize more easily to new data38,40. We will use meta-learning to fit multiple different QM datasets with slightly different properties. To our knowledge, meta-learning for MLIPs has not been previously carried out, although it has been used in other areas of science50,51,52.
The meta-learning algorithm we have chosen to fit multiple datasets for MLIPs is called Reptile39. Reptile works by repeatedly sampling a task (a dataset), performing a limited number of optimization steps on the task and then updating the weights of the machine learning model towards the new weights. Reptile was chosen over other meta-learning algorithms such as MAML37 as Reptile is simpler to implement and therefore more likely to be adopted by the wider community. A comparison of methods such as MAML for interatomic potentials will therefore be left to future work.
Reptile is described in Algorithm 2 with a visual illustration also given. The algorithm works by separating the training data into distinct learning problems (tasks). An individual task is selected and multiple optimization steps are performed. The parameters of the model are then updated. A new task is then selected and the procedure is repeated multiple times. This moves the model to a region of parameter space where it can readily move between the different datasets present. The parameters can vary between datasets in the Reptile algorithm. As such, inconsistencies between datasets can be overcome as the Reptile algorithm does not require the same functional form for every dataset.
Throughout this work, the k = 1 result is used as comparison point. This is because when k = 1 the algorithm becomes equivalent to stochastic gradient descent on the expected loss over all the training tasks39. This is referred to as joint training in Ref. 39 At k = 1, the algorithm is not expected to account for differences in the QM theory but still uses all the information present from the datasets.
Interatomic potential
In this work, we have used the NN architecture implemented in torchANI with the same structure as the ANI-1x model10,31. However, the meta-learning techniques described are not specific to this form of model and there is no reason that they could not be applied to other machine learning models that employ similar iterative solvers.
The hyperparameters used for the ANI potential are the same as those used for previous training to the ANI-1x and ANI-1ccx datasets, see Ref. 31 for more details.
Datasets
Aspirin
Aspirin structures were produced by molecular dynamic simulations at 300K, 600K and 900K. Density Functional based Tight Binding (DFTB) was used to perform the MD simulations and a total of 400 structures were created for each temperature. QM calculations of the energies and forces were then performed on these structures with three levels of theory: DFT with the ωB97x exchange-correlation function and 6-31G* basis set, DFT with Becke, 3-parameter, Lee-Yang-Parr (B3LYP) exchange-correlation functions and def2-TZVP basis set and Hartree-Fock with the def2-SVP basis set for 300K, 600K and 900K, respectively. These datasets were used to pre-train a molecular potential. The pre-trained potential was then refit to a new dataset of MD configuration at the Møller-Plesset (MP2) level of theory with the def2-SVP basis set (a more accurate level of theory). The training dataset for refitting used 400 MD configurations sampled at 300K whilst the test set contained structures at 300K,600K and 900K. A batch size of 8 was used for training.
QM9
The QM9 dataset contains over 100,000 equilibrium structures for small organic molecules with up to 9 heavy atoms53. In Ref. 13, the QM9 dataset was recalculated with 76 different exchange-correlation functionals and 3 basis sets13.
Multiple Organic Molecules
Seven separate datasets were chosen to fit a potential to organic molecule potential that could be easily re-trained to new data. The seven datasets used for meta-learning were chosen to cover both diverse regions of chemical space and multiples levels of theory – including the accurate recreation of dispersion effects. The chemical space covered included reactive paths and biologically and pharmacologically relevant structures. Whilst ANI-1x does cover a large number of conformations for organic molecules, it has limitations. This is demonstrated by Fig. 1 and Supplementary Fig. 1. Figure 1 demonstrates how the additional datasets increase the size of the molecules and range of energies included. The E0 energy is calculated using linear fitting an then subtracted from each dataset. The minimum energy for each dataset is then shifted to zero. Whilst it is not covered in this work as we use the ANI potential, including larger molecules in datasets may be increasingly important for newer generations of interatomic potentials that include message passing and describe longer length scales54,55. Figure S1 shows the distribution of uncertainty for the ANI-1x potential across the dataset space. Whilst ANI-1x dz, ANI-1x tz, GEOM and QMugs have similar probability distributions, QM7-x and Transition-1x contain larger uncertainties. Transition-1x contains reactive structures that are not contained in the original dataset and therefore higher uncertainties are expected. For QM7-x, there are also higher uncertainties and this may be due to the different sampling techniques used.
A property that is not shown in Table 1 is the software used for the DFT calculations. Even when the same level of theory is used, we can expect different software to give slightly different results. This will cause further discrepancies between the datasets as a variety of codes are employed. For example, although Transition-1x and ANI-1x are calculated at the same level of theory, Transition-1x is calculated with the ORCA program whilst ANI-1x is calculated with Gaussian56,57. Additionally, other inputs to QM calculations may cause further problems. For example, changing the convergence criteria of a calculation will alter the accuracy of the output and introduce additional noise to the result.
The individual description and justification for including each dataset used is as follows:
-
QM9 - This dataset contains a diverse range of 76 functionals and 3 basis sets for small equilibrium organic molecules13.
-
ANI-1x - This is a large dataset of small (up to 8 heavy atoms) organic molecules generated with active learning methods14.
-
QMugs - This dataset includes the largest molecules with up to 100 heavy atoms. It specializes in including drug-like molecules17
-
GEOM - This is the largest dataset and contains both large molecules and drug-like molecules15.
-
QM7-x - This is also a large dataset of small (up to 7 heavy atoms) organic molecules but has dispersion accurately described with many-body dispersion16.
-
Transition-1x - This datasets includes minimum energy paths for 12,000 reactions18.
-
ANI-1ccx - This dataset contains coupled cluster level theory calculations for a subset of the ANI-1x dataset14.
Other datasets considered for inclusion include SPICE, PubChemQC-PM6 and Tensormol58,59. However, with the existing datasets a sufficient representation of chemical space is covered. It is also worth noting that retraining to recreate the specific properties of the excluded datasets would also be quickly possible with the meta-learning potential.
Meta-learning hyperparameter optimization
There are three parameters in the Reptile algorithm. These control the number of steps (k) taken at each optimization step, how the parameters are updated (ϵ) from the task’s individual NN parameters and the maximum number of epochs used for retraining. The number of epochs was investigated to see whether restricting the training improved accuracy by ensuring the potential remained close to the meta-learned potential or if longer retraining improved results. For a detailed discussion of the hyper parameters chosen when fitting to the seven separate datasets, see Section S1.2. The ϵ value used throughout this work is ϵ = 1 whilst the k value is changed depending on the problem. The maximum number of epochs used for retraining for the meta-learning algorithm with k > 1 is restricted to 150 epochs.
Stages of fitting for the organic molecule datasets
In the first iteration, 100,000 structures were taken randomly from the ANI-1x, QMugs, GEOM, QM7-x and Transition-1x datasets. For QM9, 10,000 structures were used for each level of theory. This is restricted as 276 levels of theory exist, and each theory level samples different structures in the QM9 dataset. After the first iteration, the highest error structures were added to the next iteration60. The cutoffs used for adding structures are described in Supplementary Section 1.6. This process was repeated 3 times. A diagram of the process is show in Supplementary Fig. 3.
Data availability
The data used in this work is publically accessible alongside previously published works.
Code availability
The code required to perform the work shown will be made available of request.
References
Isayev, O. et al. Universal fragment descriptors for predicting properties of inorganic crystals. Nat. Commun. 8, 15679 (2017).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Mikulskis, P., Alexander, M. R. & Winkler, D. A. Toward interpretable machine learning models for materials discovery. Adv. Intell. Syst. 1, 1900045 (2019).
Pilania, G. Machine learning in materials science: from explainable predictions to autonomous design. Comput. Mater. Sci. 193, 110360 (2021).
Ouyang, R., Curtarolo, S., Ahmetcik, E., Scheffler, M. & Ghiringhelli, L. M. Sisso: a compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates. Phys. Rev. Mater. 2, 083802 (2018).
Jha, D. et al. Elemnet: deep learning the chemistry of materials from only elemental composition. Sci. Rep. 8, 17593 (2018).
Nandy, A. et al. Computational discovery of transition-metal complexes: from high-throughput screening to machine learning. Chem. Rev. 121, 9927–10000 (2021).
Keith, J. A. et al. Combining machine learning and computational chemistry for predictive insights into chemical systems. Chem. Rev. 121, 9816–9872 (2021).
Zhuo, Y., Mansouri Tehrani, A. & Brgoch, J. Predicting the band gaps of inorganic solids by machine learning. J. Phys. Chem. 9, 1668–1673 (2018).
Smith, J. S., Isayev, O. & Roitberg, A. E. ANI-1: an extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 8, 3192–3203 (2017).
Faber, F. A., Lindmaa, A., von Lilienfeld, O. A. & Armiento, R. Machine learning energies of 2 million elpasolite (ABC2D6) crystals. Phys. Rev. Lett. 117, 135502 (2016).
Magedov, S., Koh, C., Malone, W., Lubbers, N. & Nebgen, B. Bond order predictions using deep neural networks. J. Appl. Phys. 129, 064701 (2021).
Nandi, S., Vegge, T. & Bhowmik, A. Multixc-qm9: large dataset of molecular and reaction energies from multi-level quantum chemical methods. Sci. Data 10, 783 (2023).
Smith, J. S. et al. The ani-1ccx and ani-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci. Data 7, 134 (2020).
Axelrod, S. & Gómez-Bombarelli, R. GEOM, energy-annotated molecular conformations for property prediction and molecular generation. Sci. Data 9, 185 (2022).
Hoja, J. et al. Qm7-x, a comprehensive dataset of quantum-mechanical properties spanning the chemical space of small organic molecules. Sci. Data 8, 43 (2021).
Isert, C., Atz, K., Jiménez-Luna, J. & Schneider, G. Qmugs, quantum mechanical properties of drug-like molecules. Sci. Data 9, 273 (2022).
Schreiner, M., Bhowmik, A., Vegge, T., Busk, J. & Winther, O. Transition1x - a dataset for building generalizable reactive machine learning potentials. Sci. Data 9, 779 (2022).
Schütt, K. T., Arbabzadah, F., Chmiela, S., Müller, K.-R. & Tkatchenko, A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 8, 13890 (2017).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 134, 074106–14 (2011).
Unke, O. T. et al. Machine learning force fields. Chem. Rev. 121, 10142–10186 (2021).
Mueller, T., Hernandez, A. & Wang, C. Machine learning for interatomic potential models. J. Chem. Phys. 152, 050902 (2020).
Bartók, A. P., Kermode, J., Bernstein, N. & Csányi, G. Machine learning a general-purpose interatomic potential for silicon. Phys. Rev. X 8, 041048 (2018).
Noé, F., Tkatchenko, A., Müller, K.-R. & Clementi, C. Machine learning for molecular simulation. Ann. Rev. Phys. Chem. 71, 361–390 (2020).
Frenkel, D. & Smit, B. Understanding Molecular Simulation 2nd edn (Academic Press, Inc., USA, 2001).
Weiner, S. J. et al. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 106, 765–784 (1984).
Jorgensen, W. L. & Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110, 1657–1666 (1988).
Bartlett, R. J. & Musiał, M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 79, 291–352 (2007).
Tran, R. et al. The open catalyst 2022 (oc22) dataset and challenges for oxide electrocatalysts. ACS Catal. 13, 3066–3084 (2023).
Smith, J. S. et al. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 10, 2903 (2019).
Chen, M. S. et al. Data-efficient machine learning potentials from transfer learning of periodic correlated electronic structure methods: Liquid water at AFQMC, CCSD, and CCSD(T) accuracy. J. Chem. Theory Comput. 19, 4510–4519 (2023).
Taylor, M. E. & Stone, P. Transfer learning for reinforcement learning domains: a survey. J. Mach. Learn. Res. 10, 1633–1685 (2009).
Pan, S. J. & Yang, Q. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 22, 1345–1359 (2010).
Zaverkin, V., Holzmüller, D., Bonfirraro, L. & Kästner, J. Transfer learning for chemically accurate interatomic neural network potentials. Phys. Chem. Chem. Phys. 25, 5383–5396 (2023).
Kovács, D. P. et al. Linear atomic cluster expansion force fields for organic molecules: Beyond RMSE. J. Chem. Theory Comput. 17, 7696–7711 (2021).
Finn, C., Abbeel, P. & Levine, S. Model-agnostic meta-learning for fast adaptation of deep networks. In Proceedings of the 34th International Conference on Machine Learning, 1126–1135 (2017).
Hospedales, T., Antoniou, A., Micaelli, P. & Storkey, A. Meta-learning in neural networks: a survey. IEEE Trans. Pattern Anal. Mach. Intell. 44, 5149–5169 (2022).
Nichol, A., Achiam, J. & Schulman, J. On first-order meta-learning algorithms. Preprint at https://arxiv.org/abs/1803.02999 (2018).
Huisman, M., van Rijn, J. N. & Plaat, A. A survey of deep meta-learning. Artif. Intell. Rev. 54, 4483–4541 (2021).
Bommasani, R.et al. On the opportunities and risks of foundation models. Preprint at https://arxiv.org/abs/2108.07258 (2022).
OpenAI. GPT-4 technical report. Preprint at https://arxiv.org/abs/2303.08774 (2023).
Cole, D., Mones, L. & Csányi, G. A machine learning based intramolecular potential for a flexible organic molecule. Faraday Discuss. 224, 247–264 (2020).
St. John, P. C., Guan, Y., Kim, Y., Kim, S. & Paton, R. S. Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost. Nat. Commun. 11, 2328 (2020).
Shao, Y., Dietrich, F. M., Nettelblad, C. & Zhang, C. Training algorithm matters for the performance of neural network potential: A case study of adam and the kalman filter optimizers. J. Chem. Phys. 155, 204108 (2021).
Fu, X.et al. Forces are not enough: Benchmark and critical evaluation for machine learning force fields with molecular simulations., Transactions on Machine Learning Research, 2835–8856, (2023)
Han, X. et al. Pre-trained models: Past, present and future. AI Open 2, 225–250 (2021).
Hu*, W.et al. Strategies for pre-training graph neural networks. In International Conference on Learning Representations (2020). https://openreview.net/forum?id=HJlWWJSFDH.
Hendrycks, D., Lee, K. & Mazeika, M. Using pre-training can improve model robustness and uncertainty. In Proceedings of the 36th International Conference on Machine Learning, vol. 97, 2712–2721 (PMLR, 2019).
Sun, Y. et al. Fingerprinting diverse nanoporous materials for optimal hydrogen storage conditions using meta-learning. Sci. Adv. 7, eabg3983 (2021).
Nie, J., Wang, N., Li, J., Wang, K. & Wang, H. Meta-learning prediction of physical and chemical properties of magnetized water and fertilizer based on lstm. Plant Methods 17, 119 (2021).
Wang, J., Zheng, S., Chen, J. & Yang, Y. Meta learning for low-resource molecular optimization. J. Chem. Inf. 61, 1627–1636 (2021).
Ramakrishnan, R., Dral, P. O., Rupp, M. & von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, 140022 (2014).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022).
Lubbers, N., Smith, J. S. & Barros, K. Hierarchical modeling of molecular energies using a deep neural network. J. Chem. Phys. 148, (2018).
Neese, F., Wennmohs, F., Becker, U. & Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 152, 224108 (2020).
Frisch, M. J.et al. Gaussian 16 Revision C.01. (Gaussian Inc. Wallingford CT, 2016).
Nakata, M., Shimazaki, T., Hashimoto, M. & Maeda, T. Pubchemqc pm6: data sets of 221 million molecules with optimized molecular geometries and electronic properties. J. Chem. Inf. 60, 5891–5899 (2020).
Yao, K., Herr, J. E., Toth, D., Mckintyre, R. & Parkhill, J. The tensormol-0.1 model chemistry: a neural network augmented with long-range physics. Chem. Sci. 9, 2261–2269 (2018).
Smith, J. S., Nebgen, B., Lubbers, N., Isayev, O. & Roitberg, A. E. Less is more: sampling chemical space with active learning. J. Chem. Phys. 148, 241733 (2018).
Acknowledgements
This work was supported by the United States Department of Energy (US DOE), Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division under Triad National Security, LLC (‘Triad’) contract grant no. 89233218CNA000001 (FWP: LANLE3F2). A. E. A. Allen and S. Matin also acknowledge the Center for Nonlinear Studies. Computer time was provided by the CCS-7 Darwin cluster at LANL. LAUR-23-27568.
Author information
Authors and Affiliations
Contributions
Conceptualization (AA, NL, SM, JS, RM, ST, KB); Data curation (AA); Formal analysis (AA); Investigation (AA); Methodology (AEAA, NL, SM, JS, RM, ST, KB); Software (AA); Visualization (AA); Writing - original draft (AA); Writing - review and editing (AA, NL, SM, JS, RM, ST, KB). Supervision (NL, RM, KB).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Allen, A.E.A., Lubbers, N., Matin, S. et al. Learning together: Towards foundation models for machine learning interatomic potentials with meta-learning. npj Comput Mater 10, 154 (2024). https://doi.org/10.1038/s41524-024-01339-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-024-01339-x
- Springer Nature Limited