
Abstract
On the basis of first-principles calculations, we investigate the electronic and magnetic properties of 1T phase chromium sulfide halide CrXY (X = O, S, Se; Y = Cl, Br, I) monolayers in CrCl2 structure with the P\(\overline{3}\)m1 space group. Except for the CrOI monolayer, all CrXY monolayers are stable and ferromagnetic semiconductors. Our results show that the ferromagnetic coupling is dominated by the kinetic exchange between the empty eg-orbital of Cr atoms and the p-orbital of anions under the three-fold rotational symmetry. In this context, the coupling strength allows for being greatly enhanced by turning the nature of Cr–X bonds, i.e., increasing the covalent contribution of the bonds by minimizing the energy difference of the coupled orbitals. As we illustrate for the example of CrOY, the Curie temperature (Tc) is nearly tripled by substituting O by S/Se ion, eventually reaching the highest Tc in CrSeI monolayer (334 K). The high stabilities and Curie temperature manifest these monolayer ferromagnetic materials feasible for synthesis and applicable to 2D spintronic devices.
Similar content being viewed by others
Introduction
The discovery of monolayer magnetic material CrI31 opens the door to the development of two-dimensional (2D) spintronic devices aiming for ultralow energy consumption, ultrafast device operation, and ultrahigh density information storage2,3,4. For practical applications, it is highly desired to enhance the strength of magnetic exchange interaction such that the magnetic device can be operated at room temperature (300 K). However, up to now, the Curie temperatures of 2D ferromagnetic semiconductors reported in experiments are much lower than room temperature, e.g., the Tc of CrI3 and CrGeTe3 are 45 K1 and 20 K5, respectively. Thus, it remains a challenging task of searching 2D ferromagnetic semiconductors with high Tc6,7,8,9,10.
Generally, the magnetic cations in transition-metal semiconductors are separated by nonmagnetic anions. Magnetic couplings mainly involve kinetic exchange mechanism, or the so-called superexchange interaction, i.e., the magnetic exchange of d-electron-bearing cations arises from virtual hopping between the cation d-orbital and anion p-orbital. In crystals, the symmetry properties not only determine whether the virtual hopping is allowed, but also decide the sign of magnetic exchange interaction, i.e., being of ferromagnetic (FM) or antiferromagnetic (AFM) exchange11,12,13. According to the Goodenough–Kanamori–Anderson rule, when the interacting cation–anion–cation path makes an angle of 90°, the magnetic coupling of cations prefers FM exchange11,14,15. Besides, the strength of this virtual hopping is related to the covalent admixture amplitude of p–d bonds from perturbation theory \(\left\langle p\right\vert {{{{\boldsymbol{H}}}}}_{{\rm{crystal}}}\left\vert d\right\rangle /{{{\Delta }}}_{pd}\), where the Δpd is the energy difference of the p- and d-orbital level16. Thus, it suggests that the FM exchange interaction in semiconductors could be realized by carefully choosing the symmetry of the crystal field and its strength is associated with the admixture level of p- and d-orbital, i.e., the covalent character of p–d bonds.
In this work, based on first-principles calculations, we investigate a series of semiconducting ferromagnets, 1T phase chromium sulfide halide CrXY (X = O, S, Se; Y = Cl, Br, I) monolayer. Both the calculated phonon spectra and molecular dynamics (MD) simulations indicate that the CrXY monolayer is thermodynamically stable except for the CrOI monolayer. Given the nearly 90° Cr–X/Y–Cr interacting path and the three-fold rotational symmetry, the magnetic interaction between Cr ions favors FM exchange, which is confirmed by our energetic estimations of various magnetic configurations. Furthermore, our results show that the FM coupling is mainly contributed from the hybridization between the empty eg-orbital of Cr atoms and the p-orbital of anions. Using a simple Hamiltonian model, we reveal a general mechanism that FM coupling in CrXY monolayers can be significantly enhanced by reducing the energy difference between p- and d-orbitals, especially for p- and eg-orbitals. As a result, by minimizing the energy difference of the eg- and p-orbital level, which is achieved by substituting O by S/Se, the covalent character of the hybridization is greatly enhanced, yielding the increasement of the Tc from 49 K to 334 K (CrSeI).
Results and discussion
Geometrical configuration and stabilities
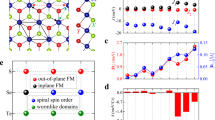
The 1T phase chromium sulfide halide CrXY monolayer is constructed by substituting one of the chalcogen atom (X) by a halogen atom (Y) in one atomic layer of CrX2, as shown in Fig. 1a. Each Cr atom is coordinated by three chalcogen atoms and three halogen atoms, resulting in a strongly distorted octahedron. The dynamic and thermal stabilities of the CrXY monolayer are studied by calculating phonon spectra and performing molecular dynamics (MD) simulations, respectively. In the calculated phonon spectra (Supplementary Fig. 1), no imaginary phonon mode is observed except for the CrOI monolayer, suggesting that the CrXY monolayers are dynamically stable. Hereafter, the CrOI monolayer will not be discussed in the following. To confirm the thermal stability, we perform MD simulations of the CrXY monolayers at 300 K for 20 ps. As revealed by MD snapshots (the inserts in Supplementary Fig. 2), the surface morphology is basically unchanged during the simulations, implying that those monolayers are thermally stable. Furthermore, we have investigated mechanical stability by calculating the elastic constants. Supplementary Table 1 shows that all CrXY monolayers satisfy the Born conditions17 (C11, C22, and C44 > 0, and C11C22 - C\({}_{21}^{2}\,\) > 0), suggesting that they are mechanically stable.

a 1T phase chromium sulfide halide CrXY monolayer crystal structure and the magnetic exchange paths. b Four different magnetic spin ordering configurations are involved. The blue, yellow and green balls represent the Cr atom, chalcogen atom (X) and halogen atom (Y), respectively.
Magnetic ground state
To address the magnetic ground state of CrXY monolayers, we compare the energies of several typical magnetic configurations, including non-collinear magnetic structure (FM, AFM-1, AFM-2 and AFM-3, shown in Fig. 1b)18. Table 1 shows that the energy of FM state in each system is lower than the energies of all AFM states. The net magnetic moment of all 2D CrXY monolayers is about 3μB/f.u., indicating a d3 electron configuration in high spin state. Furthermore, we calculate the total energy as a function of the magnetic propagation vector q = (qx, qy) by utilizing the generalized Bloch theorem (gBT)19. As shown in Fig. 2a and Supplementary Fig. 3, the minimum of the curve is located at q = (0, 0), indicating that the magnetic ground states of all 2D CrXY monolayers are FM state.

a Energy dependence on the magnetic propagation vector q, (b) density of state (DOS) of the CrSeI monolayer. The valence band maximum (VBM) energy is set to zero.
Figure 2b and Supplementary Fig. 4 show the calculated spin-dependent density of states (DOS) of the CrXY monolayers, with the coordinates based on the octahedron of CrX3Y3 (the inset of Fig. 2b). All CrXY monolayers are FM semiconductor, and Cr-3d orbitals split into the triplet orbitals t2g and doublet orbitals eg in the octahedral crystal field. Meanwhile, the lower t2g-orbitals are half occupied, and the eg-orbitals also have significant electron occupation, indicating a sizable interaction with the p-orbitals of the anion.
For realizing the long-range FM ordering in CrXY monolayers, the magnetic anisotropy is an essential prerequisite20. The magnetic anisotropy energy (MAE) is calculated by comparing different spin directions, the energies differences for different spin directions are shown in Supplementary Fig. 5. Except for CrSeCl and CrSeBr monolayers, the energies minimum of the spin direction are in the x–y plane, other CrXY monolayers are along the out-of-plane direction. All CrXY monolayers exhibit magnetic anisotropy, demonstrating that long-range FM ordering can emerge at certain temperatures.
Next, we calculate the magnetic exchange interaction parameters Jex by means of the four-state method21,22, which includes the first-nearest-neighbor (\({J}_{1}^{ex}\)), second-nearest-neighbor (\({J}_{2}^{ex}\)) and third-nearest-neighbor (\({J}_{3}^{ex}\)) (Fig. 1a). As shown in Table 2, the \({J}_{1}^{ex}\) is dominated and positive, which is crucial in determining the FM ordering. Besides, it is worth noting that when substituting O ion by S/Se ion the \({J}_{1}^{ex}\) is greatly enhanced. (Other U choices of 1, 1.5, 2, and 2.5 eV are also presently tested and qualitatively render the same results, as shown in Supplementary Table 2)
Ferromagnetic superexchange mechanism
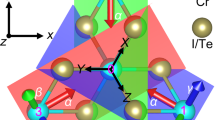
Given the angle of ∠Cr–X/Y–Cr being close to 90°, the superexchange interaction between Cr ions in CrXY monolayer prefers FM exchange11,14,15. Besides, because of the three-fold rotational symmetry, the three p-orbitals of anion are split into singlet orbital (l/√3)(px + py + pz) defining as pσ and doublet orbitals being of (l/√2)(px - py) and (1/√6)(pz - px - py), the coordinates referring to the coordinate axes parallel to the lines connecting the anion to the three neighboring cations. The singlet orbital pσ has a large overlap with the eg-orbitals of the cations, i.e., there is a sizable hybridization between the pσ- and eg-orbitals of all three surrounding X/Y ions, which can be inferred from the wave function depicted in Supplementary Fig. 6a. For each pair of Cr-X/Y, this hybridization leads to exchange polarization. If the spins of the three cations are all parallel to each other (FM ordering), the energy due to the exchange polarization would be the lowest, since the singlet orbital pσ overlaps them equally15.
To interpret the enhanced \({J}_{1}^{ex}\) when substituting O ion by S/Se ion, we consider a model to describe the superexchange interaction between two Cr ions via the anion in-between. For simplicity, this model involves one p-orbital of the anion, and two d-orbitals for each Cr ions, in which the lower orbital is half occupied and the higher one is empty referring to the t2g and eg orbitals, respectively. The Hamiltonian is given as:
where the first two terms \(\hat{H}\)M and \(\hat{H}\)L are the on-site energy terms for the transition metal and anion ligand, respectively, the third term \(\hat{H}\)U, describes the Coulomb repulsion between two electrons in d-orbital (the repulsion between electrons in the p-orbital is neglected), the last term \(\hat{H}\)t represents the hybridization between p- and d-orbitals and reads
where tpμ represent the hopping between p- and μ-orbitals, l and r represent the two transition-metal sites, μ is the d-orbital index (μ = α, β), c† and c are the creation and annihilation operators, respectively.
Next, we identify the possible p–d hopping channels (tpμ) by following the interatomic Slater-Koster integrals23. Given that the ∠Cr–X/Y–Cr is close to 90° and only one p-orbital is considered in the superexchange path, the d electrons in one Cr ion can only reach the other Cr via the eg-p-t2g hopping channel (Fig. 3a). While for the d electrons from the same orbital couple with each other via the t2g-p-t2g or the eg-p-eg hopping channel, there is no way that two consecutive hopping processes are possible, see Fig. 3b,c. Taking into consideration the t2g and eg orbitals being of the half occupied and the empty eg orbital, respectively, the exchange coupling mediated by the eg-p-t2g hopping channel is FM, and the AFM is forbidened by Pauli principle.

Superexchange channels between two cations via an intermediate anion: (a) eg-p-t2g, (b) t2g-p-t2g and (c) eg-p-eg. d Schematic diagrams of orbital evolution and superexchange interaction. Red and green bars represent t2g- and eg-orbitals, respectively. Δ1, Δ2 and ΔCF are the energy difference between eg- and p-orbitals, t2g- and p-orbitals and eg- and t2g-orbitals, respectively. Process I, II and III represent the electron hopping between p↑- and eg↑-orbitals, p↓- and t2g↓-orbitals and p↑- and t2g↑-orbitals, respectively. Δex is the exchange splits for Cr-d orbitals.
As shown in Fig. 3d, there are two fourth-order processes for the case of FM order on transition-metal cations: first with two electrons jumping away from the p-orbital of anion to eg- and t2g-orbitals, and two electrons coming back (i.e. the hopping process I and II); second with one electron from p-orbital hopping to eg-orbital on one side, then the electron from t2g-robital on the other side hopping to p-orbital, and two electrons coming back (i.e. the hopping process I and III). Then, the effective Hamiltonian for FM order is (for the details of derivation see the Supplementary materials)
where Δ1 = ε\({}_{{e}_{g}\uparrow }\)-εp↑ (the energy difference between eg- and p-orbitals), Δ2 = ε\({}_{{t}_{2g}\downarrow }\)-εp↓ (the energy difference between t2g- and p-orbitals) and ΔCF = ε\({}_{{e}_{g}\uparrow }\)-ε\({}_{{t}_{2g}\uparrow }\) (the energy difference between eg- and t2g-orbitals due to the crystal field (CF)), t1 and t2 represent the hopping between p- and eg-orbitals and p- and t2g-orbitals, respectively, and Ud is the Coulomb repulsion between two electrons in d-orbital.
In the case of AFM only second-order processes are possible (i.e. the hopping process I or II). And the effective Hamiltonian for AFM order is
According to Hex = HFM - HAFM = −JS1S2 (for Cr3+, S1 = S2 = 3/2), we arrive at
Since the energy difference terms (Δ1, Δ2, and ΔCF) are larger than 0, the \({J}_{1}^{ex}\) is positive, i.e., the superexchange is FM ordering, which is in line with our analysis of exchange polarization. The corresponding parameters of Δ and t can be further extracted by projecting the Kohn–Sham states on the atomic Wannier functions of Cr-3d and X/Y-p orbitals24. Compared with the minior changes of \({({t}_{1}{t}_{2})}^{2}\) as substituting the anion (X/Y) by the heavier congener (see Supplementary Table 3), there are sizable reductions of Δ1, Δ2 and ΔCF in the denominator of Eq. (5) [Fig. 4a, b], playing a decisive role in determining the strength of FM exchange interaction. Particularly, as replacing O by S (Se), the Δ1 changes from around 2 eV to around (less than) 1 eV, which accounts for the great enhancement of \({J}_{1}^{ex}\). Moreover, because the Δ1 and Δ2 of X atom are less than those of Y atom, we conclude that the superexchange path of Cr–X–Cr plays a major contribution to the spin coupling.

Energy difference between eg- and p-orbitals (Δ1), t2g- and p-orbitals (Δ2) and eg- and t2g-orbitals (Δ3) for (a) different chalcogen (X) and (b) halogen atoms (Y).
Given the smallest magnitude of Δ1 among the energy difference terms in the denominator (Figs. 3d, 4), we quantitatively investigate the evolution of \({J}_{1}^{ex}\) as a function of Δ1 with the onsite Coulomb repulsion Ud = 5 eV and the hopping integrals t1 = t2 = -0.5 eV. As shown in Fig. 5, \({J}_{1}^{ex}\) increases significantly as decreasing Δ1. However, when Δ1 is around 1 eV, the corresponding \({J}_{1}^{ex}\) reaches almost 30 meV, which is larger than that obtained from the DFT calculation (~11 meV). This discrepancy can be attributed to neglecting the interorbital Coulomb repulsion (\({U}_{d}^{{{{\rm{Inter}}}}}\)) when the eg and t2g orbitals are occupied at the same time, leading to the Δ1 in Eq. 5 being replaced by an effective \({{{\Delta }}}_{1}^{{{{\rm{eff}}}}} \sim {{{\Delta }}}_{1}+{U}_{d}^{{{{\rm{Inter}}}}}\). Thus, if we consider Δ1 > 1 eV, e.g. in the range of [1.5, 3.5], the corresponding \({J}_{1}^{ex}\) obtained from this model are in good agreement with the calculated results (inset in Fig. 5), indicating the rationality and validity of this mode. Besides, by plotting the differential charge density of the CrOCl, CrSCl and CrSeCl monolayers in Fig. 6, we can identify that the electronic density in CrSCl and CrSeCl is large in the interstitial region between Cr and S/Se ion, implying the increased covalent contribution in the Cr–S/Se bond with respect to that in Cr–O bond, which is a synergy of the variation in t and Δ. It should be also noticed that substituting Cl by the late halogen elements gives rise to relatively weak changes in the covalent character of Cr–Y bonds (see Supplementary Fig. 6b), yielding a limited effect on tuning the \({J}_{1}^{ex}\).

Evolution of \({J}_{1}^{ex}\) as a function of Δ1 with Δ2 = 5 eV, ΔCF = 2.5 eV, Ud = 5 eV.

Differential charge density of (a) CrOCl, (b) CrSCl and (c) CrSeCl monolayers, where yellow and cyan regions denote charge accumulation and depletion with the isosurface being set to 0.06 e/Å3. The red arrow indicates the electron in the center of the Cr–X bond.
Estimation of curie temperature
To determine the exact Curie temperature (Tc) in CrXY monolayers, we perform Monte Carlo simulations with the Metropolis algorithm based on the fully anisotropic Heisenberg model by mcsolver package25, using a 32 × 32 × 1 matrix, the Hamiltonian reads
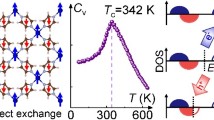
where spins are treated as classical vectors that can rotate and point to any space orientations. The Curie temperature can be accurately extracted from the peak of the specific heat capacity for the CrOCl and CrOBr monolayers as shown in Fig. 7a. In order to verify the reliability of this method, we have calculated the Tc of the CrI3 monolayer by taking the magnetic exchange parameters \({J}_{1}^{ex}\) and \({J}_{2}^{ex}\) from refs. 26,27,28, and our results are in good agreement with those references (see Supplementary Table S4). From the Fig. 7b, the predicted Curie temperatures of CrSY and CrSeY monolayers approach or exceed the room temperature, owing to the larger \({J}_{1}^{ex}\). And Tc can be further enhanced under approximate tensile strains, 10% tensile stress can further raise the Tc of the CrSeI monolayer to 409 K.

a Evolution of the Monte Carlo specific heat capacity of the CrOCl and CrOBr monolayers, the inset in (a) is the magnetic moment with the change of the temperature. b Curie temperature (Tc) in CrXY monolayers.
In summary, we discover a series of the 2D FM semiconductor CrXY (X = O, S, Se; Y = Cl, Br, I) in CrCl2 structure. Our study shows that the FM coupling is due to the hybridization between the empty eg-orbital of Cr atoms and the p-orbital of anions under the three-fold rotational symmetry. This mechanism allows for the strategy of strengthening the FM coupling by increasing the covalent hybridization contribution of the Cr–X bond in CrXY. As we unveiled in the CrSY and CrSeY monolayers, utilizing this strategy the Tc could be greatly enhanced to room temperature. This high Tc renders these CrXY monolayers feasible candidates for 2D ferromagnetic materials and spintronic devices.
Methods
First-principles calculations
The spin-polarized first-principles calculations have been performed based on the projector augmented wave (PAW) pseudopotentials29 implemented in the Vienna ab initio simulation package (VASP)30,31. The generalized gradient approximation in the Perdew–Burke–Ernzerh form32,33 is used for treating the exchange-correlation effect, and the GGA + U method34 is employed to properly describe the strong electron correlation effect of the Cr-3d orbitals (U = 3.0 eV)35,36,37. A cutoff energy of 500 eV is adopted for the plane wave expansion. The convergence criteria of geometrical optimization are 10−5 eV and 0.01 eV Å−1 for energy and force, respectively. For electronic self-consistent calculations, the energy and force convergence criteria are further decreased to 10−7 eV and 0.001 eV Å−1, respectively. To avoid interactions between two periodic units, a vacuum space of 15 Å is employed. A Monkhorst-Pack k-point mesh of 10 × 10 × 1 and 15 × 15 × 1 are utilized to the structural optimization and the calculation of the electronic and magnetic properties for the unit cells, respectively. The phonon dispersion is calculated using a 4 × 4 × 1 supercell with the finite displacement method by the PHONOPY software38,39. To investigate the thermal stability, we carry out isobaric-isothermal (NPT) ensemble molecular dynamics (MD) simulation at 300 K with the total simulation time of 20 ps and a time step of 2 fs by using Forcite code40.
Data availability
The data supporting the findings of this study are available within this article and its Supplementary Information. Additional data that support the findings of this study are available from the corresponding author on reasonable request.
References
Huang, B. et al. Layer-dependent ferromagnetism in a van der Waals crystal down to the monolayer limit. Nature 546, 270–273 (2017).
Wolf, S. A. et al. Spintronics: a spin-based electronics vision for the future. Science 294, 1488–1495 (2001).
Žutić, I., Fabian, J. & Sarma, S. D. Spintronics: fundamentals and applications. Rev. Mod. Phys. 76, 323 (2004).
Fert, A. Nobel lecture: origin, development, and future of spintronics. Rev. Mod. Phys. 80, 1517 (2008).
Gong, C. et al. Discovery of intrinsic ferromagnetism in two-dimensional van der waals crystals. Nature 546, 265–269 (2017).
Miao, N. H., Xu, B., Zhu, L. G., Zhou, J. & Sun, Z. M. 2D intrinsic ferromagnets from van der Waals antiferromagnets. J. Am. Chem. Soc. 140, 2417–2420 (2018).
Xiao, T. T., Wang, G. & Liao, Y. Theoretical prediction of two-dimensional CrOF sheet as a ferromagnetic semiconductor or a half-metal. Chem. Phys. 513, 182–187 (2018).
Jiao, J. Y. et al. 2D Magnetic Janus semiconductors with exotic structural and quantum-phase transitions. J. Phys. Chem. Lett. 10, 3922–3928 (2019).
Kabiraj, A., Kumar, M. & Mahapatra, S. High-throughput discovery of high Curie point two-dimensional ferromagnetic materials. J. Phys. Chem. Lett. 6, 35 (2020).
Muhammad, I., Ali, A., Zhou, L. G., Zhang, W. & Wong, P. K. J. Vacancy-engineered half-metallicity and magnetic anisotropy in CrSI semiconductor monolayer. J. Phys. Chem. Lett. 909, 164797 (2022).
Anderson, P. W. New approach to the theory of superexchange interactions. Phys. Rev. 115, 2 (1959).
Anderson, P. W. Theory of magnetic exchange interactions: exchange in insulators and semiconductors. Solid State Phys 14, 99–214 (1963).
Anderson, P. W., A Career in Theoretical Physics 113–130 (World Scientific, 1994).
Goodenough, J. B. An interpretation of the magnetic properties of the perovskite-type mixed crystals La1−xSrxCoO3−λ. J. Phys. Chem. Solids 6, 287–297 (1958).
Kanamori, J. Superexchange interaction and symmetry properties of electron orbitals. J. Phys. Chem. Solids 10, 87–98 (1959).
Sawatzky, G. A., Geertsma, W. & Haas, C. Magnetic interactions and covalency effects in mainly ionic compounds. J. Magn. Magn. Mater. 3, 37–45 (1976).
Wang, J. H., Yip, S., Phillpot, S. R. & Wolf, D. Crystal instabilities at finite strain. Phys. Rev. Lett. 71, 4182 (1993).
Ni, J. Y. et al. Giant biquadratic exchange in 2D magnets and its role in stabilizing ferromagnetism of NiCl2 monolayers. Phys. Rev. Lett. 127, 247204 (2021).
Sandratskii, L. M. Noncollinear magnetism in itinerant-electron systems: theory and applications. Adv. Phys. 47, 91–160 (1998).
Mermin, N. D. & Wagner, H. Absence of ferromagnetism or antiferromagnetism in one- or two-dimensional isotropic heisenberg models. Phys. Rev. Lett. 17, 1133 (1966).
Xiang, H. J., Lee, C., Koo, H. J., Gong, X. G. & Whangbo, M. H. Magnetic properties and energy-mapping analysis. Dalton Trans. 42, 823–853 (2013).
Šabani, D., Bacaksiz, C. & Milošević, M. V. Ab initio methodology for magnetic exchange parameters: generic four-state energy mapping onto a Heisenberg spin Hamiltonian. Phys. Rev. B 102, 014457 (2020).
Slater, J. C. & Koster, G. F. Simplified LCAO method for the periodic potential problem. Phys. Rev 94, 1498 (1954).
Pizzi, G. et al. Wannier90 as a community code: new features and applications. J. Phys. Condens. Matter 32, 165902 (2020).
Liu, L. et al. Magnetic switches via electric field in BN nanoribbons. Appl. Surf. Sci. 480, 300–307 (2019).
Lu, X. B., Fei, R. X. & Yang, L. Curie temperature of emerging two-dimensional magnetic structures. Phys. Rev. B 100, 205409 (2019).
Rehman, M. U. et al. Quantum anomalous Hall effect by coupling heavy atomic layers with CrI3. Phys. Rev. B 100, 195422 (2019).
Webster, L. & Yan, J. A. Strain-tunable magnetic anisotropy in monolayer CrCl3, CrBr3, and CrI3. Phys. Rev. B 98, 144411 (2018).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Furthmller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Perdew, J. P. et al. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 46, 6671 (1993).
Perdew, J. P. & Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 45, 13244 (1992).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA + U study. Phys. Rev. B 57, 1505 (1998).
Solovyev, L. V., Dederichs, P. H. & Anisimov, V. I. Corrected atomic limit in the local-density approximation and the electronic structure of d impurities in Rb. Phys. Rev. B 50, 16861 (1994).
Wang, L., Maxisch, T. & Ceder, G. Oxidation energies of transition metal oxides within the GGA + U framework. Phys. Rev. B 73, 195107 (2006).
Huang, C. X. et al. Toward intrinsic room-temperature ferromagnetism in two-dimensional semiconductors. J. Am. Chem. Soc. 140, 11519–11525 (2018).
Gonze, X. & Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 55, 10355 (1997).
Togo, A., Oba, F. & Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 78, 134106 (2008).
Rappe, A. K., Casewit, C. J., Colwell, K. S., Goddard III, W. A. & Skiff, W. M. Uff, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035 (1992).
Acknowledgements
The research is supported by the Science Challenge Project (Grant No. TZ2016003-1-105), the Tianjin Natural Science Fundation (Grant No. 20JCZDJC00750), and the Fundamental Research Funds for the Central Universities, Nankai University (Grant Nos. 63211107 and 63201182)
Author information
Authors and Affiliations
Contributions
H.-R.Z. performed the DFT calculations, compiled the figures and wrote the manuscript. H.-R.Z., B.S. and X.Z. conceived and analyzed Hamiltonian model. B.S. and X.Z. proposed and supervised the manuscript. All authors contributed to the analysis and discussion of the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41524_2023_1013_MOESM1_ESM.pdf
Supplementary information for: Enhancing ferromagnetic coupling in CrXY (X = O, S, Se; Y = Cl, Br, I) monolayers by turning the covalent character of Cr-X bonds
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, HR., Shao, B. & Zuo, X. Enhancing ferromagnetic coupling in CrXY (X = O, S, Se; Y = Cl, Br, I) monolayers by turning the covalent character of Cr-X bonds. npj Comput Mater 9, 56 (2023). https://doi.org/10.1038/s41524-023-01013-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-023-01013-8
- Springer Nature Limited