
Abstract
We develop a computationally efficient scheme to accurately determine finite-temperature band gaps for metal halide perovskites belonging to the class ABX3 (A = Rb, Cs; B = Ge, Sn, Pb; and X = F, Cl, Br, I). First, an initial estimate of the band gap is provided for the ideal crystalline structure through the use of a range-separated hybrid functional, in which the parameters are determined non-empirically from the electron density and the high-frequency dielectric constant. Next, we consider two kinds of band-gap corrections to account for spin-orbit coupling and thermal vibrations including zero-point motions. In particular, the latter effect is accounted for through the special displacement method, which consists in using a single distorted configuration obtained from the vibrational frequencies and eigenmodes, thereby avoiding lengthy molecular dynamics. The sequential consideration of both corrections systematically improves the band gaps, reaching a mean absolute error of 0.17 eV with respect to experimental values. The computational efficiency of our scheme stems from the fact that only a single calculation at the hybrid-functional level is required and that it is sufficient to evaluate the corrections at the semilocal level of theory. Our scheme is thus convenient for the screening of large databases of metal halide perovskites, including large-size systems.
Similar content being viewed by others

Introduction
Metal halide perovskite solar cells have shown remarkable progress in power conversion efficiency, which has been boosted up to 25% within only a few years1. Approximately 2000 perovskites2 can be synthesized but their suitability for photoelectric devices strongly depends on their electronic band gap. In particular, this property varies with the specific phase in which these materials occur, going from the highly symmetric cubic phase at high temperatures to structures of lower symmetry at lower temperatures. Given the large variety of available perovskites, it is important to develop a computational tool to screen large databases in search of an optimal material. Such a tool should provide band gaps at finite temperature in a computationally efficient way without compromising on accuracy. In the case of cubic halide perovskites that have small unit cells, Wiktor et al. have shown that it is possible to achieve an accurate description of the experimental band gaps by combining, on the one hand, high-level many-body perturbation theory for the electronic structure and, on the other hand, ab initio molecular dynamics to account for the finite temperature, the two schemes implying extensive computational resources of comparable order3. Next, Bischoff et al. further demonstrated that the accuracy by which the electronic structure is described could be preserved through the use of computationally more advantageous nonempirical hybrid functionals4. However, this development needs to be accompanied with a similar reduction of computational cost for treating the effects of finite temperature in order to deploy such methodology to a large set of perovskite materials in an efficient way.
Density functional theory5,6 based on semilocal approximations for the exchange and correlation energy represents a powerful tool for predicting ground state properties in materials sciences. However, it is well-known that such semilocal approximations generally underestimate the electronic band gap7. In contrast with this general notion, several previous studies focusing on perovskites have reported that the semilocal functional proposed by Perdew, Burke, and Ernzerhof (PBE) gives band gaps in good agreement with experimental data8,9,10. This agreement should be considered accidental because of the neglect of various effects3, such as spin-orbit coupling (SOC), nuclear quantum effects (NQEs), and thermal vibrations. Many-body perturbation theory in the GW approximation11 is currently recognized as the most accurate scheme for band-gap calculations, especially in its self-consistent quasiparticle formulation including vertex corrections in the screening12,13,14,15. However, such calculations are computationally highly demanding and cannot be envisaged as an efficient tool for screening large databases.
Hybrid functionals like the global PBE016,17 or the range-separated HSE18,19 are obtained by admixing a fraction of Fock exchange to the semilocal exchange potential and can serve as valuable alternatives to GW calculations. These hybrid functionals are defined through fixed mixing parameters, and thereby generally fail in providing accurate band gaps for a large class of materials20,21,22. It has been shown that higher accuracy can be achieved through material-specific dielectric-dependent hybrid (DDH) functionals14,23,24,25,26,27,28. Alkauskas et al.23 and Marques et al.24 linked the incorporated amount of Fock exchange to the inverse high-frequency dielectric constant 1/ϵ∞. Next, range-separated DDH functionals were developed in which 1/ϵ∞ determines the fraction of Fock exchange in the long range (LR), while retaining a suitable fraction of Fock exchange in the short range (SR)25,29,30. Very recently, further developments led to the suggestion of two kinds of nonempirical range-separated DDH functionals26,27. In the first one (denoted DD-RSH-CAM) the parameters are determined through the first-principles calculation of the dielectric screening function26, while in the second one (denoted DSH) the parameters are obtained through combining metallic and dielectric screening27. These two advanced DDH schemes have been shown to yield band gaps of accuracy comparable to state-of-the-art GW calculations for a large variety of materials26,27, but at a significantly lower computational cost.
To achieve high accuracy at finite temperature, it is further necessary to account for electron-phonon interactions, which have been found to significantly affect the band gap through the zero-point motions and the thermal vibrations3. Widely used supercell methods to estimate the electron-phonon interaction are based on the adiabatic approximation and include statistical sampling through Monte-Carlo31,32 and molecular dynamics methods3,33,34. Within the context of the adiabatic approximation, one also finds the so-called special displacement method (SDM), in which only one single optimal configuration of the atomic positions is sufficient to evaluate the band-gap renormalization due to NQEs and thermal vibrations35,36. For polar materials, Poncé et al. pointed out that the adiabatic approximation leads to a divergence at small momentum transfer37, but Zacharias and Giustino remarked that the observed divergency is not intrinsic to the adiabatic approximation36. In the SDM, the divergence is found not to build up35,36,38,39, because the periodic boundary conditions effectively short-circuit the long-range electric field associated with longitudinal optical phonons35. Recent reports show that it is necessary to resort to the nonadiabatic formulation in the evaluation of the zero-point phonon renormalization in the case of infrared-active materials37,40,41,42. Nonadiabatic effects are usually included through a generalized Fröhlich model40,42, which in its one-band formulation can also be used for an estimation of the correction39,41,43. Furthermore, the challenge of calculating band gaps of metal halide perovskites is exacerbated by other issues associated with thermal vibrations, such as anharmonic effects44, strong dependence of spin-orbit coupling3 and phonon renormalization38,41,45 on the adopted electronic-structure theory, and cross-coupling between spin-orbit coupling and phonon renormalization3,46.
In this work, we achieve accurate band-gap predictions for metal halide perovskites at finite temperature in a highly efficient way by combining dielectric-dependent hybrid functionals for the electronic structure with the special displacement method for the nuclear quantum effects and the thermal vibrations. To highlight the advantage of using dielectric-dependent hybrid functionals, our investigation also includes standard hybrid functionals, such as PBE0 and HSE06. Our best band-gap predictions are obtained by applying the following procedure. We start by calculating initial band-gap estimates with the dielectric-dependent hybrid functional for the pristine crystalline structure. Next, two corrections are applied. The first correction results from spin-orbit coupling effects. The second correction accounts for the nuclear quantum effects and the thermal vibrations. In this case, the special displacement method is applied and nonadiabatic effects are estimated through the one-band Fröhlich model. The band gap of the supercell structure with the special displacements is extracted through a procedure similar to the Tauc analysis of experimental spectra47. We demonstrate that both corrections can be determined at the semilocal level of theory, which does not lead to any significant loss of accuracy with respect to hybrid-functional results of higher level and thus only implies a moderate computational effort. The sequential inclusion of the two corrections leads to band gaps that progressively improve with respect to available experimental references, thereby confirming the validity of our approach.
This manuscript is organized as follows. In Results, we first describe the model structures and the calculation of the dielectric constants. We then report our band-gap predictions focusing on the corrections due to spin-orbit coupling and to electron-phonon interactions. The discussion is further developed in Discussion. In Methods, we briefly outline the main theoretical methodologies used in this work, i.e., the construction of the dielectric-dependent hybrid functional and the special displacement method. We also give the calculation details.
Results
Structural models
Our general motivation is to predict band gaps at room temperature for metal halide pervoskites. When various structures of a given material exist at room temperature, we take under consideration the available competitive structures to demonstrate the reliability of our approach upon structural variation. For benchmark purposes, we also consider materials at higher temperatures when a detailed experimental characterization is available.
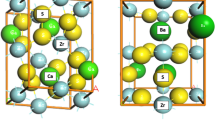
The materials under consideration in this work all have the structural formula ABX3, where A and B are metal atoms and X is a halide atom. These materials come in different structures identified by the prefix β, γ, δ, R, and M, which indicate that their phase is tetragonal, orthorhombic with corner-sharing octahedra, orthorhombic with edge-sharing octahedra, rhombohedral, and monoclinic, respectively. Within this group, all the structures with corner-sharing BX6 octahedra (R, γ, and β) are metal halide perovskites, while the other structures (δ and M) do not formally belong to this class. Representative atomic structures covering all space groups considered are illustrated in the Supplementary Fig. 1.
In this study, we do not consider cubic phases because they are generally unstable at 300 K, the targeted temperature for photovoltaic materials. The instability results from soft phonon modes46,48, which can be related to phase transitions48. This indicates that a harmonic description of the phonons would be inappropriate. Indeed, cubic structures are generally subject to important anharmonic effects44,49, which cause the excess free energy surface to change as a function of temperature49. The cubic phases at high temperature correspond to average structures, which differ noticeably from the local atomic-scale description3,49. For instance, the average B-X-B bond angle corresponds to 180∘ for the cubic structure, but in molecular dynamics simulations the distribution is peaked at a lower angle (~168∘, ref. 3). These structural changes depend on temperature and have noticeable effects on the band gap. Furthermore, the cross-coupling between SOC and thermal effect has been found to be sizable in cubic systems3,46. However, our test calculations on noncubic δ-CsPbI3 and R-CsPbF3 show that the error associated to such cross-coupling effects is ~0.1 eV (see the Supplementary Table 1). For these reasons, cubic structures would require a different theoretical treatment than the one proposed in this work.
Here, we make use of the experimentally determined structures given in Supplementary Table 2. These structures are used in the calculations of the high-frequency dielectric constant and of the band gap. Since these structures are taken from experiment, they already include the thermal expansion effect corresponding to the temperature at which their structure has been characterized (cf. discussion in ref. 3).
Dielectric constants
The high-frequency dielectric constants used in the DSH functional are calculated through finite electric fields50. For fixed atomic positions, we calculate the variation of the polarization ΔP as a function of the electric field (cf. Supplementary Fig. 2 for δ-CsPbI3). More specifically, \(\Delta P=P^{\prime} -{P}^{{{{\rm{0}}}}}\), where \(P^{\prime}\) and P0 are the polarizations with and without the applied electric field. The dependence is close to linear up to a critical value, above which the calculation no longer converges50. For all materials, we use in this work a value of 0.001 a.u. for the electric field and determine the high-frequency dielectric constant through
where V and Eϵ are the volume of the supercell and the electric field, respectively.
The dielectric constants ϵ∞ calculated at the PBE level are reported in Table 1. The values of the dielectric constants used in this work correspond to the average over the three principal directions of the dielectric tensor. In these perovskite materials, the dielectric constant in a given direction remains generally close to this average value (Supplementary Table 3). For instance, the material showing the largest spread is δ-RbGeI3, for which we found 4.93, 5.81, and 4.75, with an average of 5.16. Our calculated values show good agreement with previous PBE + U results from ref. 51, which have been integrated as benchmarks in the Materials Project52. However, there are only few benchmarks for materials belonging to M and γ phases. Therefore, to further validate our calculation of dielectric constants, we take γ-RbGeI3 and M-CsSnCl3 as representatives of theses phases and calculate ϵ∞ with the higher-level DSH functional that we derived. We obtain 5.09 and 3.15, which show differences of at most 7% compared to the plain PBE results of 5.10 and 3.38, respectively. This error is noticeably smaller than the typical mean absolute percentage error of 15% found for PBE results compared with experiment26. Furthermore, the effect of this error on the band gap is to a large extent attenuated because ϵ∞ enters through its inverse into the hybrid functional26. It has also been shown that the DSH functional with PBE values for ϵ∞ is generally more accurate for band-gap predictions than a DSH functional with self-consistently determined dielectric constants27,53. Hence, we take ϵ∞ values calculated at the PBE level to derive the parameter αLR = 1/ϵ∞ of the DSH functionals used in this work.
The inverse screening lengths μ of these materials vary between 0.67 and 0.82 bohr−1, with an average value of 0.73 bohr−1 (cf. Table 1). This value is close to the average result of 0.71 bohr−1 that Chen et al. found for a large set of materials26. To carry out a more detailed comparison, we consider in the Supplementary Table 4 the μ values for a selection of materials as obtained with three methods, namely Thomas-Fermi, DD-RSH-CAM, and DSH. We observe that DSH and DD-RSH-CAM give close values of μ for most materials, which result in similar band-gap estimates. At variance, the Thomas-Fermi values of μ generally differ more significantly, particularly for wide band-gap materials. These deviations generally lead to inaccuracies as far as the calculated band gaps are concerned. As an example, the reader is referred to the case of LiF discussed in the Supplementary Table 4.
Band gaps
The band structures of the class of materials considered in this work, both perovskites and non-perovskites, have been heavily investigated in the literature through a variety of electronic structure methods going from semilocal to GW methods54,55,56,57,58. The calculated band gaps typically vary by as much as a factor of two depending on the method. In our study, we first determine the fundamental band gap of the ternary ABX3 perovskites with PBE, PBE0(0.25), HSE06, and DSH (Table 2), including neither spin-orbit effects nor coupling to phonons. As reference, we also list in Table 2 band gaps calculated through high-level QSGW under the same assumptions54,59,60. As expected, the functional PBE strongly underestimates the band gaps showing the worst mean absolute error (MAE) of 1.30 eV and the worst mean absolute relative error (MARE) of 55%. The hybrid functional HSE06 opens the band gap with respect to the PBE value by 0.50 to 1.05 eV, and the hybrid functional PBE0(0.25) further opens the band gap by about 0.7 eV, in accord with general considerations61. We remark that the MAEs with respect to the accurate QSGW values progressively decrease when adopting in sequence PBE, HSE06, PBE0(0.25), and DSH (cf. Table 2 and Fig. 1). In particular, DSH yields a MAE of 0.22 eV and a MARE of 9%. To further support the quality of the DSH description, we perform for β-CsSnI3 a comparison between the DSH band structure calculated here and the GW band structure obtained by Huang and Lambrecht54. As shown in the Supplementary Fig. 3, the band gap occurring at the Z point is well reproduced and the overall trends of the band structure are very similar. This level of agreement between hybrid functionals and GW matches that found for other materials in the literature62.

We next analyze the general band-gap properties of the materials considered comparing different phases. As shown in Table 2, the R, γ, and β phases all have direct band gaps, while the δ phases show an indirect band gap. For the monoclinic phases, the band gaps can be either direct or indirect. However, due to the flatness of the bands54, the difference between the direct and the indirect bands gaps is generally not large. For instance, in the case of M-CsSnF3, we find an indirect band gap of 6.59 eV to be compared with the smallest direct band gap of 6.94 eV. Additionally, we remark that the δ phases generally have larger band gaps than the corresponding β or γ phases. This can be readily understood by their band structures. To illustrate this property, we calculate the band structures of β-CsSnI3, γ-CsSnI3, and δ-CsSnI3 with the PBE functional (cf. Supplementary Fig. 4). It is found that the valence bands of δ-CsSnI3 are much flatter bands than those of β-CsSnI3 and γ-CsSnI3, which can be related to the distance between nearest neighbor halogen atoms. This provides an explanation for the systematically wider band gaps found for the δ phase compared to the other phases.
Considering chemical variations within the same symmetry class, we first notice that the calculated band gap \({E}_{{{{\rm{bare}}}}}\) for all classes decreases for variations of the halide atom going from Cl to I while keeping the composition in terms of metal atoms unmodified. For instance, \({E}_{{{{\rm{bare}}}}}^{\gamma {{{\mbox{-}}}}{{{\text{CsPbCl}}}}_{3}} > {E}_{{{{\rm{bare}}}}}^{\gamma {{{\mbox{-}}}}{{{\text{CsPbBr}}}}_{3}} > {E}_{{{{\rm{bare}}}}}^{\gamma {{{\mbox{-}}}}{{{\text{CsPbI}}}}_{3}}\). This property can be understood invoking the electronegativity, which reduces from Cl to I. Since the character of the valence band is dominated by the halide atoms54,63,64, such a downward move in the periodic table results in an upward shift of the valence band and consequently in a narrower band gap. Second, we consider band-gap variations when keeping the A and X composition unmodified and varying the B atom. In this case, the band gap of Pb-based compounds is always larger than the corresponding Sn-based counterpart. This effect can also be attributed to a reduction of electronegativity going from Pb (2.33) to Sn (1.96). In the absence of any change associated with the halide atom, the smaller electronegativity of Sn leads to an upward shift for both the VBM and CBM64. However, Tao et al.64 found that the replacement of Pb with Sn results in a larger shift for the VBM than for the CBM because of their different ratios of s and p character. Therefore, smaller band gaps are found for Sn-based compounds. Since the electronegativity of Ge (2.01) falls in between those of Pb and Sn, we infer the following trend: \({E}_{{{{\rm{bare}}}}}^{\delta {\mbox{-}{{\text{RbPbI}}}}_{3}} \,>\, {E}_{{{{\rm{bare}}}}}^{\delta {\mbox{-}{{\text{RbGeI}}}}_{3}} \,>\, {E}_{{{{\rm{bare}}}}}^{\delta {\mbox{-}{{\text{RbSnI}}}}_{3}}\). Third, when changing the A atom, we do not have a uniform rule for the band-gap variation. Indeed, we find \({E}_{{{{\rm{bare}}}}}^{\delta {\mbox{-}{{\text{RbPbI}}}}_{3}} <\, {E}_{{{{\rm{bare}}}}}^{\delta {\mbox{-}{{\text{CsPbI}}}}_{3}}\), but we also find \({E}_{{{{\rm{bare}}}}}^{\delta {\mbox{-}{{\text{RbSnI}}}}_{3}} \,>\, {E}_{{{{\rm{bare}}}}}^{\delta {\mbox{-}{{\text{CsSnI}}}}_{3}}\). This effect can be related to the fact that the A-atom states lie far away from the VBM and CBM, and thus do not influence their energy levels directly54,63,64.
In the following, the calculated band gaps are benchmarked with respect to nine materials for which a careful experimental characterization of the band gap is available. This experimental set of ABX3 materials comprises eight perovskites and one non-perovskite. The B site is occupied by Ge, Sn, and Pb atoms, while the X site is occupied by Cl, Br, and I atoms. The A site is solely occupied by Cs atoms, but this site is known not to affect to band gap in a significant way54,63,64. The band gaps covered by this experimental set of materials go from 1.25 to 4.46 eV, which is the relevant range for photovoltaic materials. Overall, these materials offer a diversified set to benchmark our method.
Many of the materials considered contain heavy elements, which are expected to give significant SOC effects. To estimate SOC corrections, we consider three different functionals: PBE, PBE0(0.25), and DSH. The band-gap variations are given in Table 3. We observe similar variations for the three functionals studied. The largest differences between PBE and DSH are found for R-CsPbF3 and γ-CsPbF3 and amount to only 0.10 and 0.09 eV. In a study comprising 103 materials but not including perovskites, Huhn et al. also found that the SOC corrections calculated with PBE and HSE06 are overall very similar, with most differences falling below 60 meV and the most notable one being 189 meV65. However, we remark that Wiktor et al. found that the SOC corrections on the band gap are underestimated by as much as 0.3 eV for Pb-based cubic phases3, indicating that SOC effects need careful evaluation for high-symmetry structures containing Pb atoms. Hence, we conclude that the PBE functional is sufficiently accurate to estimate SOC corrections for all considered materials in this work, none of which shows the cubic symmetry.
The calculated SOC corrections can be found in Fig. 2(a) and Table 4. These corrections always lead to a band-gap reduction with respect to \({E}_{{{{\rm{bare}}}}}\). The largest corrections correspond to Pb-based compounds and reach values around −1 eV for γ-CsPbCl3 and γ-CsPbBr3. The largest correction for a Sn-based compound amounts to ~−0.4 eV and is found for γ-CsSnI3. We remark that the SOC corrections do not only depend on composition but also on the underlying atomic structure. Indeed, in the case of CsPbBr3, we find −0.37 and −1.02 eV for the δ and γ structure, respectively. This indicates that SOC effects may be large and highly nontrivial.

Partial band-gap corrections a ΔESOC and b ΔEZPR+T, together with c the full band gap correction ΔEfull = ΔESOC + ΔEZPR+T for all the materials in this work.
We then obtain the band gaps ESOC by adding the SOC corrections to \({E}_{{{{\rm{bare}}}}}\) (cf. Table 4). For the subset of materials for which experimental estimates are available, the accuracy of the corrected band gaps improves significantly, with the MAE reducing from 0.57 to 0.26 eV. Correspondingly, the MARE drops from 27% to 11%. These errors clearly illustrate the importance of considering SOC effects for achieving accurate band gaps.
Next, we focus on the coupling to phonons, which affect the band gap through ZPR and thermal vibrations. For the ZPR and the vibrational properties, we use experimental lattice constants at the temperatures given in the Supplementary Table 2. In this way, thermal expansion effects are already accounted for. For room temperature, we take T = 300 K in this work. All the modes of the structures considered in this work show positive frequencies.
To obtain the band-gap renormalization caused by zero-point motion and thermal vibrations, we use the special displacement method35 described in the Methods section. The supercell sizes adopted in this work contain between 160 and 625 atoms (cf. Supplementary Table 5) and have been chosen in order to achieve accuracies better than 0.05 eV. We checked this explicitly for all selected supercells by performing the same calculation with smaller supercells, leading to differences smaller than 0.05 eV for the band-gap predictions.
The SDM does not account for the long-range Fröhlich coupling resulting from the nonadiabatic treatment36,37,39,40,41,42,43. To obtain an estimate for this contribution, we use the one-band Fröhlich model (see Supplementary Note 1)43. For a series of perovskites belonging to the class of materials under investigation in our work, we obtain estimates ranging between −0.05 and −0.13 eV, with a mean value of −0.09 eV (see Supplementary Table 6). As will be seen in the following, the error resulting from the neglect of this effect is smaller than the final MAE claimed in our work (0.17 eV). Hence, it can be concluded that the long-range Fröhlich coupling coming from the nonadiabatic treatment of the electron-phonon interaction is not susceptible to affect in a significant way the accuracy of our results for metal halide perovskite materials.
The SDM provides supercells with distorted atomic structures, which we use to extract the band gap. More specifically, these disordered supercells simulating the effect of finite temperature cannot be characterised with the same Brillouin zone as the ideal material, because of the lack of translational symmetry. To conform with the practice in the experimental determination of optical band gaps at finite temperature47, we, therefore, rely on extrapolations of the wings in the electronic density of states (EDOS) to obtain the VBM and CBM. For simplicity, we here perform a linear extrapolation of the band wings34,66.
In order to perform reliable extrapolations of the band wings, the calculated eigenvalues in the vicinity of the band edges should be sufficiently dense. This condition is not guaranteed for all materials when using the cp2k code, which solely uses the Γ point. In some cases, the states in vicinity of the band edge are sparsely distributed and do not connect with the band wing upon reasonable Gaussian smearing. It is then necessary to use denser k-point meshes or alternatively larger supercells. We illustrate this issue for an ideal structure of γ-CsSnI3 in Fig. 3(a). First, we apply the code vasp67,68 to the primitive unit cell and establish a converged benchmark by increasing the k-point sampling of the Brillouin zone (cf. Supplementary Fig. 5). Then, we compare the converged reference shown in Fig. 3(a) with a cp2k calculation obtained with a 6 × 4 × 6 supercell and find good agreement for the band gap within less than 0.005 eV. Hence, the band gap can be obtained accurately provided a sufficiently large supercell is used. In the Supplementary Table 5, we give suitable supercell sizes to obtain converged band gaps for the materials considered in this work. Once a sufficiently large supercell is ensured, the EDOS is determined for the ideal structure and for two displaced structures accounting for ZPR and ZPR+T, as illustrated for M-RbSnF3 in Fig. 3(b). The linear extrapolation of the band wings then allows us to extract the VBM [Fig. 3(c)] and the CBM [Fig. 3(d)]. Such an analysis is performed for each material (cf. Supplementary Figs. 6 and 7).

a EDOS calculated within PBE with vasp for a primitive cell of γ-CsSnI3 using a k-point mesh of 16 × 16 × 16 and a Gaussian smearing of σ = 0.05 eV, compared with a 6 × 4 × 6 supercell calculation with cp2k using the Γ point and σ = 0.15 eV. b EDOS for the ideal structure and for the displaced structures accounting for ZPR and ZPR+T (T = 300 K) in the case of M-RbSnF3 (σ = 0.15, PBE). c and d show the valence and conduction edges, respectively. Dashed lines indicate the linear extrapolations used to determine the VBM and CBM. The extracted band edges are given for the cases defined by the color code in b.
The use of hybrid functionals for the EDOS with a dense k-point mesh or with a large supercell would undermine the efficiency of our band-gap determinations. There are reports in the literature with ZPR calculations showing a dependence on the adopted electronic-structure method38,41,45. Variations are particularly important for silicon45 and diamond38 and might amount up to ~0.25 eV. For other materials like MgO and LiF, the variation between functionals is much smaller (~0.05 eV)41. Since it is difficult to develop a rationale for understanding the size of this dependence, we explicitly investigate whether thermal corrections for perovskite materials can be accurately evaluated with the semilocal PBE functional. For this, we select M-RbSnF3 and M-CsSnF3, which show noticeable differences between the band gaps calculated with PBE0(0.25) and DSH (cf. Table 2). We check the effect of the functional by extracting the ZPR+T band-gap corrections from the EDOS obtained with PBE, PBE0(0.25), and PBE0(0.40) (see Table 5 and Supplementary Fig. 8). The functional PBE0(0.40) can be taken as representative of DSH for M-RbSnF3 and M-CsSnF3, since the bare band gaps calculated with these two functionals differ by less than 0.08 eV. Additionally, we use PBE and PBE0(0.25) to test R-CsGeCl3, which shows the largest band gap renormalization due to ZPR + T in our work, i.e., of 0.4 eV (cf. Table 6). In all cases studied, we find that the corrections ΔEZPR+T obtained with PBE coincide with those obtained with the hybrid functionals within at most 0.03 eV. Thus, this justifies the use of the PBE functional for estimating the effect of the band-gap corrections due to ZPR and thermal vibrations in the case of perovskite materials.
The band-gap renormalizations due to zero-point motion and thermal vibrations are given in Table 6 and are illustrated in Fig. 2(b). In particular, we also give the corrections at T = 0 K corresponding to the ZPR. The compounds containing the light F atoms show the most prominent ZPR effects, with the largest one found for M-RbSnF3 and amounting to −0.20 eV. The next group of compounds showing sizable ZPRs are those containing Cl atoms, among which R-CsGeCl3 shows the most significant value of −0.14 eV. Increasing the temperature from 0 to 300 K produces further band-gap renormalization. Together with the ZPR, this leads to ΔEZPR+T corrections ranging between −0.40 and 0.23 eV (Table 6), which cannot be neglected when aiming at high accuracies. We account for the combined effect of SOC and phonons by determining the two band-gap renormalizations separately, as these effects have been found to give a negligible cross coupling (see Supplementary Table 1). Adding ΔEZPR+T to ESOC, we then obtain the band gap estimates Etheory, which further improve the comparison with experiment leading to a MAE of 0.17 eV and a MARE of 6% (cf. Table 4).
The band-gap renormalization due to thermal vibrations can be compared with previous work in the literature. The band-gap variation of γ-CsPbBr3 between 0 and 300 K has been investigated through constant-volume molecular dynamics simulations69 yielding a shift of 0.01 eV in good agreement with our estimate of ΔET = 0.02 eV (cf. Table 6). Moreover, for large-size nanocrystals of γ-CsPbI3, temperature-dependent luminescence spectra show that the observed band gap remains invariant with temperature70. This behavior stems from the combined effect of lattice expansion and phonon renormalization. To establish a connection with our work, we estimate the correction due to lattice expansion using the experimental thermal lattice-expansion coefficient of 0.39 × 10−4 K−171. We find a band-gap increase of 0.07 eV over a thermal range of 300 K, which almost perfectly compensates the corresponding band-gap reduction of 0.06 eV due to thermal vibrations (cf. ΔET in Table 6). Hence, our correction is fully consistent with the null result observed in the experiment70.
Our method relies on the harmonic approximation for describing the vibrational properties of the investigated materials. Patrick et al. performed a careful study including anharmonic effects in both cubic and noncubic CsSnI344. To evaluate possible contributions coming from anharmonic effects, we applied our harmonic scheme to the noncubic β-CsSnI3 and γ-CsSnI3 at 380 and 300 K, respectively, finding band-gap renormalizations of 0.23 and 0.11 eV due to ZPR + T. These values are very close to the respective results of 0.16 and 0.11 eV found by Patrick et al. including anharmonic effects. This suggests that anharmonic effects only lead to small corrections for noncubic perovskites. However, we emphasize that anharmonic effects can be larger in the cubic phases because the structure then undergoes noticeable distortion moving away from the highly symmetric structure, which only holds on average49.
The correction ΔEZPR+T can either increase or reduce the band gap. Most materials in our selection exhibit the conventional Varshni effect, i.e., their band gap decreases with increasing temperature (red shift)72. The ZPR and the band gap reduction due to thermal vibrations then contribute in the same direction with corrections being as significant as −0.40 eV in the case of R-CsGeCl3. At variance, some materials, most particularly the Sn-based γ and β phases, show a band gap opening with increasing temperature (blue shift). This unusual effect is referred to as anomalous band gap shift and has experimentally been observed for a variety of hybrid organic-inorganic lead halide perovskites73.
In the case of structures composed of octahedral units, the origin of the band-gap renormalizations can be understood in terms of B-X bond-length and B-X-B bond-angle variations (see Supplementary Table 7). Indeed, it is well-known that reducing the B-X bond lengths or the B-X-B bond angles generally leads to lower band gaps due to enhanced orbital coupling74,75,76. Practically all octahedral structures in our selection show reduced bond angles and increased bond lengths when going from 0 to 300 K (cf. Supplementary Table 7). These trends oppositely impact the band gap with a combined effect that can either lead to band-gap opening or to band-gap closure. More specifically, for the rhombohedral structures, the effect due to the large bond-angle reductions (5∘–10∘) apparently dominates, leading to overall band-gap closing. The larger the bond-angle reduction, the lower the band gap. For γ phases, the bond-angle reductions are generally smaller (1∘–4∘) and their effect is similar to the effect due to the bond-length increases, resulting together in small band-gap variations, either red-shifted or blue-shifted. The structural variations of the δ phases are generally less significant, with bond-angle reductions and bond-length increases amounting to at most 1∘ and 0.02 Å, respectively. Nevertheless, their combined effect still yields quite sizable band-gap reductions, the largest being 0.33 eV in the case of δ-CsPbBr3.
Discussion
In Fig. 4, we show how the calculated corrections progressively improve the theoretical band gaps. We here consider all the materials for which experimental band gap values are available. The corrections due to SOC and ZPR + T are added in sequence to the bare band gaps calculated with the DSH hybrid functional. Moreover, our best estimates for the fundamental band gaps Etheory are compared with optical band gaps from experiment. We hereby neglect excitonic effects, which are estimated to be on the order of 10 to 100 meV for this class of materials77,78,79,80. For instance, photoluminescence experiments on γ-CsSnI3 suggest a binding energy of 18 meV78. Calculations based on the Bethe-Salpeter equation also give very small exciton binding energies for CsGeX3 (X = Cl, Br, I), on the order of at most ~1 meV80.

Band gaps obtained with the DSH functional \({E}_{{{{\rm{bare}}}}}\) (yellow triangles), after SOC correction ESOC (purple circles), and additionally including ZPR and thermal effects Etheory (green hexagons). The MAEs and MAREs are given with respect to the experimental values (red squares).
The residual deviations in Fig. 4 between our theoretical estimates and the experimental values should be assigned to the intrinsic limitations of the adopted theory. The fact that the errors are not systematic reinforces this point of view. In fact, neglected effects such as exciton binding energies, polar corrections due to LO phonon coupling, anharmonic vibrational effects, errors resulting from the use of PBE for SOC and phonon renormalizations, and cross-coupling between SOC and temperature effects, have all been demonstrated to affect the results to a lower extent than the residual error. Residual deviations, such as encountered for R-CsGeBr3, γ-CsPbCI3, and M-CsSnCl3 should therefore not be attributed to one particular effect. It is likely that the largest residual errors ranging up to 0.49 eV for an individual perovskite results from limitations of the adopted electronic-structure method. It should be remarked that even state-of-the-art GW calculations show non-systematic behavior with individual errors ranging up to 0.44 eV15.
As seen in Fig. 4, the agreement with experiment systematically improves when applying corrections due to ΔESOC and ΔEZPR+T in sequence. On average, ΔESOC and ΔEZPR+T amount to −0.36 and −0.15 eV, respectively, corresponding to an average global correction Efull of 0.51 eV (cf. Fig. 2). The MAEs of \({E}_{{{{\rm{bare}}}}}\), ESOC, and Etheory are 0.57, 0.26, and 0.17 eV with respect to experiment, with respective MAREs of 27%, 11%, and 6% (see also Table 4). The finally achieved accuracy of 0.17 eV (6%) informs us that the applied corrections are essential to achieve this level of agreement with experiment, since they are of the same size or larger on average. Furthermore, the achieved accuracy is comparable to that of state-of-the-art GW calculations for extended sets of materials12,13,15,81.
To support the use of the material-specific functional DSH, we carry out a comparison starting from standard functionals, such as PBE, HSE06, and PBE0(0.25). As discussed, our band-gap corrections can all accurately be calculated using the PBE functional. The use of a different functional then only affects the calculation of the bare band gap. The detailed results of these calculations are given in the Supplementary Tables 8, 9, and 10 for PBE, HSE06, and PBE0(0.25), respectively, and can be directly compared with the DSH results given in Table 4. To assess the performance of all the investigated functionals, we have summarized in Table 7 their respective MAEs and MAREs for the calculated band gaps with respect to the experimental values. We notice that for the PBE and HSE06 functionals the agreement with experimental band gaps steadily deteriorates upon including the corrections. In particular, the \({E}_{{{{\rm{bare}}}}}\) from HSE06 already shows relatively low MAE and MARE of 0.36 eV and 14%, respectively. However, this result should be considered accidental and due to error cancellation. The results in Table 7 also indicate that the performance of PBE0(0.25) for the materials investigated is rather good (MAE of 0.28 eV; MARE of 11%) showing systematic improvement upon the inclusion of the corrections. As seen in Table 7, DSH outperforms all the other functionals with a MAE of 0.17 eV and a MARE of 6%. We stress that the best agreement between theory and experiment is obtained when all the various contributing physical effects are properly estimated.
In hindsight, it is of interest to question whether the approach developed is indeed superior to simple band-gap predictions made on the basis of trivial PBE calculations. To address this issue, we develop a simple model based on the PBE band gap and test it on the nine materials for which experimental band gaps are available. From Supplementary Fig. 9, one infers that the relation between PBE and experimental band gaps is approximately linear. To obtain a model prediction, we then fit the data with a linear function: \({E}_{{{{\rm{gap}}}}}^{{{{\rm{model}}}}}=1.39\cdot {E}_{{{{\rm{gap}}}}}^{{{{\rm{PBE}}}}}+0.47\) eV. Such a model prediction yields a MAE of 0.30 eV with a maximum error as high as 0.92 eV. These errors correspond to the data fitted and are susceptible to increase when a larger set of materials is taken under consideration. Nevertheless, the MAE value and the maximum error of this simple model are already noticeably worse than the MAE of 0.17 eV and maximal error of 0.49 eV found with the scheme introduced in this work. This provides further support to the validity of our scheme.
We remark that the present methodology has here successfully been applied to the large class of noncubic metal halide perovskite materials. To validate this methodology to this class of materials, we verified that the consideration of anharmonicity, cross-coupling between SOC and phonon effects, and nonadiabatic effects in phonon renormalization due to long-range polar interactions, all lead to negligible contributions to the band gap. Thus, it should be understood that the present methodology can reach the high level of accuracy found in our work only to extent that these conditions hold.
To summarize, we have investigated the fundamental band gaps for a set of inorganic halide perovskites through the use of various functionals. To ensure a meaningful comparison with experiment, we have included band-gap corrections due to spin-orbit coupling and to thermal vibrations including zero-point motions. In particular, we use the dielectric-dependent hybrid functional DSH with parameters fixed through the dielectric function. Among the functionals considered, the functional DSH stands out providing a high level of accuracy compared to experimental band gaps (MAE = 0.17 eV; \({{{\rm{MARE}}}}=6\)%). The achieved accuracy relies on the consideration of both corrections, which lead to a step-by-step improvement of the agreement with experiment. Our final accuracy is comparable to the state of the art as far as band-gap estimates are concerned.
Additionally, our scheme is designed to be computationally efficient. The band gap calculations with the hybrid functional are performed only once for a single crystalline structure for each material. We demonstrate that both band-gap corrections due to spin-orbit coupling and to thermal vibrations including zero-point motions can accurately be determined with the computationally more convenient semilocal PBE functional. In particular, the effects due to zero-point motions and thermal vibrations are estimated through the special displacement method, which gives the band-gap correction through a one-shot calculation, thereby speeding up the effort with respect to more lengthy molecular dynamics simulations. Consequently, our scheme is not only highly accurate but also provides band gaps in a computationally efficient way. Thus, these features make our approach convenient for material screening procedures82,83,84 in which large databases of metal halide perovskites are taken under consideration.
Methods
Range-separated dielectric-dependent hybrid functional
We use a range-separated hybrid functional formulation in which the parameters are determined from the dielectric response of the system. In such a scheme, the Coulomb potential is partitioned into a short-range (SR) and a long-range (LR) component through an error function18,25,26,27,53,85:
where μ is the range-separation parameter. Next, the SR and LR components appearing in the nonlocal exchange potential are separately decomposed into nonlocal Fock and semilocal PBE86 exchange terms through admixtures defined by αSR and αLR:
Numerous widely used density functionals can be recovered from Eq. (3). For example, irrespective of the choice of μ, the semilocal functional PBE (ref. 86) is found for αSR = αLR = 0, whereas the global hybrid functional PBE0 (ref. 16) can be obtained by setting αSR = αLR = 0.25. Similarly, the two recently proposed DDH functionals, i.e., DD-RSH-CAM26 and DSH27, also belong to this class of functionals and can be found by setting αSR = 1 and αLR = 1/ϵ∞. However, these two DDH schemes differ in the way the parameter μ is set. In DD-RSH-CAM, μ is derived from a fit to the dielectric function calculated through first-principles linear response4,26, while in DSH this parameter is approximated through an empirical expression that can be evaluated analytically27. As we show above, the two proposed values of μ are nevertheless rather close (see Supplementary Table 4). Therefore, for speeding up the computational effort, we adopt in this work the dielectric-dependent functional denoted DSH in ref. 27 with the empirical expression for μ proposed therein.
More specifically, the empirical expression for μ proposed in ref. 27 stems from the nonlocal screened Coulomb potential derived by Shimazaki and Asai87:
Here, \({\tilde{k}}_{{{{\rm{TF}}}}}^{2}={k}_{{{{\rm{TF}}}}}^{2}\left[{\left({\epsilon }_{\infty }-1\right)}^{-1}+1\right]/\alpha\) and \({k}_{{{{\rm{TF}}}}}=2\root 6 \of {(3n/\pi )}\) is the Thomas-Fermi screening length, which depends on the valence charge density n. Following refs. 25,27, we include the d electrons in the valence density when the valence band is dominated by d orbitals, namely for Ge, Sn, Pb, Br, and I. The coefficient α = 1.563 is considered to be independent of material88,89. Equation (4) can be well approximated by the error-function expression87,
provided one sets \(\mu =2{\tilde{k}}_{{{{\rm{TF}}}}}/3\). In Eqs. (4) and (5), the first term considers only the SR contribution that corresponds to metallic screening, whereas the second term considers the full-range (FR) of the Coulomb interaction representing dielectric screening27. Therefore, the nonlocal Fock exchange found by Shimazaki and Asai can be expressed as27:
To use this expression as nonlocal exchange in a hybrid functional formulation, one needs to add a compensating semilocal exchange in the long range27. Thus, one obtains
This expression is formally the same as the one obtained by Chen et al. following an alternative derivation path26. This expression is also consistent with Eq. (3), through which it can be obtained by setting αSR = 1 and αLR = 1/ϵ∞. Hence, we use in this work the exchange potential given in Eq. (7) with \(\mu =2{\tilde{k}}_{{{{\rm{TF}}}}}/3\).
Special displacement method
Based on the theory introduced by Williams90 and Lax91 (WL) and the use of the harmonic approximation, the temperature-dependent band gap can be expressed as35:
where the product runs over all modes ν, and Q is used to indicate collectively the configuration defined by the normal coordinates Qν. This expression can be readily understood as the thermal average of \({E}_{{{{\rm{gap}}}}}^{Q}\) with weights \({\prod }_{\nu }\exp [-{Q}_{\nu }^{2}/(2{\sigma }_{\nu ,{T}}^{2})]/\sqrt{2\pi }{\sigma }_{\nu ,{T}}\), in which σν,T is a spatial Gaussian broadening given by
with \({l}_{\nu }=\sqrt{\hslash /(2{M}_{{{{\rm{p}}}}}{\omega }_{\nu })}\) and \({n}_{\nu ,T}={\{\exp \left[\hslash {\omega }_{\nu }/\left({k}_{{{{\rm{B}}}}}T\right)\right]-1\}}^{-1}\) for the zero-point vibrational amplitude and the Bose-Einstein distribution, respectively. Mp and ων denote the mass of the proton and the frequency of the νth normal mode. The broadening σν,T accounts for both the zero-point and thermal effects, and is denoted as σZPR+T. For T → 0 and nν,T→0, the broadening only results from the zero-point amplitude through
The average in Eq. (8) for a temperature T can be obtained by considering a single distorted atomic configuration, which is created by displacing the atoms by an amount of Δτkα from the equilibrium structure35:
where ν runs over all the non-translational modes, α indicates a Cartesian direction x, y or z, Mk is the mass of the kth nucleus, and ekα,ν is the vibrational eigenmode of the νth normal mode. We note that depending on whether we use σν,T from Eq. (9) or from Eq. (10), we either describe the ZPR and thermal effects together or just the ZPR effects alone. From the study of Zacharias and Giustino35, we infer that the band gap calculated for the distorted structure defined by the displacements in Eq. (11) reproduces the thermally averaged band gap defined in Eq. (8) within an accuracy of about 50 meV when the adopted supercell contains 150 atoms or more35. We consider this level of accuracy sufficient for the purpose of our work.
The special displacement method allows one to obtain band-gap renormalizations due to phonon-coupling within the context of the adiabatic approximation35,36. The problematic limit at small momentum transfers in the summation over the Brillouin zone37 is effectively handled in our supercell calculations by considering larger and larger supercells. We systematically considered supercells of varying size (containing a number atoms ranging between 160 to 625) and obtained constant results within 0.05 eV. In practice, the frequencies remain always finite and the ω0 of the recipe of Zacharias and Giustino36 could be taken to correspond to the lowest frequency found for the series of considered supercell sizes. For any lower ω0, the results would remain unchanged and the obtained values correspond to a finite result. However, the adiabatic approximation does not account for the long-range polar coupling to the longitudinal optical phonons, which results from a nonadiabatic treatment43. In this work, this effect has been estimated through the one-band Fröhlich model (see the Band gap Section and Supplementary Note 1).
Calculation details
The DSH band gaps of the ideal crystalline systems are determined through the implementation of DD-RSH-CAM functionals26 available in the Quantum-espresso software suites92. We use fully-relativistic pseudopotentials generated by the optimized norm-conserving Vanderbilt pseudopotential scheme93 to account for spin-orbit coupling. All the calculations are carried out with the stringent set of the Pseudo Dojo (available at http://www.pseudodojo.org) to ensure band gaps converged within 0.1 eV. A kinetic plane-wave cutoff of 100 Ry is used for all materials together with sufficiently dense k-point grids. In the band-gap calculations, we use unit cells with atomic structures and lattice parameters taken from experiment (cf. Supplementary Table 2). The high-frequency dielectric constant ϵ∞ is calculated at the PBE level through the application of finite electric fields50,94 and is used to fix the parameter αLR of the DSH functional.
For the application of the special displacement method35,36, we use the cp2k suite of codes95, which comprises an efficient tool for determining the vibrational frequencies and modes through finite differences. The core-valence interactions are described through Goedecker-Teter-Hutter (GTH) pseudopotentials96,97. We use double-zeta basis sets of MOLOPT quality98. The plane-wave energy cutoff for the electron density is set to 600 Ry to ensure the convergence of the total energy. The vibrational frequencies ων and eigenmodes ekα,ν required for the distorted structure specified in Eq. (11) are obtained through finite displacements of 0.01 Å from fully relaxed atomic positions. We use Γ-point sampling with the supercells defined in Supplementary Table 5. As an alternative scheme for obtaining vibrational properties, the non-diagonal approach proposed by Lloyd-Williams et al.99 could be used to further reduce the involved computational cost.
The band gaps are determined from linear extrapolations of the EDOS obtained for the supercells specified in the Supplementary Table 5. The cp2k results are systematically obtained at the PBE level of theory, with the exception of the test results in Table 5, which required the use of hybrid functionals. In this case, the auxiliary density matrix method (ADMM) is employed to speed up the calculations100.
Our efficient band-gap calculations contain several parts that contribute to the overall computational cost. The first contribution results from the determination of the dielectric constant, but the cost of this calculation is not significant since it is carried out at the PBE level. The first important contribution comes from the band-gap calculation for the ideal crystalline system with the DSH functional. Since the cost of this calculation is easy to evaluate, we use it here to set the unit. The calculation of the SOC correction is carried out at the PBE level and is negligible on the scale of our cost unit. The second important contribution comes from the determination of the vibrational properties at the PBE level. The corresponding cost of this part depends on the number atoms considered, but it is comparable to the cost unit for systems containing 270 atoms. For some materials, the EDOS needs to be determined with rather large supercells, but still at the PBE level. For the largest case considered (2880 atoms), this part gives a cost that amounts to half a cost unit and is thus quite negligible in most cases101,102,103. All together, the dominant costs for the band-gap determination result from the vibrational properties and from the DSH hybrid-functional calculation in comparable amounts.
Data availability
The structure used in the paper can be found in the materials cloud: https://archive.materialscloud.org/record/2022.35.
Code availability
The relevant codes in this study are available from the corresponding authors upon reasonable request.
References
Jeong, J. et al. Pseudo-halide anion engineering for α-FAPbI3 perovskite solar cells. Nature 592, 381–385 (2021).
Filip, M. R. & Giustino, F. The geometric blueprint of perovskites. Proc. Natl. Acad. Sci. 115, 5397–5402 (2018).
Wiktor, J., Rothlisberger, U. & Pasquarello, A. Predictive determination of band gaps of inorganic halide perovskites. J. Phys. Chem. Lett. 8, 5507–5512 (2017).
Bischoff, T., Wiktor, J., Chen, W. & Pasquarello, A. Nonempirical hybrid functionals for band gaps of inorganic metal-halide perovskites. Phys. Rev. Mater. 3, 123802 (2019).
Hohenberg, P. & Kohn, W. Inhomogeneous electron gas. Phys. Rev. 136, B864 (1964).
Kohn, W. & Sham, L. J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133 (1965).
Sham, L. J. & Schlüter, M. Density-functional theory of the energy gap. Phys. Rev. Lett. 51, 1888 (1983).
Even, J., Pedesseau, L., Jancu, J.-M. & Katan, C. Importance of spin-orbit coupling in hybrid organic/inorganic perovskites for photovoltaic applications. J. Phys. Chem. Lett. 4, 2999–3005 (2013).
Yin, W.-J., Yan, Y. & Wei, S.-H. Anomalous alloy properties in mixed halide perovskites. J. Phys. Chem. Lett. 5, 3625–3631 (2014).
Demchenko, D. et al. Optical properties of the organic-inorganic hybrid perovskite CH3NH3PbI3: Theory and experiment. Phys. Rev. B 94, 075206 (2016).
Hedin, L. New method for calculating the one-particle green’s function with application to the electron-gas problem. Phys. Rev. 139, A796 (1965).
Shishkin, M., Marsman, M. & Kresse, G. Accurate quasiparticle spectra from self-consistent GW calculations with vertex corrections. Phys. Rev. Lett. 99, 246403 (2007).
Chen, W. & Pasquarello, A. Accurate band gaps of extended systems via efficient vertex corrections in GW. Phys. Rev. B 92, 041115 (2015).
Bischoff, T., Reshetnyak, I. & Pasquarello, A. Band gaps of liquid water and hexagonal ice through advanced electronic-structure calculations. Phys. Rev. Res. 3, 023182 (2021).
Tal, A., Chen, W. & Pasquarello, A. Vertex function compliant with the ward identity for quasiparticle self-consistent calculations beyond GW. Phys. Rev. B 103, L161104 (2021).
Perdew, J. P., Ernzerhof, M. & Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 105, 9982–9985 (1996).
Burke, K., Ernzerhof, M. & Perdew, J. P. The adiabatic connection method: a non-empirical hybrid. Chem. Phys. Lett. 265, 115–120 (1997).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened coulomb potential. J. Chem. Phys. 118, 8207–8215 (2003).
Krukau, A. V., Vydrov, O. A., Izmaylov, A. F. & Scuseria, G. E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 125, 224106 (2006).
He, J. & Franchini, C. Screened hybrid functional applied to 3d0→3d8 transition-metal perovskites LaMO3 (M= Sc–Cu): Influence of the exchange mixing parameter on the structural, electronic, and magnetic properties. Phys. Rev. B 86, 235117 (2012).
Chen, W. & Pasquarello, A. Band-edge levels in semiconductors and insulators: Hybrid density functional theory versus many-body perturbation theory. Phys. Rev. B 86, 035134 (2012).
Franchini, C. Hybrid functionals applied to perovskites. J. Condens. Matter Phys. 26, 253202 (2014).
Alkauskas, A., Broqvist, P. & Pasquarello, A. Defect levels through hybrid density functionals: Insights and applications. Phys. Status Solidi B 248, 775–789 (2011).
Marques, M. A., Vidal, J., Oliveira, M. J., Reining, L. & Botti, S. Density-based mixing parameter for hybrid functionals. Phys. Rev. B 83, 035119 (2011).
Skone, J. H., Govoni, M. & Galli, G. Nonempirical range-separated hybrid functionals for solids and molecules. Phys. Rev. B 93, 235106 (2016).
Chen, W., Miceli, G., Rignanese, G.-M. & Pasquarello, A. Nonempirical dielectric-dependent hybrid functional with range separation for semiconductors and insulators. Phys. Rev. Mater. 2, 073803 (2018).
Cui, Z.-H., Wang, Y.-C., Zhang, M.-Y., Xu, X. & Jiang, H. Doubly screened hybrid functional: an accurate first-principles approach for both narrow-and wide-gap semiconductors. J. Phys. Chem. Lett. 9, 2338–2345 (2018).
Zhang, M.-Y., Cui, Z.-H., Wang, Y.-C. & Jiang, H. Hybrid functionals with system-dependent parameters: Conceptual foundations and methodological developments. WIREs: Comput. Mol. Sci. 10, e1476 (2020).
Refaely-Abramson, S. et al. Gap renormalization of molecular crystals from density-functional theory. Phys. Rev. B 88, 081204 (2013).
Refaely-Abramson, S., Jain, M., Sharifzadeh, S., Neaton, J. B. & Kronik, L. Solid-state optical absorption from optimally tuned time-dependent range-separated hybrid density functional theory. Phys. Rev. B 92, 081204 (2015).
Monserrat, B., Drummond, N., Pickard, C. J. & Needs, R. Electron-phonon coupling and the metallization of solid helium at terapascal pressures. Phys. Rev. Lett. 112, 055504 (2014).
Patrick, C. E. & Giustino, F. Quantum nuclear dynamics in the photophysics of diamondoids. Nat. Commun. 4, 1–7 (2013).
Pan, D., Wan, Q. & Galli, G. The refractive index and electronic gap of water and ice increase with increasing pressure. Nat. Commun. 5, 1–6 (2014).
Wiktor, J., Reshetnyak, I., Ambrosio, F. & Pasquarello, A. Comprehensive modeling of the band gap and absorption spectrum of bivo4. Phys. Rev. Mater. 1, 022401 (2017).
Zacharias, M. & Giustino, F. One-shot calculation of temperature-dependent optical spectra and phonon-induced band-gap renormalization. Phys. Rev. B 94, 075125 (2016).
Zacharias, M. & Giustino, F. Theory of the special displacement method for electronic structure calculations at finite temperature. Phys. Rev. Res. 2, 013357 (2020).
Poncé, S. et al. Temperature dependence of the electronic structure of semiconductors and insulators. J. Chem. Phys. 143, 102813 (2015).
Karsai, F., Engel, M., Flage-Larsen, E. & Kresse, G. Electron–phonon coupling in semiconductors within the gw approximation. New J. Phys. 20, 123008 (2018).
Zacharias, M., Scheffler, M. & Carbogno, C. Fully anharmonic nonperturbative theory of vibronically renormalized electronic band structures. Phys. Rev. B 102, 045126 (2020).
Miglio, A. et al. Predominance of non-adiabatic effects in zero-point renormalization of the electronic band gap. Npj Comput. Mater. 6, 1–8 (2020).
Shang, H., Zhao, J. & Yang, J. Assessment of the mass factor for the electron–phonon coupling in solids. J. Phys. Chem. C 125, 6479–6485 (2021).
Engel, M. et al. Zero-point renormalization of the band gap of semiconductors and insulators using the projector augmented wave method. Phys. Rev. B 106, 094316 (2022).
Nery, J. P. & Allen, P. B. Influence of Fröhlich polaron coupling on renormalized electron bands in polar semiconductors: Results for zinc-blende gan. Phys. Rev. B 94, 115135 (2016).
Patrick, C. E., Jacobsen, K. W. & Thygesen, K. S. Anharmonic stabilization and band gap renormalization in the perovskite CsSnI3. Phys. Rev. B 92, 201205 (2015).
Monserrat, B. Correlation effects on electron-phonon coupling in semiconductors: Many-body theory along thermal lines. Phys. Rev. B 93, 100301 (2016).
Saidi, W. A., Poncé, S. & Monserrat, B. Temperature dependence of the energy levels of methylammonium lead iodide perovskite from first-principles. J. Phys. Chem. Lett. 7, 5247–5252 (2016).
Tauc, J., Grigorovici, R. & Vancu, A. Optical properties and electronic structure of amorphous germanium. Phys. Status Solidi B 15, 627–637 (1966).
Huang, L.-y. & Lambrecht, W. R. Lattice dynamics in perovskite halides CsSnX3 with X= I, Br, Cl. Phys. Rev. B 90, 195201 (2014).
Cannelli, O. et al. Atomic-level description of thermal fluctuations in inorganic lead halide perovskites. J. Phys. Chem. Lett. 13, 3382–3391 (2022).
Umari, P. & Pasquarello, A. Ab initio molecular dynamics in a finite homogeneous electric field. Phys. Rev. Lett. 89, 157602 (2002).
Petousis, I. et al. High-throughput screening of inorganic compounds for the discovery of novel dielectric and optical materials. Sci. Data 4, 1–12 (2017).
Jain, A. et al. Commentary: The materials project: A materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Liu, P., Franchini, C., Marsman, M. & Kresse, G. Assessing model-dielectric-dependent hybrid functionals on the antiferromagnetic transition-metal monoxides MnO, FeO, CoO, and NiO. J. Condens. Matter Phys. 32, 015502 (2019).
Huang, L.-y. & Lambrecht, W. R. Electronic band structure, phonons, and exciton binding energies of halide perovskites CsSnClx, CsSnBrx, and CsSnIx. Phys. Rev. B 88, 165203 (2013).
Yu, Z.-L. et al. Oriented tuning the photovoltaic properties of γ-RbGeX3 by strain-induced electron effective mass mutation. J. Phys. D: Appl. Phys. 50, 465101 (2017).
Ghaithan, H. M., Alahmed, Z. A., Qaid, S. M., Hezam, M. & Aldwayyan, A. S. Density functional study of cubic, tetragonal, and orthorhombic CsPbBr3 perovskite. ACS Omega 5, 7468–7480 (2020).
Nyayban, A., Panda, S., Chowdhury, A. & Sharma, B. I. First principle studies of rubidium lead halides towards photovoltaic application. Mater. Today Commun. 24, 101190 (2020).
Su, Y., Song, K.-K., Zhong, M., Shi, L.-B. & Qian, P. Stability and phonon-limited mobility for CsSnI3 and CsPbI3. J. Alloys. Compd. 889, 161723 (2021).
Huang, L.-y. & Lambrecht, W. R. Electronic band structure trends of perovskite halides: Beyond Pb and sn to Ge and Si. Phys. Rev. B 93, 195211 (2016).
McKechnie, S. et al. Dynamic symmetry breaking and spin splitting in metal halide perovskites. Phys. Rev. B 98, 085108 (2018).
Komsa, H.-P., Broqvist, P. & Pasquarello, A. Alignment of defect levels and band edges through hybrid functionals: Effect of screening in the exchange term. Phys. Rev. B 81, 205118 (2010).
Deák, P. et al. Choosing the correct hybrid for defect calculations: A case study on intrinsic carrier trapping in β-Ga2O3. Phys. Rev. B 95, 075208 (2017).
He, X., Qiu, Y. & Yang, S. Fully-inorganic trihalide perovskite nanocrystals: a new research frontier of optoelectronic materials. Adv. Mater. 29, 1700775 (2017).
Tao, S. et al. Absolute energy level positions in tin-and lead-based halide perovskites. Nat. Commun. 10, 1–10 (2019).
Huhn, W. P. & Blum, V. One-hundred-three compound band-structure benchmark of post-self-consistent spin-orbit coupling treatments in density functional theory. Phys. Rev. Mater. 1, 033803 (2017).
Ambrosio, F., Miceli, G. & Pasquarello, A. Redox levels in aqueous solution: Effect of van der Waals interactions and hybrid functionals. J. Chem. Phys. 143, 244508 (2015).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Boziki, A., Dar, M. I., Jacopin, G., Grätzel, M. & Rothlisberger, U. Molecular origin of the asymmetric photoluminescence spectra of CsPbBr3 at low temperature. J. Phys. Chem. Lett. 12, 2699–2704 (2021).
Tang, B. et al. High stability and temperature-dependent photoluminescence of orthorhombic CsPbIx perovskite nanoparticles. Adv. Opt. Mater. 8, 2000498 (2020).
Haeger, T., Heiderhoff, R. & Riedl, T. Thermal properties of metal-halide perovskites. J. Mater. Chem. C 8, 14289–14311 (2020).
Varshni, Y. P. Temperature dependence of the energy gap in semiconductors. Physica 34, 149–154 (1967).
Dar, M. I. et al. Origin of unusual bandgap shift and dual emission in organic-inorganic lead halide perovskites. Sci. Adv. 2, e1601156 (2016).
Coduri, M. et al. Band gap engineering in MASnBr3 and CsSnBr3 perovskites: Mechanistic insights through the application of pressure. J. Phys. Chem. Lett. 10, 7398–7405 (2019).
Prasanna, R. et al. Band gap tuning via lattice contraction and octahedral tilting in perovskite materials for photovoltaics. J. Am. Chem. Soc. 139, 11117–11124 (2017).
Knutson, J. L., Martin, J. D. & Mitzi, D. B. Tuning the band gap in hybrid tin iodide perovskite semiconductors using structural templating. Inorg. Chem. 44, 4699–4705 (2005).
Filip, M. R., Eperon, G. E., Snaith, H. J. & Giustino, F. Steric engineering of metal-halide perovskites with tunable optical band gaps. Nat. Commun. 5, 1–9 (2014).
Chen, Z. et al. Photoluminescence study of polycrystalline CsSnI3 thin films: Determination of exciton binding energy. J. Lumin. 132, 345–349 (2012).
Baranowski, M. et al. Exciton binding energy and effective mass of CsPbCl3: a magneto-optical study. Photonics Res. 8, A50–A55 (2020).
Dias, A., Lima, M. P. & Da Silva, J. L. Role of structural phases and octahedra distortions in the optoelectronic and excitonic properties of CsGeX3 (X= Cl, Br, I) perovskites. J. Phys. Chem. C 125, 19142–19155 (2021).
Leppert, L., Rangel, T. & Neaton, J. B. Towards predictive band gaps for halide perovskites: Lessons from one-shot and eigenvalue self-consistent gw. Phys. Rev. Mater. 3, 103803 (2019).
Im, J. et al. Identifying Pb-free perovskites for solar cells by machine learning. Npj Comput. Mater. 5, 1–8 (2019).
Yan, Q. et al. Solar fuels photoanode materials discovery by integrating high-throughput theory and experiment. Proc. Natl. Acad. Sci. U.S.A. 114, 3040–3043 (2017).
Pilania, G. et al. Machine learning bandgaps of double perovskites. Sci. Rep. 6, 1–10 (2016).
Skone, J. H., Govoni, M. & Galli, G. Self-consistent hybrid functional for condensed systems. Phys. Rev. B 89, 195112 (2014).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Shimazaki, T. & Asai, Y. Band structure calculations based on screened fock exchange method. Chem. Phys. Lett. 466, 91–94 (2008).
Bechstedt, F., Del Sole, R., Cappellini, G. & Reining, L. An efficient method for calculating quasiparticle energies in semiconductors. Solid State Commun. 84, 765–770 (1992).
Cappellini, G., Del Sole, R., Reining, L. & Bechstedt, F. Model dielectric function for semiconductors. Phys. Rev. B 47, 9892 (1993).
Williams, F. E. Theoretical low temperature spectra of the thallium activated potassium chloride phosphor. Phys. Rev. 82, 281 (1951).
Lax, M. The Franck-Condon principle and its application to crystals. J. Chem. Phys. 20, 1752–1760 (1952).
Giannozzi, P. et al. Quantum espresso: a modular and open-source software project for quantum simulations of materials. J. Condens. Matter Phys. 21, 395502 (2009).
Hamann, D. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 88, 085117 (2013).
Souza, I., Íniguez, J. & Vanderbilt, D. First-principles approach to insulators in finite electric fields. Phys. Rev. Lett. 89, 117602 (2002).
Hutter, J., Iannuzzi, M., Schiffmann, F. & VandeVondele, J. Cp2k: atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 15–25 (2014).
Goedecker, S., Teter, M. & Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 54, 1703 (1996).
Hartwigsen, C., Gœdecker, S. & Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from h to rn. Phys. Rev. B 58, 3641 (1998).
VandeVondele, J. & Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 127, 114105 (2007).
Lloyd-Williams, J. H. & Monserrat, B. Lattice dynamics and electron-phonon coupling calculations using nondiagonal supercells. Phys. Rev. B 92, 184301 (2015).
Guidon, M., Hutter, J. & VandeVondele, J. Auxiliary density matrix methods for Hartree- Fock exchange calculations. J. Chem. Theory Comput. 6, 2348–2364 (2010).
Lin, Z.-G., Tang, L.-C. & VandeVondele, J. Auxiliary density matrix methods for Hartree-Fock exchange calculations. J. Chem. Theory Comput. 6, 2348–2364 (2010).
Krishnamoorthy, T. et al. Lead-free germanium iodide perovskite materials for photovoltaic applications. J. Mater. Chem. A 3, 23829–23832 (2015).
Voloshinovskii, A., Myagkota, S., Pidzyrailo, N. & Tokarivskii, M. Luminescence and structural transformations of CsSnCl3 crystals. J. Appl. Spectrosc 60, 226–228 (1994).
Acknowledgements
The authors acknowledge useful interactions with W. Chen, S. Falletta, M. Zacharias, and R. Ouyang. Support from the Swiss National Science Foundation (SNSF) is acknowledged under Grant No. 200020-172524. The calculations have been performed at the Swiss National Supercomputing Center (CSCS) (grant under project ID s879 and s1123) and at SCITAS-EPFL.
Author information
Authors and Affiliations
Contributions
A.P. and H.W. designed this project. H.W. performed most calculations with the help from T.B. and P.G. for some materials. A.T. carried out calculations using VASP. H.W. and A.P. wrote the manuscript. All authors discussed data and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Tal, A., Bischoff, T. et al. Accurate and efficient band-gap predictions for metal halide perovskites at finite temperature. npj Comput Mater 8, 237 (2022). https://doi.org/10.1038/s41524-022-00869-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-022-00869-6
- Springer Nature Limited