
Abstract
The interactions between electrons and phonons play the key role in determining the carrier transport properties in semiconductors. In this work, comprehensive investigations on full electron–phonon (el–ph) couplings and their influences on carrier mobility and thermoelectric (TE) performances of 2D group IV and V elemental monolayers are performed, and we also analyze the selection rules on el–ph couplings using group theory. For shallow n/p-dopings in Si, Ge, and Sn, ZA/TA/LO phonon modes dominate the intervalley scatterings. Similarly strong intervalley scatterings via ZA/TO phonon modes can be identified for CBM electrons in P, As, and Sb, and for VBM holes, ZA/TA phonon modes dominate intervalley scatterings in P while LA phonons dominate intravalley scatterings in As and Sb. By considering full el–ph couplings, the TE performance for these two series of monolayers are predicted, which seriously downgrades the thermoelectric figures of merits compared with those predicted by the constant relaxation time approximation.
Similar content being viewed by others

Introduction
Two-dimensional (2D) materials offer challenging opportunities for a wide range of applications in nanoscale devices. Among the extraordinary properties in 2D materials, carrier transport properties play the key role in determining the performance in microelectronic, optoelectronic and thermoelectric devices. Until now, extensive studies have been devoted on carrier transport properties in 2D materials, which are dominantly determined by the interaction between electrons and phonons1,2. Recently, it has been reported that the electron–phonon couplings can in turn efficiently limit the phonon transport properties and reduce the lattice thermal conductivities in graphene3, MoS2, and PtSSe4. In the simplest way, the electron–phonon (el–ph) interaction can be understood as originated from the electrostatic potential (known as deformation potential) generated by exciting a phonon in the crystal, which, in turn, affects all carriers directly. Subsequently, the deformation potential approximation (DPA) method based on the intravalley coupling between electrons and long-wavelength longitudial acoustic (LA) phonon modes5,6, was widely used to estimate or understand the carrier mobilities in many non-polar 2D semiconductors including graphene7, phosphorene8 and transition metal dichalcogenide (TMD)9. However, in some polar or highly anisotropic systems, the DPA method usually misestimates the intrinsic carrier mobility, in which the coupling from the longitudinal optical (LO) phonon modes described as the Fr\(\ddot{o}\)hlich interaction, and the direction-dependent contributions can not be neglected. For example, as previously reported2,10,11, the scatterings from flexural ZA phonons contribute dominantly to the total scattering rates in group IV elemental monolayers (silicene, germanene, and stanene) due to the lack of σh-symmetry in buckled honeycomb structure belonging to D3d point group, which fails the DPA method. Therefore, depending on the specific materials and the situation of the underlying mechanism, systematically computing energy- and momemtum-dependent el–ph interactions is necessary to accurately predict phonon-assisted processes.
Inspired by the extraordinary properties of graphene, the group IV elemental materials of silicene (Si), germanene (Ge), and stanene (Sn) as the promising alternatives, have attracted tremendous interests. Studies show that such monolayers all possess Dirac fermions similar to graphene, high mechanical flexibility and high electron mobility, leading to the potential applications in batteries and topological devices12,13. Monolayer Sn was predicted to possess an intrinsic spin-orbit coupling (SOC) gap ~100 meV, which is ideal to realize the quantum spin Hall (QSH) effect at room temperature14,15. Recently, Jian et al. successfully synthesized large-scale and high-quality stanene on Sb (111), providing a good platform to study interesting phenomena such as QSH effect16. In contrast to 2D group IV materials, 2D group V elemental materials of phosphorene (P), arsenene (As), and antimonene (Sb) with the same buckled honeycomb structure are semiconductors with wide band gaps, probably leading to the applications in transistors with high on/off ratios17 or optoelectric devices operating in the visible range18. Recent works reported that, the surface plasmon resonance sensor based on β-phosphorene/MoS2 heterojunction possesses significantly higher sensitivity compared with conventional or graphene-based SPR sensors19, and the monolayer arsenene has the potential to serve as anode materials in high-performance Magnesium-ion batteries20. Interestingly, the β-antimonene undergoes a transformation from a topological semimetal to a topological insulator at 22 bilayer, then to QSH phase at 8 bilayers and finally to a topological trivial semiconductor at 3 or thinner bilayers21.
Herein, based on the first principles method, we systematically investigate the full el–ph coupling effects in 2D group IV and V materials. These two series of 2D materials indeed possess a broad range of carrier mobilities, which may lead to various applications. Compared with group IV counterparts, group V materials are more buckled, strengthening the overlap of the pz orbitals, thus may lead to the enhancement of the interaction between the electrons and ZA phonons. The conduction band and valence band for group V materials are flatter than group IV materials, which is beneficial to satisfy the energy conservation condition and further increases the el–ph coupling near the Fermi level. To further investigate the roles of different phonon modes, we also derive in details the selection rule for the full el–ph scatterings in these two series of materials. Based on the group-theory analysis, we find that, the LA phonons play a dominant role in the hole scattering via acoustic phonon modes in arsenene and antimonene, which is in contrast to the previous report that group V materials have the same scattering mechanism as group IV materials22. For monolayer phosphorene, arsenene, and antimonene, despite the different bands and locations of valence-band maximum (VBM), LA phonon modes with A irreducible representation (irreps) of C2 group near Γ point dominate the intravalley scatterings due to the identical symmetry and irreps of the intial and final electronic states. However, phosphorene is the only material with degenerate VBM where the intervalley scattering from one VBM to another is dominated by ZA and TA phonon modes. The VBM of monolayer arsenene and antimonene locates at Γ point without degeneracy, hence the intravalley scattering becomes dominant. Finally, by considering the full el–ph scatterings, an accurate prediction of the carrier transport properties and thermoelectric performances of these two series of materials are achieved.
Results and discussion
Optimized configurations and band structures
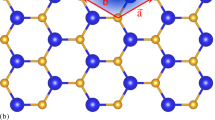
Compared with the planar geometry of graphene, 2D group IV and V crystals have buckled structure with two sublayers. The top and side views of configurations of the 2D group IV materials (silicene, germanene, stanene) and group V materials (phosphorene, arsenene, antimonene) are shown in Fig. 1a. The optimized lattice constants a, the distance between two sublayers h, bond angle θ and bond length d are shown in Supplementary Table 1 which are consistent with previous reports23,24,25,26.

a Atomic structure of monolayer buckled hexagonal structure. b Electron localization function of silicene and phosphorene.
In graphene, the 2s orbital is hybridized with px and py orbitals to form three planar σ bonds via sp2 hybridization, and the remaining pz orbital forms π bonding between adjacent C atoms to guarantee the planarity27. However, when the bond length increases, the pz–pz overlapping decreases and the weaker π bonding can not ensure the stability of the planar geometry. By buckling, the planner sp2 bonding is dehybridized to form sp3-like hybridization to enhance the overlapping between pz orbitals28. Figure 1b shows the calculated electronic localization function (ELF) for silicene and phosphorene. The ELF is a position-dependent function with values ranging from 0 to 1. ELF = 1 reflects the maximum probability to find the localized electrons while ELF = 0.5 means the electron-gas-like behavior29. The strength of the π bond of 2D group V materials is much higher than their group IV counterparts. The one more electron in P, As and Sb increases the degeneracy of electronic states, leading to the unstable state according to the Jahn–Teller theorem, and by a high degree of buckling the degeneracy can be relieved, resulted from the broken symmetry30. Hence, compared with group IV materials, the group V counterparts possess larger buckling heights, smaller lattice constants and bond angles, which together increase the overlap of the pz orbitals.
In order to further investigate the strength of chemical bonding, we also calculate the bulk (BH) and shear moduli (GH) for group IV and group V materials, which determine the ductile-brittle nature of materials by Pugh’s ratio BH/GH31. According to Pugh’s rule, a small Pugh’s ratio indicates brittle nature of a material. As shown in Supplementary Table 1, the Pugh’s ratios for phosphorene, arsenene, and antimonene are all lower than their buckled group IV counterparts, indicating that the high degree of buckling in group V materials strengthens the π bonding. The trend of Pugh’s ratios BH/GH(Si) < BH/GH(Ge) < BH/GH(Sn) and BH/GH(P) < BH/GH(As) < BH/GH(Sb) indicates that increasing bond length d lowers the bonding strength by decreasing orbital overlap for the same group materials, compensating the effect from the increase of buckling.
The calculated band structures of silicene, germanene, and stanene are shown in Supplementary Fig. 1a–c. All these group IV materials are semimetals with a Dirac-cone located at the K point, and the SOC effect opens the band gaps of 1.48 meV, 23 meV, 72 meV, respectively. Based on the band structures projected by the atomic orbitals, the band structure near the conduction band minimum CBM (C1) and valence band maximum VBM (V1) are dominantly contributed from pz orbitals. However, as shown in Supplementary Fig. 1d–f, as a comparison, the group V materials are all semiconductors with sizable indirect band gaps. The calculated band gaps without (with) SOC are 1.94 eV (1.94 eV), 1.59 eV (1.47 eV), 1.26 eV (0.99 eV) for phosphorene, arsenene, and antimonene respectively. The CBM locates close to the middle of the Γ–M, which is mainly contributed from px and pz orbitals, and the VBM locates at Γ point for arsenene and antimonene which is dominantly contributed from px and py orbitals, while for phosphorene the VBM locates along Γ–K direction mainly contributed by pz orbitals. The multiple local valleys and peaks, which may play important roles in intervalley el–ph scattering, are labeled as C2, C3 and V2, V3, and their difference from CBM or VBM are shown as \({E}_{{C}_{2}}\), \({E}_{{C}_{3}}\), \({E}_{{V}_{2}}\), \({E}_{{V}_{3}}\) in Table 1. For instance, the energy difference between V1 and V2 valleys (\({E}_{{V}_{2}}\)) is 50 meV in phosphorene.
Full el–ph scatterings: selection rules and carrier mobilities
The calculated carrier mobilities at 300 K along armchair (a) and zigzag (b) directions for these two series of 2D materials, based on the modified DPA theory32, are shown in Table 2, accompanied with effective mass m*, deformation potential constant El, elastic modulus C2D, and the relaxation time τ defined as τ = μm*/e, which is determined by the interaction between carriers and LA phonons. The deformation potential constant El reflects the coupling strength of electron and long-wavelength LA phonons, defined as ΔECBM(VBM)/(δl/l0)33, in which ΔECBM(VBM) is the shift of the VBM and CBM energy level with respect to the lattice deformation δl. The elastic modulus C2D is related to the interatomic forces. Hence, both El and C2D are influenced by the overlap of atomic orbitals. The values of El and C2D of 2D group V materials are larger than their group IV counterparts due to the enhanced overlap of atomic orbitals by buckling. The obtained mobilities for group IV materials based on the DPA method are comparable to graphene in the order of 105–106 cm2 V−1 s−1, and those of group V materials are in the range of about 10–1000 cm2 V−1 s−1. However, as mentioned above, the DPA method may fail for the band-convergence systems such as the group IV and V elemental crystals shown in Table 2, therefore the full el–ph scatterings should be investigated to accurately predict the carrier transport properties. According to Eq. (1), the carrier mobility is mainly determined by the electron velocities and the relaxation times τ.
The calculated el–ph scatterings for CBM electrons via acoustic and optical phonon modes for these two series of 2D materials are shown in Fig. 2 and Supplementary Fig. 4, respectively, and the calculated el–ph scatterings for VBM holes via acoustic and optical phonon modes for these two series of 2D materials are shown in Fig. 3 and Supplementary Fig. 5, respectively. All the scattering rates are calculated at 300 K within ~0.4 eV. Obviously, the calculated scattering rates for group V materials are much larger than their group IV counterparts, in consistency with the behaviors of deformation potential constant El and elastic modulus C2D mentioned above, which is probably resulted from two mechanisms: (i) The enhanced pz–pz orbitals overlapping enlarges the el–ph matrix element \({g}_{mn\nu }\left({{{\bf{k}}}},{{{\bf{q}}}}\right)\) described in Eq. (2) due to the stronger sensitivity to the atomic positions. (ii) The band structures of group V materials near Fermi level are much flatter than the those of group IV materials, which is more beneficial to satisfy the energy conservation conditions in the scattering processes. Starting from the CBM, with the increase of electronic energy, the scattering rates increase gradually for group IV materials while dramatically for group V materials especially for phosphorene, which possesses a wide and nearly flat region near the C1 valley as shown in Supplementary Fig. 1d, leading to about 4 times larger in the scattering rate at E = 0.07 eV than those in arsenene and antimonene.

The scattering rate of electrons in the C1 valley with energies within ~0.4 eV. The contribution from three acoustic branches and the total scattering rate considering optical branches are demonstrated.

The scattering rate of holes in the V1 peak with energies within ~0.4 eV.
Due to the small phonon energies, for monolayer Sn, As, and Sb, since the energy of CBM electrons is well smaller than \({E}_{{C}_{2}}\), only the intravalley scattering and intervalley scatterings between degenerate C1 valleys are allowed for shallow dopings. When the energy of C1-pocket electrons is higher than \({E}_{{C}_{2}}\), C1–C2 intervalley scattering is allowed, and the scattering rate increases abruptly as a result. In particular, for monolayer Sb, when the energy of C1-pocket electrons is higher than \({E}_{{C}_{3}}\), both C1–C2 and C1–C3 intervalley scatterings are allowed, leading to a further increase of the scattering rates, as shown in Fig. 2f and Supplementary Fig. 4f.
The mode- and energy-resolved el-–ph scattering rates of V1-peak holes for these two series of materials at 300 K within ~0.4 eV are shown in Fig. 3 and Supplementary Fig. 5. The behaviors of the scattering rates of holes for silicene and germanene are similar to their electron case, due to the Dirac-cone dispersions for both CBM and VBM carriers. For stanene, as the hole energy descreases to \({E}_{{V}_{2}}\), the scattering rate experiences a sharp rise, implying the activation of V1–V2 intervalley scatterings. The intravalley and V2–V1 intervalley scatterings are also allowed for V2-peak holes with energy lower than \({E}_{{V}_{2}}\), as shown in Fig. 3c and Supplementary Fig. 5c. For the hole scatterings in phosphorene, due to the flat valence band near Fermi level, also reflected in the effective mass as listed in Table 2, the scattering rates of V1-peak holes with energy near Fermi level are very high compared with those for monolayer As and Sb. When the hole energy is lower than \({E}_{{V}_{2}}\), the V1–V2 intervalley scattering are activated and the scattering rates further increase up to ~3.7 × 1015 s−1 near −0.05 eV. Figure 4a shows the contour map of the valence band in Brillouin zone for phosphorene. It is clear that the energy difference between V1 and V2 peaks is rather small, which is beneficial for satisfying the conservation of energy. Figure 4b shows the scattering rates of holes via ZA phonons in the valence band in the Brillouin zone, which reveals that, holes at the junction of the two peaks undergo strong scatterings, due to the typical quadratic dispersion of ZA mode and flat electronic dispersion.

Contour map of a the valence band and b the scattering rate of phosphorene as a function of wave vector k at first Brillouin zone.
The respective mode-resolved scatterings rates for electrons at CBM and holes at VBM for these two elemental crystals are shown in Supplementary Figs. 6 and 7. For group IV elemental monolayers (Si, Ge, Sn) with shallow n- and p-dopings, the intravalley scattering rates for CBM electrons or VBM holes via TO and LO phonon modes are significantly larger than those via acoustic phonon modes, and ZO modes also contribute significantly in monolayer Ge. Overall, the intervalley scatterings rates significantly surpass the intravalley scatterings, and the contributions from ZA, TA, and LO phonon modes dominate the intervalley scatterings. However, the situations for group V elemental monolayers (P, As, Sb) are different, especially for intravalley scatterings. For intravalley scatterings in monolayer P, the contributions from ZO, LA, and TA phonon modes dominate. In monolayer As, the contributions from TO, LO, LA, and TA phonon modes dominate, and in monolayer Sb, the contributions from LA, ZO, and LO phonon modes dominate. Similarly, intervalley scatterings for CBM electrons are much stronger than intravalley scatterings in group V monolayers, and the contributions from ZA and TO modes dominate. For el–ph scatterings for VBM holes in group V elemental crystals, ZA and TA modes dominate interpeak scatterings in monolayer P, as shown in Supplementary Fig. 7. As shown in Supplementary Figs. 1 and 7, since the global VBM located at Γ point is well above the other local VBMs, interpeak scatterings are not considered in monolayer As and Sb for shallow p-dopings.
In order to further clarify the underlying mechanisms behind electron–phonon interactions, based on the group-theory analysis, the selection rules in terms of the symmetry of systems for the interaction between phonons and electrons are derived. The electron–phonon interaction depends on the electronic wave functions of the initial states ϕi(k), final states ϕf(k + q) and the coupled phonon with the eigenvector q described by the phonon-induced deformation potential δq,νU34,35,36. Based on the Fermi’s golden rules, in order to calculate the scattering probability from initial state ϕi(k) to final states ϕf(k + q) of electrons or holes via a specific phonon mode, we only need to obtain the core matrix, which could be expressed as37
where the perturbation potential \(\delta U={\sum }_{l,a}{q}_{l,a}\frac{\partial U({r}_{i})}{\partial {q}_{l,a}}\), in which ql,a is the ion displacement of the a atom in the l-th unit cell, and ri is the electron coordinate. Therefore, the expression for the selection rules for such scattering process \({{{\mathcal{M}}}}\) based on the group theory is written as,
where Γi/f represents the irreps for electron initial/final states, and Γph denotes the symmetry of the corresponding phonon involved in this process. The involved electrons and phonons must obey the rules of momentum conservation and the compatibility relation between little groups, since they possibly locate at different points in the Brillouin Zone. non null/null means the allowed/forbidden transition channels. Due to the lack of in-plane symmetry σh in monolayer Si, Ge, Sn of group IV and monolayer P, As, Sn of group V, their electronic and phonon symmetry is lowered considerably compared to plane graphene.
For simplification without loss of generality, we only discuss on the cases for el–ph scatterings via acoustic phonon modes, and the cases for those via optical phonon modes can be discussed in a similar way. As shown in Supplementary Fig. 1 and Supplementary Table 2, for the intrapeak scatterings of holes in monolayer Si, Ge, and Sn, the hole states in the V1 peak along K–M and K–Γ involved in the intrapeak scatterings transform with the A and B irreps respectively, both belonging to the little group C2. Mathematically, it is allowed for the initial states and final states to belong to the same irreps in the el–ph scattering, i.e. A \(\rightleftharpoons\) A or B \(\rightleftharpoons\) B, but the contribution of such scattering channel is negligible compared to the scatterings with different irreps for initial and final states in the real systems we studied here. Because, as shown in Supplementary Fig. 1a–c, the energy difference of the scattering channel A \(\rightleftharpoons\) B is smaller than that for the scattering channel of A \(\rightleftharpoons\) A or B \(\rightleftharpoons\) B, leading to the larger density of states of the former scattering channel, which is beneficial for the el–ph scattering in real systems. Therefore, for intrapeak scatterings in monolayer Si, Ge, and Sn, we only need to consider the initial and final states transforming with the A and B irreps, i.e. A \(\rightleftharpoons\) B processes. As shown in Supplementary Table 2, in monolayer Si, Ge, and Sn, both ZA and TA modes near Γ point transform with the B irreps, while LA modes transform with A irreps. Hence, restricted by the selection rules shown as Eq. (2), the hole scatterings from \({V}_{1}^{\prime}\) to \({V}_{1}^{^{\prime\prime} }\) in monolayer Si, Sn, and Ge via the out-plane ZA and in-plane TA modes are allowed, due to the non null of B ⊗ B ⊗ A, as shown in Supplementary Table 2. The analysis based on the selection rules is in good agreement with the numerical results shown as Fig. 3a–c. It should be noticed that, some phonon modes locating near Γ point transform with the irreps belonging to the little group of CS, but it is reasonable to exclude them since the initial and final states of carriers are with C2 symmetry, which are forbidden to be coupled to the phonon modes with CS group considering the incompatibility relation between C2 and CS38.
Furthermore, as shown in Fig. 5, for the interpeak scatterings of holes in group IV monolayer materials, since the degenerate VBMs are located at six corners in the Brillouin Zone, there are at least three different interpeak-scattering channels, including the scattering to the nearest, next-to-nearest and opposite-side K points. However, the interpeak scattering to the next-to-nearest VBMs is actually forbidden since the phonon modes involved are with CS symmetry, which is incompatible to the C2 symmetry. Therefore, only TA modes are allowed restricted by the selection rules and conservation of momentums, as shown in Supplementary Table 4, which is consistent with previous work2. Moreover, as shown in Supplementary Tables 3 and 5, since the CBM and VBM points touch at the K points forming the so-called Dirac points in group IV elemental crystals, therefore, in the same way, the intravalley scatterings for CBM electrons scattered via the ZA and TA phonon modes are allowed, and the intervalley scatterings for CBM electrons between degenerate CBMs via TA modes are allowed. It should be noted that, although ZA phonon mode is allowed in intravalley/intrapeak el–ph scatterings for group IV elemental crystals obeying the selection rule, the calculated scattering rates for intravalley/intrapeak el–ph scatterings via ZA phonon mode as shown in Supplementary Figs. 6a–c and 7a–c are indeed trivial. This disagreement can be understood by the fact that, CBM/VBM carriers with linear dispersion as shown in Supplementary Fig. 1 can not scatter to each other via ZA phonon modes with quadratic dispersion near Γ point, restricted by the energy conservation.

The transitional K-path of intravalley scattering in VBM, and intervalley scattering from VBM to V2 or V3 valley for monolayer (a) Si, (b) Ge, (c) Sn, (d) P, (e) As and (f) Sb. The contour lines of energy range from − 0.40 eV to 0.00 eV below Fermi level. Each K path is denoted by acoustic phonon modes which are involved in concordant electron–phonon interactions allowed by selection rules. Red crosses reprensent forbidden transition due to lack of compability between relative irreducible representations.
As shown Fig. 5 and Supplementary Table 4, for the interpeak scatterings of the VBM holes for monolayer P, As and Sb, the situation is quite different due to the different locations of VBMs. For monolayer As and Sb, since the VBM locates at Γ point, which lacks of degeneracy in the Brillouin Zone, the interpeak scatterings between VBMs no longer exist for the VBM holes at shallow p-dopings. As a result, only LA mode dominates the el–ph scatterings of VBM holes for monolayer As and Sb, since the initial and final states of VBM holes for monolayer Sb and As near the Γ point transform with A irreps belonging to the little group C2, which is in good agreement with the mode-resolved scattering rates of VBM holes for monolayer As and Sb as shown in Fig. 3e, f, respectively. However, special care should be taken for interpeak scatterings of VBM holes for monolayer P, since the degeneracy of the VBM points in the Brillouin Zone for monolayer P is six, as shown in Fig. 5d. Three interpeak-scattering channels to the nearest neighboring VBMs are possible for VBM holes, and the little groups for the initial and final hole states are C2. However, the phonon involved in the interpeak scattering to the secondly nearest neighboring K point transforms with the little group of CS, incompatible to C2, therefore this scattering channel is forbidden. Thus, in monolayer P, the scatterings of VBM holes to the nearest and the thirdly nearest neighboring VBMs are allowed, and respectively dominated by the TA and ZA phonon modes. The selectrion-rule analysis is in good agreement with the numerical results shown in Fig. 3d.
Similarly, as shown in Supplementary Fig. 1 and Supplementary Table 3, the CBMs of group V materials (monolayer P, As, and Sb) locate at the points along Γ–M with the little group CS, and thus the corresponding irreps of the initial states and final states of CBM are A′. The acoustic phonon modes of ZA, TA, and LA near Γ points and along Γ–M transform with the A′, A″, and A′ irreps, respectively as shown in Supplementary Fig. 3a–c. Therefore, restricted by the selection rules, both ZA and LA modes are allowed in the intravalley scatterings of CBM electrons for monolayer P, As, and Sb. For the intervalley scatterings of electrons between degenerate CBMs, the ZA and LA modes are allowed, as shown in Supplementary Table 5. And Fig. 6 gives the transitional paths of electrons scattered by phonons in conduction bands.

The transitional K-path of intravalley scattering in CBM, and intervalley scattering from CBM to C2 valley for monolayer a Si, b Ge, c Sn, d P, e As, and f Sb. The contour lines of energy range from 0.00 eV to 0.40 eV above Fermi level. Each K path is denoted by acoustic phonon modes which are involved in concordant electron–phonon interactions allowed by selection rules. Red crosses reprensent forbidden transition due to lack of compability between relative irreducible representations.
Based on the calculated full el–ph materix elements, the temperature-dependent and mode-resolved relaxation time and the subsequent carrier mobilites along special directions can be calculated according to Eqs. (1) and (2), and the calculated mobilities for these two series of 2D elemental materials are shown in Fig. 7a, b, respectively. The calculated mobilities at 300 K, by a comparison with those calculated by the DPA method are shown in Table 2 as well. By comparison, three distinctions about the carrier mobilities of 2D group IV and group V materials can be identified: (1) Generally, the results by the DPA method are overestimated by 2–3 orders in magnitude due to the only consideration of LA phonons and neglecting the intervalley scattering process; (2) The hole mobilities of group V materials calculated by these two methods are in roughly good agreement except phosphorene owing to the dominant LA phonons-holes coupling in monolayer β-Sb and As; (3) The obvious anisotropy in carrier transport for group V materials obtained by the DPA method no longer exists in the results considering full el–ph coupling.

The calculated carrier mobility for electrons and holes for a group IV and b group V materials considering full electron–phonon coupling with temperature ranging from 100 K to 500 K.
Thermoelectric performance
The thermoelectric (TE) performance of a material is quantified by a dimensionless figure of merit zT(zT = S2σT/(κe + κL)), where S represents the Seebeck coefficient, σ is the electron conductivity, and κe/L is the electronic/lattice thermal conductivity. High TE performance needs high zT value of materials with excellent electrical transport properties and poor thermal transport properties simultaneously. However, optimization of zT values in the TE materials is always challenging due to the inter-correlations of the parameters, and optimizing one leads to deteriorating the other. In recent years, several optimization strategies have been proposed to enhance successfully zT values in TE materials, and among them, the strategies of the dimension confinement and band convergence attract broad interests owing to the successful discovery of layered TE materials with extremely high zT values39,40,41. Furthermore, considering the appropriate band gaps and heavy atoms in arsenene, stanene, and antimonene, high zT values can be expected in these two series of 2D elemental materials. As reported previously42, antimonene has the zT value of 2.15 at room temperature and can be further enhanced to 2.90 with strain engineering, predicted by the DPA and constant relaxation time approximation (CRTA) methods. For arsenene, different constant relaxation times lead to ten times deviation in zT values26, implying the importance of the accurate calculations of relaxation time in the zT values for a material.
Here, we also calculate the TE and the related transport properties of these two series of 2D elemental crystals by considering full el–ph coupling. Figure 8a shows the calculated Seebeck coefficient S for the group IV materials as a function of chemical potential Ef at 300 K using band- and momentum-dependent relaxation times considering full el–ph couplings. For comparison, the results using the CRTA method are also presented. As we know, the behavior of the Seebeck coefficient can be understood by the Mott relation43,44,45,
Where N(E) and τ(E) are the energy-dependent density of states (DOS) and electronic relaxation time, respectively. The Seebeck coefficient can be separated into the band term and the scattering term. A large enhancement of the DOS near the Fermi level leads to a high S. The scattering term indicates that S is also related to the logarithmic energy derivatives of τ(E), which is inversely proportional to the scattering rates and usually ignored in TE community. In the CRTA method, the scatterings term is equal to zero considering the definition as shown in Eq. (3).

The a–c seebeck coefficients d–f electrical conductivity g–i zT value as a function of chemical potential at 300 K for silicene, germanene, and stanene.
Considering the n-type systems (Ef > 0), the scattering rates of electrons gradually increase as shown in Fig. 2a–c, therefore the scattering term is positive and considerably large, compared with the zero scattering term in the CRTA method. Moreover, when the scattering term exceeds the band term, S even undergoes an abnormal sign-reversal. Hence, regarding the relaxation time as a constant for group IV materials brings significant errors to S. However, for the group V materials as shown in Fig. 9a, the energy-dependent relaxation time influences little on the Seebeck coefficient resulted from the overwhelming band term in the Seebeck coefficient. The maximum Seebeck coefficient of group V materials is ~1500 μVK−1, which is nearly 8 times larger than the group IV materials and many traditional bulk TE materials including Bi2Te3 (215 μVK−1)46, PbTe (185 μVK−1)47, and SnSe (~510 μVK−1)48.

The a–c seebeck coefficients d–f electrical conductivity g–i zT value as a function of chemical potential at 300 K for phosphorene, arsenene, and antimonene.
For the electrical conductivity σ as shown in Figs. 8b and 9b, near Ef = 0, σ reaches ~1 × 105 Ω−1 m−1, ~1 × 103 Ω−1 m−1, ~1 × 104 Ω−1 m−1 for silicene, germanene, and stanene respectively, nearly 1, 4, and 3 orders smaller in magnitude than those calculated using the CRTA method respectively. The σ for n-type group V materials are nearly overestimated by an order of magnitude. For a p-type system, the DPA method gives a better estimation of σ for arsenene, and antimonene compared with phosphorene. As mentioned above, in phosphorene the intervalley scatterings via ZA phonons are strong which fails the DPA method.
The total thermal conductivities is the sum of the electronic contribution κe and the lattice contribution κL, i.e. κ = κe + κL. The electronic thermal conductivity κe exhibits similar trend with σ for both DPA and the full el–ph scattering methods. According to the Wiedemann–Franz law, κe = LσT, where L is the Lorenz number, increasing σ leads to the increase of κe. The lattice thermal conductivities κL for these two series of 2D elemental crystals are chosen from our previous reports49.
The results of dimensionless figure of merit zT are shown in Figs. 8c and 9c. For group IV materials, due to the highly overestimated σ by the CRTA method, despite the relatively small S, the maximum zT value by the CRTA method are in the range of 1.4–1.7, which is much larger than many traditional thermoelectric materials, e.g., Bi2Te3 (1.2)46, PbTe (0.30)50, SnSe (0.70)51. However, the zT values for group V materials predicted by considering full el–ph coupling are only 0.02–0.2, much smaller than those predicted by using the DPA method. For example, the p-type phosphorene was reported as a good room-temperature thermoelectric material by the CRTA method with zT = 0.48, but the zT value decreases to 0.008 when considering full el–ph coupling. The maximum zT value at 300 K in antimonene reaches ~0.4, which is 5 times smaller than that predicted by the CRTA method, and is the highest zT among these materials. As shown in Supplementary Fig. 8, when the temperature rises, the maximum zT value for antimonene at 700 K reaches 1.3, which is comparable to conventional TE materials such as PbSe (1.1 at 900 K)52, Cu2Se (1.5 at 1000 K)53, and PbTe (1.64 at 770 K)54.
In summary, using first principles calculations, we systematically investigate the effects of the electron–phonon couplings in 2D honeycomb group IV and group V materials with D3d symmetry. High buckling increases the overlap between pz orbitals and enhances the interaction between carriers and ZA phonons. The less dispersive band structure of group V materials further enhance the electron–phonon coupling due to easier satisfaction of energy conservation condition. We find that the D3d symmetry is not a sufficient condition for ZA phonon scattering to dominate. Based on our results of selection rules, the symmetry of transition process only permits LA phonon to be involved in intravalley scattering within VBM in monolayer phosphorus, arsenene and antimonene. However, TA and ZA modes from intervalley scattering between degenerate VBM is dominant in phosphorus and far beyond LA modes. Yet only LA modes dominate in monolayer arsenene and antimonene since no intervalley scattering between VBM at Γ point due to lack of degeneracy. By comparison to the carrier mobilities calculated by the DPA and the full el–ph couplings, respectively, we find the DPA method always overestimates carrier mobilities except hole mobilities in monolayer β-Sb and As. Furthermore, we evaluate the TE performance of all these materials with the constant relaxation time obtained from the DPA method, and band- and momentum-resolved relaxation rates respectively, which indicate that the TE properties of materials may be seriously misestimate with constant relaxation time approximation due to neglecting the intervalley scatterings or intravalley scatterings via phonons other than LA modes.
Methods
Electron–phonon coupling
We carry out density functional theory (DFT) using the QUANTUM ESPRESSO code55 with the local density approximation (LDA)56. Norm-conserving pseudopotentials (NCPP) method with a kinetic energy cutoff of 90 Ry are used to perform the self-consistent DFT calculation. A vacuum space of 28 Å; is set along the perpendicular direction to eliminate the interlayer interacions due to the periodic boundary conditions. For the phonon dispersion, the coarse Monkhorst-Pack k-mesh and q-mesh for all the materials discussed are taken as 16 × 16 × 1 and 8 × 8 × 1 respectively. The Wannier interpolation method is used to generate the ultradense fine grid to describe the processes of electron–phonon scattering accurately in the Wannier9057 and EPW code58,59. Accordingly, much fine k and q meshes, 240 × 240 × 1 and 120 × 120 × 1 can be used to calculate the el–ph coupling matrix guaranteeing the numerical convergence of the intrinsic carrier mobility. In the Boltzmann transport theory, the electron mobility can be defined as60
Where ne is the number of electrons, Ω and ΩBZ denote the volume of the unit cell and the first Brillouin zone, respectively, \({f}_{n{{{\bf{k}}}}}^{0}\) is the Fermi-Dirac distribution, vnk,α = h−1∂ϵnk/∂kα is the velocity of the single-particle electron eigenvalue ϵnk. The interaction between electron and phonon lies in the parameter τnk which is expressed as60
where the sum is over all the final band index m for electrons and all the phonon mode index ν and wavevector q. ωqν and nqν are the frequency and Bose-Einstein distribution of phonons. ϵmk+q and \({f}_{m{{{\bf{k}}}}+{{{\bf{q}}}}}^{0}\) are the electron eigenvalue and the Fermi-Dirac distribution of the final state with band index m and wavevector \({{{\bf{k}}}}^{\prime} ={{{\bf{k}}}}+{{{\bf{q}}}}\). The electron–phonon matrix element \({g}_{mn\nu }\left({{{\bf{k}}}},{{{\bf{q}}}}\right)\) is calculated using61
with ϕnk and ϕmk+q being the initial and final electronic Bloch state, respectively. ΔqνVKS is the variation of the self-consistent Kohn-Sham (KS) potential experienced by the electrons.
Electronic transport properties
We use BoltzTraP2 code based on the rigid band approach to calculate the transport parameters for arsenene and antimonene monolayers62. The temperature- and doping-dependent electrical transport properties, including carrier concentrations for holes and electrons nh/e, electronic conductivity σ, electronic thermal conductivities κel and Seebeck coefficient S are computed by solving the semiclassical Boltzmann transport equation (BTE), which can be written as63,64,65,
where Ω is the unit cell volume, f0 is the Fermi-Dirac distribution, μ is the chemical potential, D(ε) is the density of states, \({\overline{\sigma }}_{\alpha \beta }(\varepsilon )\) is the energy-dependent conductivity tensor, which can be obtained by \({\overline{\sigma }}_{\alpha \beta }(\varepsilon )=\frac{1}{N}{\sum }_{n,{{{\bf{k}}}}}{\overline{\sigma }}_{\alpha \beta }(n,{{{\bf{k}}}})\frac{\delta (\varepsilon -{\varepsilon }_{n,k})}{d\varepsilon }\), in which N is the number of sampled k points and \({\overline{\sigma }}_{\alpha \beta }(n,{{{\bf{k}}}})\) can be calculated by the formulae based on the kinetic theory, i.e., \({\overline{\sigma }}_{\alpha \beta }(n,{{{\bf{k}}}})={e}^{2}{\tau }_{n,{{{\bf{k}}}}}{v}_{\alpha }(i,{{{\bf{k}}}}){v}_{\beta }(n,{{{\bf{k}}}})\). The velocity of carrier vα,β(n, k) is defined by \({v}_{i}=\frac{1}{\hslash }\frac{\partial {\varepsilon }_{n,{{{\bf{k}}}}}}{\partial {k}_{i}}(i=\alpha ,\beta )\). In this code, the electron–phonon relaxation time is treated as both energy and direction dependent from Eq. (2).
Data availability
The data that support the findings of this study and the code for the first principles methods proposed in this work are available from the corresponding author (Hao Zhang) upon reasonable request.
Code availability
The codes in this study are available from the authors upon reasonable request.
References
Giri, A., Tokina, M. V., Prezhdo, O. V. & Hopkins, P. E. Electron–phonon coupling and related transport properties of metals and intermetallic alloys from first principles. Mater. Tod. Phys. 12, 100175 (2020).
Nakamura, Y. et al. Intrinsic charge transport in stanene: roles of bucklings and electron-phonon couplings. Adv. Electron. Mater. 3, 1700143 (2017).
Yang, X. et al. Indirect electron-phonon interaction leading to significant reduction of thermal conductivity in graphene. Mater. Tod. Phys. 18, 100315 (2021).
Liu, C. D., Yao, M. J., Yang, J., Xi, J. Y. & Ke, X. Z. Strong electron-phonon interaction induced significant reduction in lattice thermal conductivities for single-layer MoS2 and PtSSe. Mater. Tod. Phys. 15, 100277 (2020).
Long, M. Q., Tang, L., Wang, D., Wang, L. J. & Shuai, Z. G. Theoretical predictions of size-dependent carrier mobility and polarity in graphene. J. Am. Chem. Soc. 131, 17728–17729 (2009).
Long, M. Q., Tang, L., Wang, D., Li, Y. L. & Shuai, Z. G. Electronic structure and carrier mobility in graphdiyne sheet and nanoribbons: Theoretical predictions. ACS Nano 5, 2593–2600 (2011).
Xi, J. Y., Long, M. Q., Tang, L., Wang, D. & Shuai, Z. G. First-principles prediction of charge mobility in carbon and organic nanomaterials. Nanoscale 4, 4348 (2012).
Qiao, J. S., Kong, X. H., Hu, Z. X., Yang, F. & Ji, W. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat. Commun. 5, 4475 (2014).
Du, Y. P. et al. Unexpected magnetic semiconductor behavior in zigzag phosphorene nanoribbons driven by half-filled one dimensional band. Sci. Rep. 5, 8921 (2015).
Gunst, T., Markussen, T., Stokbro, K. & Brandbyge, M. First-principles method for electron-phonon coupling and electron mobility: Applications to two-dimensional materials. Phys. Rev. B 93, 035414 (2016).
Gaddemane, G., Vandenberghe, W. G. & Fischetti, M. V. Theoretical study of electron transport in silicene and germanene using full-band monte carlo simulations. In 2016 International Conference on Simulation of Semiconductor Processes and Devices (SISPAD), https://doi.org/10.1109/sispad.2016.7605219 (IEEE, 2016).
Peng, Q., Wen, X. D. & De, S. Mechanical stabilities of silicene. RSC Adv. 3, 13772 (2013).
Balendhran, S., Walia, S., Nili, H., Sriram, S. & Bhaskaran, M. Elemental analogues of graphene: silicene, germanene, stanene, and phosphorene. Small 11, 640–652 (2015).
Xu, Y. et al. Large-gap quantum spin hall insulators in tin films. Phys. Rev. Lett. 111, 136804 (2013).
Broek, B. et al. Two-dimensional hexagonal tin:ab initiogeometry, stability, electronic structure and functionalization. 2D Mater 1, 021004 (2014).
Guo, J. et al. Strain-induced band engineering in monolayer stanene on sb(111). Phys. Rev. Mater 1, 054004 (2017).
Zhu, Z. & Tománek, D. Semiconducting layered blue phosphorus: a computational study. Phys. Rev. Lett. 112, 176802 (2014).
Zhang, S. L., Yan, Z., Li, Y. F., Chen, Z. F. & Zeng, H. B. Atomically thin arsenene and antimonene: semimetal-semiconductor and indirect-direct band-gap transitions. Angew. Chem. Int. Ed. 54, 3112–3115 (2015).
Sharma, A. & Pandey, A. Blue phosphorene/MoS2 heterostructure based SPR sensor with enhanced sensitivity. IEEE Photon. Technol. Lett. 30, 595–598 (2018).
Ye, X. J. et al. Monolayer, bilayer, and heterostructure arsenene as potential anode materials for magnesium-ion batteries: A first-principles study. J. Phys. Chem. C 123, 15777–15786 (2019).
Zhang, P. F. et al. Topological and electronic transitions in a sb(111) nanofilm: the interplay between quantum confinement and surface effect. Phys. Rev. B 85, 201410 (2012).
Wang, Y. Y. et al. Many-body effect, carrier mobility, and device performance of hexagonal arsenene and antimonene. Chem. Soc. Rev. 29, 2191–2201 (2017).
Mortazavi, B., Dianat, A., Cuniberti, G. & Rabczuk, T. Application of silicene, germanene and stanene for na or li ion storage: a theoretical investigation. Electrochim. Acta 213, 865–870 (2016).
Kurpas, M., Junior, P., Gmitra, M. & Fabian, J. Spin-orbit coupling in elemental two-dimensional materials. Phys. Rev. B 100, 125422 (2019).
Balendhran, S. et al. Elemental analogues of graphene: silicene, germanene, stanene, and phosphorene. Small 11, 640–652 (2014).
Zhang, D. C., Zhang, A. X., Guo, S. D. & Duan, Y. F. Thermoelectric properties of beta-as, sb and bi monolayers. RSC Adv. 7, 24537–24546 (2017).
Cahangirov, S. et al. Two- and one-dimensional honeycomb structures of silicon and germanium. Phys. Rev. Lett. 102, 236804 (2009).
Akturk, E. et al. Single and bilayer bismuthene: stability at high temperature and mechanical and electronic properties. Phys. Rev. B 94, 014115 (2016).
Silvi, B. & Savin, A. Classification of chemical-bonds based on topological analysis of electron localization functions. Nature 371, 683–686 (1994).
Jahn, H. A. & Teller, E. Stability of polyatomic molecules in degenerate electronic states - I—orbital degeneracy. Proc. R. Soc. Lond. 161, 220–235 (1937).
Shao, H. Z. et al. First-principles study on the elastic properties of cu2gese3. EPL (Europhysics Letters) 113, 26001 (2016).
Lang, H. F., Zhang, S. Q. & Liu, Z. R. Mobility anisotropy of two-dimensional semiconductors. Phys. Rev. B 94, 235306 (2016).
Herring, C. & Vogt, E. Transport and deformation potential theory for many-valley semiconductors with anisotropic scattering. Phys. Rev. 101, 944–961 (1956).
Malard, L. M. et al. Group-theory analysis of electrons and phonons in n -layer graphene systems. Phys. Rev. B 79, 125426 (2009).
Jiang, J. et al. Intensity of the resonance raman excitation spectra of single-wall carbon nanotubes. Phys. Rev. B 71, 205420 (2005).
Castro Neto, A. H. & Guinea, F. Electron-phonon coupling and raman spectroscopy in graphene. Phys. Rev. B 75, 045404 (2007).
Chu, Y. B., Gautreau, P. & Basaran, C. Parity conservation in electron-phonon scattering in zigzag graphene nanoribbon. Appl. Phys. Lett. 105, 347 (2014).
Dresselhaus, M. S., Dresselhaus, G. & Jorio, A. Group Theory (Springer-Verlag New York, LLC, 2008).
Dresselhaus, M. S. et al. New directions for low-dimensional thermoelectric materials. Adv. Mater. 19, 1043–1053 (2007).
Pei, Y. Z. et al. Convergence of electronic bands for high performance bulk thermoelectrics. Nature 473, 66–69 (2011).
Zhao, L. D., Chang, C., Tan, G. J. & Kanatzidis, M. G. SnSe: a remarkable new thermoelectric material. Energy Environ. Sci. 9, 3044–3060 (2016).
Chen, K. X. et al. Excellent thermoelectric performance predicted in two-dimensional buckled antimonene: a first-principles study. J. Phys. Chem. C 121, 13035–13042 (2017).
Sun, P. J. et al. Large seebeck effect by charge-mobility engineering. Nat. Commun. 6, 7475 (2015).
Wei, B. P. et al. Nernst effect of the intermediate valence compound YbAl3: revisiting the thermoelectric properties. J. Phys. Condens. Matter. 27, 105601 (2015).
Liang, J. H., Fan, D. D., Jiang, P. H., Liu, H. J. & Zhao, W. Y. First-principles study of the thermoelectric properties of intermetallic compound YbAl 3. Intermetallics 87, 27–30 (2017).
Poudel, B. et al. High-thermoelectric performance of nanostructured bismuth antimony telluride bulk alloys. Science 320, 634–638 (2008).
Nielsen, M. D., Levin, E. M., Jaworski, C. M., Schmidt-Rohr, K. & Heremans, J. P. Chromium as resonant donor impurity in pbte. Phys. Rev. B 85, 045210 (2012).
Zhang, X. & Zhao, L. D. Thermoelectric materials: Energy conversion between heat and electricity. J. Materiomics 1, 92–105 (2015).
Peng, B. et al. The conflicting role of buckled structure in phonon transport of 2d group-iv and group-v materials. Nanoscale 9, 7397 (2017).
Zhang, Q. Y. et al. Effect of aluminum on the thermoelectric properties of nanostructured PbTe. Nanotechnology 24, 345705 (2013).
Wang, F. Q., Zhang, S. H., Yu, J. B. & Wang, Q. Thermoelectric properties of single-layered snse sheet. Nanoscale 7, 15962–15970 (2015).
Wang, H., Gibbs, Z. M., Takagiwa, Y. & Snyder, G. J. Tuning bands of PbSe for better thermoelectric efficiency. Energy Environ. Sci. 7, 804–811 (2014).
Liu, H. L. et al. Copper ion liquid-like thermoelectrics. Nat. Mater. 11, 422–425 (2012).
Ahn, K. et al. Enhanced thermoelectric properties of p-type nanostructured PbTe–MTe (m = cd, hg) materials. Energy Environ. Sci. 6, 1529 (2013).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 21, 395502 (2009).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Mostofi, A. A. et al. An updated version of wannier90: a tool for obtaining maximally-localised wannier functions. Comput. Phys. Commun. 185, 2309–2310 (2014).
Noffsinger, J. et al. EPW: A program for calculating the electron-phonon coupling using maximally localized wannier functions. Comput. Phys. Commun. 181, 2140–2148 (2010).
Poncé, S., Margine, E. R., Verdi, C. & Giustino, F. EPW: Electron-phonon coupling, transport and superconducting properties using maximally localized wannier functions. Comput. Phys. Commun. 209, 116–133 (2016).
Xi, J. Y., Wang, D., Yi, Y. P. & Shuai, Z. G. Electron-phonon couplings and carrier mobility in graphynes sheet calculated using the wannier-interpolation approach. J. Chem. Phys. 141, 034704 (2014).
Baroni, S., de Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001).
Madsen, G. K. H., Carrete, J. & Verstraete, M. J. Boltztrap2, a program for interpolating band structures and calculating semi-classical transport coefficients. Comput. Phys. Commun. 231, 140–145 (2018).
Madsen, G. K. H. & Singh, D. J. Boltztrap. a code for calculating band-structure dependent quantities. Comput. Phys. Commun. 175, 67–71 (2006).
Yang, J. et al. Evaluation of half-heusler compounds as thermoelectric materials based on the calculated electrical transport properties. Adv. Funct. Mater. 18, 2880–2888 (2008).
Hong, A. J. et al. Predicting high thermoelectric performance of abx ternary compounds namgx (x = p, sb, as) with weak electron-phonon coupling and strong bonding anharmonicity. J. Mater. Chem. C 4, 3281–3289 (2016).
Acknowledgements
This work is supported by the National Natural Science Foundation of China under Grants No. 11374063, 11674062 and 11404348, the National Key R&D Program of China (2017YFA0303403), the Shanghai Municipal Natural Science Foundation under Grant No. 19ZR1402900, the Natural Science Foundation of Jiangsu Province under grant No. BK20180456 and Fudan University-CIOMP Joint Fund (FC2019-006).
Author information
Authors and Affiliations
Contributions
H.Z. and H. Shao conceived and designed the study. Y. Wu and R. Zhang performed the first-principle calculations. B. Hou performed the group-theory analysis and selection rules. Y. Chen, J. Cao, C. Ma, Y. Zhang, Z. Lu, H. Zhu, and Z. Fang analyzed and discussed the results. Y. Wu, B. Hou, J. Cao, and H.Z. wrote the text of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Y., Hou, B., Chen, Y. et al. Strong electron–phonon coupling influences carrier transport and thermoelectric performances in group-IV/V elemental monolayers. npj Comput Mater 7, 145 (2021). https://doi.org/10.1038/s41524-021-00619-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-021-00619-0
- Springer Nature Limited
This article is cited by
-
Full-landscape selection rules of electrons and phonons and temperature-induced effects in 2D silicon and germanium allotropes
npj Computational Materials (2024)