
Abstract
We previously showed that each dog with chronic non-invasive sino-nasal aspergillosis (SNA) was infected with a single genotype of Aspergillus fumigatus. Here, we studied the transcriptome of this fungal pathogen and the canine host within the biofilm resulting from the infection. We describe here transcriptomes resulting from natural infections in animal species with A. fumigatus. The host transcriptome showed high expression of IL-8 and alarmins, uncontrolled inflammatory reaction and dysregulation of the Th17 response. The fungal transcriptome showed in particular expression of genes involved in secondary metabolites and nutrient acquisition. Single-nucleotide polymorphism analysis of fungal isolates from the biofilms showed large genetic variability and changes related with adaptation to host environmental factors. This was accompanied with large phenotypic variability in in vitro stress assays, even between isolates from the same canine patient. Our analysis provides insights in genetic and phenotypic variability of Aspergillus fumigatus in biofilms of naturally infected dogs reflecting in-host adaptation. Absence of a Th17 response and dampening of the Th1 response contributes to the formation of a chronic sino-nasal warzone.
Similar content being viewed by others

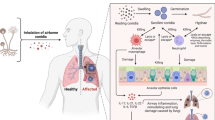

Introduction
Infections with Aspergillus fumigatus occur in a wide variety of animal species including humans, birds, cows, horses, cats, and dogs1,2. In dogs, sino-nasal aspergillosis (SNA) is the most common Aspergillus infection3. The fungus can reside in the host for an extended period of time (e.g., months) before manifesting clinical signs, which include sneezing, mucoid nasal discharge, nasal depigmentation, epistaxis, and turbinate destruction2,4. This relative long build-up time facilitates the fungus to form a fungal biofilm covering the sino-nasal area up to the frontal sinus before the disease is diagnosed2. Proper diagnosis of SNA requires a combination of computed tomography (CT), rhinoscopy, histopathology, cytology, fungal culturing, and serology2. Topical administration of azole drugs remains the most widely used and successful treatment for SNA in dogs although reports of its use are limited2,3,5. SNA has been associated with upregulation of Th1 cytokines like interleukin (IL)-8 and TNF-α6,7,8. This may indicate a localized response to the fungus to restrain the dispersal and development of an invasive infection. Yet, a high level of inflammation and incapability to clear the infection has been suggested to be due to the upregulation of anti-inflammatory molecules such as transforming growth factor-β and IL-107,8.
To date, only in vivo A. fumigatus transcriptomes are available from murine infection models9,10 but these may not be representative of natural infections. Here, we describe for the first-time transcriptomic profiling of natural A. fumigatus infections in the sinus of canine hosts using RNA sequencing. In all cases we studied the fungus that had formed a mature biofilm. We previously showed fungal heterogeneity at the level of pigmentation between isolates from a single dog as compared with isolates from human as well as indoor and outdoor substrates4. This suggested that the dog sinus is a highly selective environment inducing in-host adaptations resulting in phenotypic evolution. The fungus is expected to experience among others nutrient stress, host immune responses, and limited oxygen availability as selection pressures. The results described in this paper show that the sino-nasal mucosal surface is a warzone between A. fumigatus and the host tissue. The host fights the fungal biofilm using nutritional immunity, and inflammatory and Th1 responses, and on the other battle side, the adaptive responses of A. fumigatus enable growth of the pathogen and contribute to the suppression of the host immune response.
Results
Dog transcriptome
Read mapping of the four fungal plaques from three different dogs with SNA to the dog reference transcriptome (38019 transcripts) resulted in 8706–12578 transcripts with TPM > 1 per sample (Fig. 1A). About 44% (i.e., 6536) of the transcripts were shared between the four plaques (Fig. 1A). This number corresponds to 17% of the total transcriptome of C. lupus familiaris. The relatively low number of expressed transcripts reflects the low amount of canine RNA present in the fungal plaques (i.e., 1–17% of a total of 3–6 × 107 reads) (Supplementary data set 1). The 6536 shared transcripts were considered as the SNA dog transcriptome and were used for further analysis. Hierarchical clustering of the shared transcriptomes showed that the two samples derived from the same dog, CP8 and CP8.2, clustered together (Fig. 1B). This suggests that the four transcriptomes share many features even though the fungal pathogens have different genotypes4. GO and KEGG term analysis (Fig. 2 and Supplementary data set 1; see Goprofiler) showed significant enrichment in the shared expressed transcripts in general terms like cellular metabolic process (GO:0044237), and RNA binding (GO:0003723). In addition, immune response terms like Toll-like receptor binding (GO:0035325), cytokine production (GO:0001816), Th1 and Th2 cell differentiation (KEGG:04658), as well as osteoclast differentiation (KEGG: 03480) were overrepresented. The latter is related to bone destruction and remodeling.

Transcriptome profiles of four fungal plaques of three different dogs with SNA. Venn diagram a showing number and percentage of shared transcripts (TPM > 1) between all four fungal plaques and heatmaps with hierarchical clustering of the 6536 shared transcripts b and the 660 transcripts mapped to the InnateDB database11 c. Number of transcripts with TPM > 1 is shown under each patient code in a.

GO and KEGG term enrichment analysis of the 6536 shared expressed transcripts in four fungal plaques from three dogs with SNA. BP indicates biological process, CC cellular component and MF molecular function (for full list see Supplementary data set 1).
Ranking of the transcripts by their mean TPM revealed that the top 50 had 20 transcripts related to immune response with IL-8 as the highest ranked transcript (Supplementary data set 1). Also, high amounts of transcripts related to nutritional immunity (e.g., ferritin, and S100A8, A9, and A12) were detected.
Mapping the SNA dog transcriptome to the immune database InnateDB11 resulted in 660 hits (i.e., 11% of the 6536 shared transcripts) (Supplementary data set 1). As observed with the general SNA transcriptome (Fig. 1B), the InnateDB transcripts derived from the same dog (CP8, CP8.2) clustered (Fig. 1C). This included among others 6 interleukins (IL-1, 8, 10, 15, 16, 18, 23A), 12 interleukin receptors (IL 1R2, 2RG, 4R, 10RA, 13RA1, 15RA, 17RA/B, 18R1, 22RA2, 27RA, 31RA), seven toll-like receptors (TLR-1, 2, 4, 6, 7, 8, 10), and other important genes such as nucleotide-binding oligomerization domain containing protein 2, and Interferon gamma (Supplementary data set 1). The Bruvo genetic distance between isolates from these patients differed 0.46 as determined by STRAf4. This indicates that the host and not the genotype of A. fumigatus has strongest impact on the immune response.
We compared our RNAseq results with previously published canine microarray data describing differential gene expression of healthy dogs versus dogs with SNA6. We used a log2 fold change ±2 observed in the microarray data for the identification of differentially expressed genes. This resulted in 948 out of 5551 differentially expressed transcripts. Of these differentially expressed transcripts, 307 transcripts were also expressed in our RNAseq data (Supplementary data set 1). Enrichment analysis of these 307 differentially expressed genes showed that upregulated genes (264) were enriched in immune-related terms like superoxide anion generation (GO:0042554), phagocytosis (GO:0006909), and immune system process (GO:002376). (Supplementary data set 1). Interestingly, 15 transcripts within the top 50 highest expressed genes detected by RNAseq analysis were differently expressed in the microarray (Supplementary data set 1). Genes like S100 A12, A9, A8, and SOD2 were not differentially expressed in the microarray. In contrast, IL-8 and IDO1 were upregulated and LTF was downregulated (Fig. 3).

Comparison between the RNAseq data of this study and the microarray from6. Shared transcripts between our RNAseq data and found to be upregulated, downregulated, and not differentially expressed (log2 FC ≥ 2) in the microarray are in green, red, and gray, respectively. Transcripts mentioned in the Results and/or Discussion section are labeled.
Fungal transcriptome
A total of 9213 genes, representing 92.8% of the genes of the published Af293 genome12 were found to be expressed in the SNA samples (TPM > 1) (Supplementary data set 2). Of these genes, 8029 (~87%) were shared among all four fungal plaques. We observed a high variation of expression levels between these samples, i.e., in some cases >1000-fold. Therefore, the coefficient of variation (CV) of the log10 TPM (CV = standard deviation in four samples/mean in four samples) of the 8029 expressed genes was calculated and classified as highly stable expressed (CV < 0.05), medium stable expressed (0.5 > CV > 0.05) and highly variable expressed (CV > 0.5). These groups compromised 13, 81, and 6% of the genes, respectively.
Hierarchical clustering of the transcriptomes of shared genes in each of the fungal plaques of the three dogs revealed that fungal plaques from the same dog (CP8 and CP8.2) have a more similar expression profile than those of CP6 and CP7 (Fig. 4). Notably, the expression profiles of CP6 and CP8 are most different although the fungal isolates derived from these two plaques have the same genotype4. A similar distribution of gene expression was observed when shared expressed genes were compared that are related to secondary metabolism13 (Supplementary Fig. 1A), stress-response genes14 (Supplementary Fig. 1B), pathogen–host interaction15 (Supplementary Fig. 1C) and transcription factors (TFs) related to virulence16 and pathogenesis combined with reproduction and stress response17 (Supplementary Fig. 1D). Again, the transcriptomes of CP6 and CP8 were most distinct although these strains have the same genotype. Apparently, the host is more directive in gene expression.

Heatmap clustering expression of shared genes expressed in the fungal plaques of CP6, CP7, CP8, and CP8.2.
Fifteen GO terms were enriched within the 1046 expressed genes with low variability, most of them belonging to general categories like mRNA metabolic process (GO:0016071) and translation (GO:0006412) (Supplementary data set 1). In agreement, FunCat ontology analysis also showed enrichment in categories like translation (12.04), and ribosomal proteins (12.01.01) (Fig. 5). Enrichment of genes categorized as having a medium variability in expression showed general categories like cellular protein catabolic process (GO:0044257), cellular process (GO:0009987) and binding (GO:0005488). On the other hand, the 514 genes with highly variable expression were only enriched in the GO terms oxidation-reduction process (GO:0055114) and oxidoreductase activity (GO:0016491) (Supplementary data set 2). In the case of FunCat analysis, this group of genes showed enrichment in secondary metabolism (01.20), defense related proteins (32.05.03), resistance proteins (32.05.01) and other terms related to virulence (Fig. 5B; Supplementary data set 2).

GO a and FunCat (b; representing the first five terms) enrichment of shared expressed genes with low and high variation in expression. See Supplementary data set 2 for the complete enrichment analysis.
Whole-genome sequencing of fungal isolates
Genomic DNA of 34 isolates was sequenced with an average 23.61-fold coverage (Supplementary data set 3) and compared with the Af293 reference genome12. The variation of the genomes of the isolates ranged between 0.11% and 0.3% relative to Af293. The SNPs were regularly distributed along the 29,388,377 bp Af293 genome. Biallelic SNPs were the most prevalent variant (average number 56064), whereas multiallelic SNPs (average number 219) were much less frequently detected (Supplementary data set 3). Interestingly, the genomes of six isolates from a total of three dogs contained 2.5-fold more biallelic SNPs when compared to the other dog and environmental isolates (Fig. 6). This high-SNP number was generally distributed along the genome (Supplementary data set 3). Three out of the six isolates had been isolated from dog CP2, from which also one isolate had been obtained with a low SNP incidence (Fig. 6).

Number of biallelic SNPs per A. fumigatus isolate.
Phylogeny
Clades A–D were identified using a SNP-based phylogeny (Fig. 7). Clades B and D contain both environmental and dog isolates, whereas clades A and C contain canine isolates only. The isolates of clade A are derived from patients CP1, CP6, and CP8.2 and have a very small genetic distance based on the SNP-based phylogenetic tree. This is in agreement with genotyping based on STRAf4 (Supplementary data set 3). Clade B contained isolates of CP3 only, whereas clade C contained isolates of CP7 and CP8.2 and clade D of CP2, CP4, CP5, and CP8. In general, clustering was in line with microsatellite analysis with two exceptions. Isolates DTO 271-A9 (from CP2) and DTO 303-F3 (from CP8.2) are now in subclade D1 but were expected to be in subclade D2 and cluster A, respectively, based on the microsatellite analysis4.

SNP-based phylogenetic tree of the 26 isolates from eight SNA patients and from eight environmental isolates. The tree was inferred by using the maximum likelihood method based on the general time-reversible model. The tree with the highest log likelihood (−49,077.29) is shown and drawn to scale, with branch lengths measured in the number of substitutions per site. There was a total of 3826 nucleotide positions in the final data set. Evolutionary analyses were conducted in MEGA X71. Selected clades (A, B, C, D) and sub clades (B1, D1, D2) are depicted with letters and numbers. The dog and environmental isolates are indicated with a dog and house pictogram, respectively, whereas G, D, and E indicate the number of STRAf genotypes, dog isolates, and environmental isolates, respectively. Two isolates that were not grouped with isolates from the same STRAf genotype are denoted by a star (*). Isolates with a notable high-SNP density are denoted by a plus (+) symbol.
SNPs in secondary metabolite clusters
Non-core SNPs of dog isolates with high impact are enriched in secondary metabolic gene clusters. A missense mutation (Cys2335Ser) in Fma-PKS (Afu8g00370) (Table 1) encoding a polyketide synthase (PKS) of the fumagillin cluster (cluster 30) is present in 15 out of the 26 isolates present in clades B, C, and D but absent in clade A (CP1, CP6, and CP8.2). This nsSNP (non-synonymous SNP) is not present in two out of eight environmental isolates (i.e., DTO 060-G9 and DTO 063-C3) (Fig. 8). Fumagillin is one of the mycotoxins of A. fumigatus and whether this SNP affects the fumagillin function needs to be addressed.

SNPs present in an isolate are indicated by black shading of the cell. The origin of the isolates is depicted as a colored bar to the left.
Two missense mutations were detected in the cytochrome P450 (CYP450) monooxygenase GliC gene (Afu6g09670) (Table 1), which is involved in the gliotoxin biosynthesis pathway. Interestingly, the nsSNP 1310 A > T resulting in the Asp437Val substitution in GliC was absent in 66 A. fumigatus isolates previously described18 and is also absent in the eight environmental isolates described here. However, this nsSNP is present in 19 out of 26 dog isolates (Fig. 8). In addition, we found many SNPs in dog isolates causing missense mutations in genes encoding GliT, GliA, and GtmA that are involved in transport (GliA), detoxification of gliotoxin (GliT, GtmA), and downregulation of the expression of the gliotoxin cluster19,20,21.
Various SNPs with HIGH impact were detected in the pes3 gene (Afu5g12730) encoding a multi-modular NRP synthetase of 8515 amino acids22 (Table 1). A SNP resulting in a stop codon at amino-acid 1590 (Gly1590*) was found in the isolates belonging to cluster D1 (Fig. 7) and was absent in the environmental isolates (Fig. 8). Similarly, introduction of a stop codon at position 3299 (Trp3299*) was found in isolates of clade C.
Fumitremorgins are tremorgenic mycotoxins of A. fumigatus but their role in infection is not clear yet. Interestingly, the encoding FTM cluster is inactive in reference strain Af293 because of an Arg202Leu missense mutation in the FtmD gene (Afu8g00200)23. All fungal isolates (i.e., both environmental and dog isolates) did not have this missense mutation, indicating that these isolates might produce fumitremorgin.
Stress response and reproduction
We investigated which nsSNPs of the dog isolates are present in genes involved in reproduction and stress response (Table 1, Supplementary data set 3). One of the SNPs resulted in a stop codon at Leu87* within the orthologue of the fgaCat gene (Afu2g00200). This is a bifunctional catalase-peroxidase involved in the oxidative stress response17. Remarkably, this nonsense mutation is present in none of the environmental isolates and only in the 6 high-SNP isolates, which indicate a loss of function of this catalase-peroxidase. Additionally, we found a missense mutation in the mycelial catalase CatB/Cat1 (Afu3g02270) present only in dog isolates from CP2 (DTO 271-A9/B1/B2/B3) (Fig. 8, Table 1). Notably, 24 missense SNPs were present in the light-sensing regulator FphA (Afu6g09260), of which 21 are only present in the high-SNP isolates. This indicates a difference in light sensing between isolates and suggests that light might reach the nasal or frontal sinus24.
TFs with known roles in pathogenesis
A total of 70 missense mutations were found in 17 TFs involved in virulence (Table 1; Supplementary data set 3). However, no disruptive (stop codons) HIGH impact SNPs were present. The Ser147Pro SNP in TF CpcA was only found in dog isolates of subclade A and both isolates of CP9. SNP Ala113Thr on the hypoxia-responsive TF SrbB was present in almost all dog isolates, except for the ones of CP7 and environmental isolates (Figs. 7 and 8). PacC is involved in pH response and is important in the epithelial entry of conidia and invasive growth10. This gene had the SNP Arg529Cys and was present in all dog isolates, except for the high-SNP isolates. In addition, we found seven nsSNPs in Yap1 (Supplementary data set 3), of which five only occurred in dog isolates. Two of them (Fig. 8, Table 1) were only present in isolate DTO 271-B9 (Clade D2), suggesting that nsSNPs in this gene might have an impact on oxidative stress response (see below).
Other SNPs in genes of interest
A high impact SNP was only present in the six high-SNP isolates and results in the loss of the stop codon (*1270Gln) of Gin4 (Afu6g02300). This gene is involved in conidiation, apical compartment length, and virulence in an invertebrate and intranasal murine infection model of IA25.
Gene AceA (Afu6g07780) encodes a TF involved in the copper toxicity response and regulates the copper transporters CrpA (Afu3g12740) and Ctr2 (Afu3g08180)26. Interestingly, SNPs were found in AceA and CrpA in both environmental and dog isolates (Supplementary data set 3). In the case of CrpA (Fig. 8, Table 1), one SNP was found that was present in 18 dog isolates, and one which was only present in isolates DTO 303-E7 and E9 (clade C). The latter two dog isolates also contained a MODERATE impact nsSNP in Ctr2 (Fig. 8, Table 1). These mutations suggest that in-host adaption to copper stress occurs in dogs.
We did not find alterations in the cyp51A gene in the environmental or dog isolates, which is in accordance with our previous observations that none of these dog isolates showed resistance to three tested azoles4,27.
Micro-evolution in the host
NsSNPs previously described in 13 A. fumigatus isolates taken from a human patient with CGD over a 2 year-time frame28 were compared with the nsSNPs from dog and environmental isolates (Supplementary data set 3). We found an amino-acid change Thr214Ala in the exo-alpha-sialidase Afu4g13800. This protein has been proposed to have a role in the interaction between A. fumigatus and its host. This missense SNP is absent in the environmental isolates but present in the six high-SNP variants and in DTO 303-F1 and F2 in clade C and D, respectively (Figs. 7 and 8). In contrast28, we did not detect SNPs in Afu6g14720 encoding a putative telomere-associated RecQ helicase and which in human isolates had a broad variety of nucleotide changes (Supplementary data set 3).
Phenotyping
Dog isolates were on average more sensitive to H2O2 than the reference strain Af293 (Fig. 9). Moreover, differences in H2O2 sensitivity were observed between isolates from the same fungal plaque. Six isolates (DTO 271-A6 /E5/B4/E7/B7/B9; clades A, B1, C, D2) derived from canine patients (CP1, CP3–CP7, respectively) were very sensitive to H2O2 (inhibition zones >35 mm) as compared with Af293 (~27 mm) and the other dog isolates (inhibition zones >27 mm and <35 mm) derived from different canine patients (CP1, CP3–CP7, respectively) (Fig. 9). SNPs could not be related to H2O2 sensitivity. For example, the SNP conferring a stop codon (Leu87*) in the catalase-peroxidase fgaCat (Table 1, Supplementary data set 3) was present in isolates DTO 271-B1/B2/B3/B7/B8/B9 (clade D) but only two of them were considered as highly sensitive (inhibition halo >35 mm) (Fig. 9). A similar situation was observed for the SNPs in the conidial catalases Cat2 (Afu8g01670) and CatB/1(Afu3g0227) (Table 1).

The color of the cells corresponds to a z score of the test (red = poor growth, blue = good growth, white = average growth). Note that in the case of H2O2 the color relates to the size of the inhibition halo with a red color indicating large inhibition. Average measurements per strain are depicted in each cell. The colored bar to the right indicates the origin of the isolate.
Fungal growth at different pH values
High variation in growth was observed between dog isolates when cultured at pH 5.0, 6.5, 7.2, and 8.0. Notably, out of 27 dog isolates a total of 21 and 3 isolates showed >20% more growth at pH 8 and pH 5, respectively. In particular, growth at pH 5 for 24 h was very poor for the vast majority of fungal strains. However, after 48 h of growth the difference in colony diameters was considerably reduced when compared with the other pH values (Fig. 9). The Arg529Cys SNP found in PacC could not explain the different growth behavior of isolates. For instance, this SNP was found in several isolates but only DTO 271-B9 did not grow after 24 hours at pH 8 (Fig. 9).
Copper stress
Isolates from the environment did not grow at all on high copper medium (0.9 mM) during a 2-day period (Supplementary Fig. 2). In contrast, growth at high copper concentration was observed after 4 days in 15 out of 26 dog isolates (Fig. 9 and Supplementary Fig. 2) but not in 12 other isolates of human and environmental origin (Supplementary Fig. 2). These colonies were generally small except for DTO 303-E9 (CP7) that was similar to Af293 (Fig. 9 and Supplementary Fig. 2). Apparently, these two latter strains are the most resistant but 14 other isolates from dogs also showed resistance to high copper. SNPs causing amino-acid substitutions in the copper transporters Ctr2 (Afu3g08180) and CrpA (Afu3g12740) are present in isolate DTO 303-E9, and only in CrpA (Afu3g12740) in DTO 303-F1 (Table 1, Fig. 8) respectively. This suggests a relationship between copper resistance and the presence of the SNPs.
Discussion
Non-invasive aspergillosis in the SNA of dogs resembles human chronic non-invasive rhinosinusitis that can be caused by several fungal species such as Aspergillus species, Penicillium, Mucor, Rhizopus, and phaeohyphomycetes29. In dogs A. fumigatus is the most commonly identified etiological agent30,31. Here, we describe a transcriptomic profiling of natural A. fumigatus infections in the sinus of canine hosts using RNA sequencing. In fact, it provides the first transcriptomes resulting from natural infections in animal species with A. fumigatus. This study is challenging for several reasons. Differential gene expression cannot be assessed since we are dealing with a variable set of natural infections in dogs of different breeds. In addition, sino-nasal fungal plaques represent large three-dimensional structures that differ in location and they are most commonly found in the nasal cavity or frontal sinus. The time of infection and their micro-environments also differ. Knowledge of SNA is limited, and most studies have focused on diagnostics and treatment of dogs5,30,32,33,34, on fungal resistance27 and on the local immune response6,7,8. These studies suggested an induction of Th1 response during SNA. In addition, interleukin IL-6 was upregulated in dogs with SNA8. IL-6 is related to the Th17 response that promotes antifungal properties of neutrophils35 and is required for optimal fungal clearance at the mucosal level36,37. Microarray analysis showed no differential expression of genes involved in the Th17 response6. This may be explained by the fact that these studies used different dog breeds. The fact that these studies did not analyze individual patients but made use of pools of RNA of different patients makes it difficult to find the cause of the difference in the qPCR and microarray studies. In contrast, we did analyze individual patients enabling analysis of individual immune responses. Of the nine fungal plaques isolated over a period of 2 years, we only obtained RNA from four samples that passed the rigorous quality check required for RNAseq. The data obtained from the same dog showed high similarity in gene expression. Nevertheless, comparison of transcriptomes from more dogs are required to address genetic heterogeneity among dogs further. Our analysis are in agreement with previous qPCR studies7,8 and we found upregulation of factors involved in the Th17 response (i.e., STAT3 and the Il-23 subunit P19). Yet, expression of IL-17 was detected in only one, a Saint Bernard breed (CP7). Thus, the antifungal Th17 response seems to be affected in SNA in certain breeds. A defective Th17 response can result in reduced recruitment of neutrophils to the site of infection. However, strong or prolonged activation of the Th17 response can also cause the opposite effect resulting in an excessive inflammation and immune pathology36,38 Kynurenines may be involved in the dysregulation of the Th17 response at the mucosal surface and in the establishment of the chronic status of SNA in dogs. These molecules result from the degradation of tryptophan via indoleamine 2,3-dioxygenase (IDO1) and play a role in control and establishment of fungal infections39. Both host and A. fumigatus have IDO1 genes and can thus produce kynurenine. It has been reported that kynurenines produced by A. fumigatus can inhibit IL-17 production in human monocyte-derived macrophages40. Furthermore, the formation of host-derived kynurenine on paracoccidioidomycosis was shown to modulate the immune response such that a state of disease tolerance was created, thereby preserving host fitness without pathogen clearance39,41.
The continuous proinflammatory reaction in the dog epithelium can be stimulated by the release of S100 proteins. These proteins have pleiotropic effects on nutritional immunity and induction of proinflammatory responses42. Moreover, expression of pattern recognition receptors involved in the innate immune response to A. fumigatus like TLR (toll-like receptor) 2 and 443, Mincle (CLEC4E)44, and Dectin-1 (CLEC7A)45 accompanied by high expression of IL-8, which can be stimulated by IL-1α, hyphal antigens46,47, and Dectin-147, contributes to the continuous activation of a proinflammatory reaction and a Th1 response. The fact that IL-10 was shown to be expressed in SNA may result in a dampened Th1 response that would prevent excessive tissue damage. Moreover, it can contribute to the chronic state of the infection as suggested previously7. Furthermore, adaptive responses of A. fumigatus enable growth and contribute to the suppression of the host immune response. This includes generation of immune-modulatory metabolites, adaptation to nutritional stress, and phenotypic and genetic plasticity. For instance, full expression of the gliotoxin, neosartoricin and hexadehydroastechrome clusters was found. Gliotoxin has several immune-suppressive activities, killing of immune cells and inhibition of H2O2 production in macrophages48. Furthermore, gliotoxin together with neosartoricin can suppress the local adaptive immune response via inhibition of T-cell proliferation49. nsSNPs were found in fungal genes related with detoxification of reactive oxygen species produced by the host, and in genes involved in the pH and stress response. These host environmental factors can play a role in the selection of variants during in-host adaptation. For example, nsSNPs were found in the copper exporter CrpA and the copper importer Ctr2 in some strains that were able to grow on high concentrations of copper. In addition, expression data from the fungal plaques showed that CrpA had a highly variable expression between all four fungal plaques, which also carried isolates with the nsSNP in the mentioned gene. This suggests that high copper concentrations at the mucosal surface impose a selection of copper-resistant variants. It is not clear how high copper concentration may develop but the metal may be released from macrophages that are known to accumulate copper. Copper is a transition metal that is used by the macrophages to produce hydroxyl radicals in a Fenton reaction to kill intracellular pathogens26,50. Cytotoxic activity of secondary metabolites like gliotoxin may be responsible for release of the copper into the environment thus increasing copper concentration at the mucosal surface.
Zinc is another metal that has an important role in fungal infections, for example, host calprotectin is a heterodimer of S100A8/A9 proteins, which can sequestrate zinc, making it unavailable for the fungus51. Therefore, fungi have developed mechanisms for the uptake and homeostasis of this metal. In the case of A. fumigatus ZrfA, ZrfB, and ZrfC are zinc transporters, and AspF2 (a protein orthologue to the C. albicans zincophore PRA1)52,53. The interaction between ZrfC and AspF2 is of our interest because the former regulates the transport of zinc under alkaline conditions52, and the latter is hypothesized (in C. albicans) to bind to available zinc in the host–pathogen interphase and transfer it to Zrt153,54. In our RNAseq data, ZrfC and AspF2 were expressed in a high stable and medium stable fashion. This observation complements the fact that all fungal plaques presented a pH of 8 as determined by pH measurement directly after isolation of the plaques from the patient. We propose that alkalization of the sino-nasal area is part of the fungal strategy to colonize and counter the host nutritional immunity. Another remarkable phenotype of the fungal biofilm is the absence of asexual reproduction, the fungal plaques appear as white structures and microscopic analysis only scarcely showed some formation of conidiophores4 interestingly, expression analysis indicated no expression of the abaA gene encoding a TF that together with TFs BrlA and WetA are required for asexual reproduction55 Both brlA and wetA are medium variable expressed in the four biofilms (Supplemental material S2). These results explain why asexual reproduction is not observed. Why abaA is not expressed remains to be determined but might be linked to environmental factors in the sinus that influence asexual reproduction.
Together, our results indicate that the interaction between the developing A. fumigatus biofilm and the mucosal immune system ends up in the formation of a sino-nasal warzone. The existence of genetic variants within a fungal biofilm is expected to be the result of selection in this stressful environment of the dog’s nasal cavity and frontal sinus. Generation of individual cells with an increased frequency of mutation can be advantageous for the pathogen in a stressful environment. As such, the situation in SNA would be similar to the one encountered in chronic infections like cystic fibrosis in which hypermutable variants of Pseudomonas aeruginosa were described56,57,58. This phenomenon has previously also been described for the fungal species Candida albicans59 and Cryptococcus neoformans60,61 and this would be the first report for A. fumigatus. Increased mutation rate in our study may be owing to a defective DNA repair system as indicated by the finding that the genes involved in this system show increased incidence of biallelic non-synonymous SNPs. In addition, formation of heterokaryons in the fungal plaque may increase the presence of alternative alleles62.
Methods
Strains
A total of 34 A. fumigatus isolates were used in this study (Table 2), 8 indoor environmental isolates from the Netherlands, 26 isolates from dog patients with SNA. The 26 isolates were obtained from fungal plaques derived from 8 canine patients (CP1-8) suffering from SNA4 (Supplementary Table 1). Part of these plaques were immediately frozen in liquid nitrogen and stored at −80°C for RNA isolation and sequencing. Isolates from dogs were obtained with owner’s consent applying a standardized protocol at Veterinary Medicine, Utrecht University, The Netherlands.
RNA isolation and sequencing
Material of fungal plaques (100–200 mg) was homogenized for 5 min with two metal balls (4.76 mm in diameter) in a Tissuelyzer at 20 Hz min-1 (Qiagen, Venlo, The Netherlands) in the presence of 1 mL TRIzol (Thermo Fisher Scientific, USA). After homogenization, the mixture was incubated for 5 min at RT, followed by the addition of 200 µL chloroform and subsequent incubation at RT for 3 min. Samples were centrifuged for 10 min at 4 °C at 10,000 × g and the RNA in the aqueous phase was isolated and purified using the RNeasy RNA kit (Qiagen).
RNA sequencing was performed at GenomeScan (Leiden, The Netherlands) using Illumina NextSeq 500 according to manufacturer protocols. In brief, RNA quality was checked using the Fragment Analyzer. Libraries were made using the MEBNext Ultra Directional RNA Library PrepKit for Illumina. The size of the cDNA fragments was confirmed to be in the 300–500 bp range and 1.6 pM of DNA was used for sequencing. RNA sequence data are available in the NCBI Sequence Read Archive (SRA) under the BioProject ID PRJNA557110
RNA sequencing analysis
Quality of raw reads was checked using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), whereas cleaning and trimming were performed using Fastx-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). CanFam3.1 transcripts from Ensembl (https://www.ensembl.org/Canis_familiaris/) and A. fumigatus Af293 transcripts from AspGD (http://www.aspergillusgenome.org/) served as reference to quantify expression using Kallisto63. Transcripts per million (TPM) was used as unit of expression. Transcripts with a TPM > 1 in all four samples were considered expressed.
Functional categorization and enrichment of expressed transcripts
Gene ontology (GO) and KEGG enrichment analysis was performed using the R package gProfileR64, with best term per parent group (strong filtering). REVIGO (http://revigo.irb.hr/) was used for removing redundancy in GO terms. FunCat ontology was determined using FungiFun265. Additionally, published lists of A. fumigatus genes involved in different processes were used to query the transcriptomes (Table 3).
Comparison with reported sino-nasal aspergillosis microarray data
Expression data6 were extracted from the array express database using the R package Array express V1.42.0. Raw data were processed and reanalyzed using the RMA method contained in the R package affy V1.60.0. Expression values of each transcript corresponded to the mean value of expression of the probes targeting that particular transcript. Transcripts were considered differentially expressed when they showed a Log2 Fold change (control vs. affected dogs) > ±2 (up, downregulation, respectively).
Fungal culturing for genomic DNA isolation
Genomic DNA was isolated using a modification of the protocol of Lee et al.66. In brief, 200 µl of conidial suspensions67 were seeded in potato dextrose agar (PDA) for 7 days at 37 °C. Conidia from these cultures were inoculated into 20 mL of potato dextrose broth, followed by incubation for 3–4 days at 37 °C in an Erlenmeyer. Mycelia were harvested by filtration over three layers of sterile Miracloth (Merck, Darmstadt, Germany), washed three to four times with sterile H2O, and dried using sterile paper towel at room temperature. Samples were lyophilized, followed by pulverization using sterile mortar and pestle. Ground samples (~30 mg) were used for DNA isolation using Qiagen DNeasy PowerPlant Pro Kit following manufacturer recommendations for problematic samples. DNA concentration and quality were checked using the Qubit® assay system
Whole-genome sequencing of fungal isolates
Whole-genome sequencing was performed by the Utrecht sequencing facility. In brief, libraries were prepared using Truseq DNA Nano library and sequenced on an Illumina NextSeq 500 with150 bp pair end mid output configuration. DNA sequence data are available in the NCBI SRA under the BioProject ID PRJNA598656
Sequence analysis
Quality of raw reads was checked using fastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Cleaning and trimming were performed using Fastx-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Reads were mapped to the genome of reference strain Af293 (Genome version from AspGD: s03-m05-r07 http://www.aspergillusgenome.org/) with bowtie2 v2.2.9 using options end to end and very sensitive. SAMtools v1.3 was used for further quality control. Freebayes v0.9.10-368 with option ploidy 1 was used for variant calling. Post filtering of the vcf file was performed using vcfilter (“qual >20”, depth 5×).
Identification of variants related to A. fumigatus evolution in the host
Mutation analysis in dog isolates was performed using association tests, core/non-core SNP analysis, and variant selection and annotation per group of interest (Supplementary Fig. 3).
Variant annotation
The filtered vcf file was used as input for SnpEff v4.3k in order to predict the effect of the variants69. For downstream analysis HIGH (gain or loss of stop codon, splice region variant) or MODERATE (missense) effects were used.
Core/non-core variants
A variant that was found in all isolates of a group was considered a core variant, while variants that were different in at least one isolate were classified as non-core variant. FungiFun265 was used to perform FunCat enrichment of core/non-core variants having a HIGH impact (for example loss of start or stop codon).
A targeted search of variants affecting genes of interest
SNPs present in genes of interest13,14,15,16,17 were compared with whole-genome sequencing data of 66 isolates of A. fumigatus18. To this end, the full annotated vcf file (environmental and dog isolates) was used and SNPs with a HIGH and MODERATE effect were taken into account.
Phylogenetic tree
A phylogenetic tree was constructed based on the filtered biallelic SNPs with no missing data using the R package SNPRelate v3.770. The SNPs were pruned using thresholds reported for A. fumigatus (linkage disequilibrium threshold of 0.8 and a minor allele frequency of 0.03)18. MEGA X71 was used for the calculation of the phylogenetic tree. In brief, a multi-fasta file with the SNP per strain was aligned with MUSCLE72 and the resulting alignment was used for statistical test of best-fit model of nucleotide substitution. The generalized time-reversible (GTR) model resulted in the best fit using the corrected Akaike information criterion. A total of 3826 pruned marker SNPs was used to build a maximum likelihood phylogenetic tree based on the GTR substitution model with 1000 bootstrap replicates.
Phenotyping of A. fumigatus isolates from SNA fungal plaques
Growth of the dog isolates was assessed during oxidative (H2O2), pH and copper stress. Prior to the phenotypical tests, strains were grown at 37 °C on PDA. Spores were harvested with Milli-Q® (Millipore Corp) water and filtered through three layers of sterile Miracloth (Merck, Darmstadt, Germany). They were diluted to 1 × 108 ml−1 and stored at 4 °C for maximally 2 weeks.
pH growth test
Minimal agar medium73 with 25 mM glucose (GMMA) was titrated at pH 5.0, 6.5, 7.2, and 8.010. Wells of 12-well plates (Greiner Bio One International GmbH) were filled with 2 ml medium, inoculated with 103 conidia in the center of each well, and incubated at 37 °C. Colony diameter was measured after 24 and 48 hours using biological duplicates.
Copper stress
Copper excess agar (GMMA-Cu containing 60 mM glucose and 0.9 mM Cu2+) was prepared by dissolving 0.015 g anhydrous copper sulfate (Merck, Darmstadt, Germany) in 47 ml MM, followed by addition of 3 ml 1 M glucose. GMMA-Cu (2 ml) was added to wells of 12-well plates, inoculated with 108 conidia in the center of the wells and incubated at 37 °C up to 4 days. GMMA (containing 1.25 µM Cu2+) was used as negative control. Colony size was determined in biological triplicates.
Hydrogen peroxide stress
A two-layered agar plate (60 mm Petri dish) was used containing 100 µl 500 mM H2O2 in a 5 mm wide hole in the center. The bottom layer consisted of 10 ml GMMA and a top layer of 5 ml GMMA containing five 107 conidia (conidia were always 2 weeks old). After 16 hours of incubation, diameter of inhibition zones (as biological triplicates) was determined using ImageJ 1.52a74.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
DNA sequence data are available in the NCBI SRA under the BioProject ID PRJNA598656. RNA sequence data are available in the NCBI SRA under the BioProject ID PRJNA557110. All other relevant data are available from the corresponding author on request.
References
Seyedmousavi, S. et al. Aspergillus and aspergilloses in wild and domestic animals: a global health concern with parallels to human disease. Med. Mycol. 53, 765–797 (2015).
Elad, D. & Segal, E. Diagnostic aspects of veterinary and human aspergillosis. Front. Microbiol. 9, 1303 (2018).
Ballber, C., Hill, T. L. & Bommer, N. X. Minimally invasive treatment of sino-nasal aspergillosis in dogs. J. Vet. Intern. Med. 32, 2069–2073 (2018).
Valdes, I. D. et al. Comparative genotyping and phenotyping of Aspergillus fumigatus isolates from humans, dogs and the environment. BMC Microbiol. 18, 118-2 (2018).
Belda, B., Petrovitch, N. & Mathews, K. G. Sinonasal aspergillosis: outcome after topical treatment in dogs with cribriform plate lysis. J. Vet. Intern. Med. 32, 1353–1358 (2018).
Vanherberghen, M. et al. Analysis of gene expression in canine sino-nasal aspergillosis and idiopathic lymphoplasmacytic rhinitis: a transcriptomic analysis. Vet. Microbiol. 157, 143–151 (2012).
Peeters, D., Peters, I. R., Clercx, C. & Day, M. J. Quantification of mRNA encoding cytokines and chemokines in nasal biopsies from dogs with sino-nasal aspergillosis. Vet. Microbiol. 114, 318–326 (2006).
Peeters, D. et al. Distinct tissue cytokine and chemokine mRNA expression in canine sino-nasal aspergillosis and idiopathic lymphoplasmacytic rhinitis. Vet. Immunol. Immunopathol. 117, 95–105 (2007).
McDonagh, A. et al. Sub-telomere directed gene expression during initiation of invasive Aspergillosis. PLoS Pathog. 4, e1000154 (2008).
Bertuzzi, M. et al. The pH-Responsive PacC transcription factor of Aspergillus fumigatus governs epithelial entry and tissue invasion during pulmonary aspergillosis. PLoS Pathog. 10, e1004413 (2014).
Breuer, K. et al. InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 41, 1228 (2013).
Nierman, W. C. et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438, 1151–1156 (2005).
Khaldi, N. et al. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 47, 736–741 (2010).
Miskei, M., Karányi, Z. & Pócsi, I. Annotation of stress–response proteins in the aspergilli. Fungal Genet. Biol. 46, S105–S120 (2009).
Urban, M. et al. PHI-base: a new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 45, D604–D610 (2017).
Bultman, K. M., Kowalski, C. H. & Cramer, R. A. Aspergillus fumigatus virulence through the lens of transcription factors. Med. Mycol. 55, 24 (2017).
de Vries, R. P. et al. Comparative genomics reveals high biological diversity and specific adaptations in the industrially and medically important fungal genus Aspergillus. Genome Biol. 18, 28 (2017).
Lind, A. L. et al. Drivers of genetic diversity in secondary metabolic gene clusters within a fungal species. PLoS Biol. 15, e2003583–e2003583 (2017).
Schrettl, M. et al. Self-protection against gliotoxin-a component of the gliotoxin biosynthetic cluster, glit, completely protects aspergillus fumigatus against exogenous gliotoxin. PLoS Pathog. 6, e1000952 (2010).
Wang, D. et al. GliA in Aspergillus fumigatus is required for its tolerance to gliotoxin and affects the amount of extracellular and intracellular gliotoxin. Med. Mycol. 52, 506–518 (2014).
Dolan, S. et al. Regulation of nonribosomal peptide synthesis: bis-thiomethylation attenuates gliotoxin biosynthesis in Aspergillus fumigatus. Chem. Biol. 21, 999–1012 (2014).
O’Hanlon, K. A. et al. Targeted disruption of nonribosomal peptide synthetase pes3 augments the virulence of Aspergillus fumigatus. Infect. Immun. 79, 3978–3992 (2011).
Kato, N., Suzuki, H., Okumura, H., Takahashi, S. & Osada, H. A point mutation in ftmD blocks the fumitremorgin biosynthetic pathway in Aspergillus fumigatus strain Af293. Biosci. Biotechnol. Biochem. 77, 1061–Af1067 (2013).
Fuller, K. K., Cramer, R. A., Zegans, M. E., Dunlap, J. C. & Loros, J. J. Aspergillus fumigatus photobiology illuminates the marked heterogeneity between isolates. mBio 7, 1517 (2016).
Vargas-Muniz, J. M. et al. Dephosphorylation of the core septin, AspB, in a protein phosphatase 2a-dependent manner impacts its localization and function in the fungal pathogen Aspergillus fumigatus. Front. Microbiol. 7, 997 (2016).
Wiemann, P. et al. Aspergillus fumigatus copper export machinery and reactive oxygen intermediate defense counter host copper-mediated oxidative antimicrobial offense. Cell Rep. 19, 1008–1021 (2017).
Talbot, J. J., Kidd, S. E., Martin, P., Beatty, J. A. & Barrs, V. R. Azole resistance in canine and feline isolates of Aspergillus fumigatus. Comp. Immunol. Microbiol. Infect. Dis. 42, 37–41 (2015).
Ballard, E. et al. In-host microevolution of Aspergillus fumigatus: a phenotypic and genotypic analysis. Fungal Genet. Biol. 113, 1–13 (2018).
Mohammadi, A. et al. An investigation on non-invasive fungal sinusitis; molecular identification of etiologic agents. J. Res. Med. Sci. 22, 67 (2017).
Peeters, D. & Clercx, C. Update on canine sinonasal aspergillosis. Vet. Clin. North Am. Small Anim. Pr. 37, 901–916 (2007).
Talbot, J. J. et al. What causes canine sino-nasal aspergillosis? A molecular approach to species identification. Vet. J. 200, 17–21 (2014).
Sharman, M. J. & Mansfield, C. S. Sinonasal aspergillosis in dogs: a review. J. Small Anim. Pract. 53, 434–444 (2012).
Peeters, D., Day, M. J. & Clercx, C. An immunohistochemical study of canine nasal aspergillosis. J. Comp. Pathol. 132, 283–288 (2005).
Vanherberghen, M. et al. Cytokine and transcription factor expression by Aspergillus fumigatus-stimulated peripheral blood mononuclear cells in dogs with sino-nasal aspergillosis. Vet. Immunol. Immunopathol. 154, 111–120 (2013).
Veerdonk, F. Lvande & Netea, M. G. T-cell subsets and antifungal host defenses. Curr. Fungal Infect. Rep. 4, 238–243 (2010).
Mengesha, B. G. & Conti, H. R. The Role of IL-17 in Protection against Mucosal Candida Infections. J. Fungi (Basel) 3, https://doi.org/10.3390/jof3040052 (2017).
Dewi, I. M. W., van de Veerdonk, Frank, L. & Gresnigt, M. S. The Multifaceted Role of T-Helper Responses in Host Defense against Aspergillus fumigatus. J. fungi (Basel, Switz.) 3, 55 (2017).
Zelante, T. et al. Th17 cells in the setting of Aspergillus infection and pathology. Med. Mycol. 47, S162–S169 (2009).
Choera, T., Zelante, T., Romani, L. & Keller, N. P. A multifaceted role of tryptophan metabolism and indoleamine 2,3-dioxygenase activity in Aspergillus fumigatus–host interactions. Front. Immunol. 8, 1996 (2018).
Chai, L. et al. Anti-Aspergillus human host defence relies on type 1 T helper (Th1), rather than type 17 T helper (Th17), cellular immunity. Immunology 130, 46–54 (2010).
de Araújo, E. F. et al. The IDO–AhR Axis controls Th17/Treg immunity in a pulmonary model of fungal infection. Front. Immunol. 8, 880 (2017).
Yang, D., Han, Z. & Oppenheim, J. J. Alarmins and immunity. Immunol. Rev. 280, 41–56 (2017).
Braedel, S. et al. Aspergillus fumigatus antigens activate innate immune cells via toll-like receptors 2 and 4. Br. J. Haematol. 125, 392–399 (2004).
Yu, G. et al. Mincle in the innate immune response of mice fungal keratitis. Int. J. Ophthalmol. 11, 539–547 (2018).
Werner, J. L. et al. Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J. Immunol. 182, 4938 (2009).
Li, C. et al. Expression of dectin-1 during fungus infection in human corneal epithelial cells. Int J. Ophthalmol. 7, 34–37 (2014).
Peng, X. D. et al. Fungus induces the release of IL-8 in human corneal epithelial cells, via Dectin-1-mediated protein kinase C pathways. Int. J. Ophthalmol. 8, 441–447 (2015).
Arias, M. et al. Preparations for invasion: modulation of host lung immunity during pulmonary aspergillosis by gliotoxin and other fungal secondary metabolites. Front. Immunol. 9, 2549 (2018).
Chooi, Y. H. et al. Genome mining of a prenylated and immunosuppressive polyketide from pathogenic fungi. Org. Lett. 15, 780–783 (2013).
García-Santamarina, S. & Thiele, D. J. Copper at the fungal pathogen-host axis. J. Biol. Chem. 290, 18945–18953 (2015).
Zygiel, E. M. & Nolan, E. M. Transition metal sequestration by the host-defense protein calprotectin. Annu. Rev. Biochem. 87, 621–643 (2018).
Amich, J., Vicentefranqueira, R., Leal, F. & Calera, J. A. Aspergillus fumigatus survival in alkaline and extreme zinc-limiting environments relies on the induction of a zinc homeostasis system encoded by the zrfC and aspf2 genes. Eukaryot. Cell 9, 424–437 (2010).
Wilson, D. & Deepe, G. S. The intersection of host and fungus through the zinc lens. Curr. Opin. Microbiol. 52, 35–40 (2019).
Łoboda, D. & Rowińska-Żyrek, M. Candida albicans zincophore and zinc transporter interactions with Zn(ii) and Ni(ii). Dalton Trans. (Camb., Engl.: 2003) 47, 2646–2654 (2018).
Ojeda-López, M. et al. Evolution of asexual and sexual reproduction in the aspergilli. Stud. Mycol. 91, 37–59 (2018).
Oliver, A., Cantón, R., Campo, P., Baquero, F. & Blázquez, J. High frequency of hypermutable pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1253 (2000).
Cigana, C. et al. Genotypic and phenotypic relatedness of Pseudomonas aeruginosa isolates among the major cystic fibrosis patient cohort in Italy. BMC Microbiol. 16, 142 (2016).
Ciofu, O., Riis, B., Pressler, T., Poulsen, H. E. & Høiby, N. Occurrence of hypermutable pseudomonas aeruginosa in cystic fibrosis patients is associated with the oxidative stress caused by chronic lung inflammation. Antimicrobial Agents Chemother. 49, 2276–2282 (2005).
Healey, K. R. et al. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat. Commun. 7, 11128 (2016).
Magditch, D. A., Liu, T. B., Xue, C. & Idnurm, A. DNA mutations mediate microevolution between host-adapted forms of the pathogenic fungus Cryptococcus neoformans. PLoS Pathog. 8, e1002936 (2012).
Rhodes, J. et al. A population genomics approach to assessing the genetic basis of within-host microevolution underlying recurrent cryptococcal meningitis infection. G3 (Bethesda, Md.) 7, 1165–1176 (2017).
Zhang, J. et al. Relevance of heterokaryosis for adaptation and azole-resistance development in Aspergillus fumigatus. Proc. Biol. Sci. 286, 20182886 (2019).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Reimand, J. et al. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 44, 83 (2016).
Priebe, S., Kreisel, C., Horn, F., Guthke, R. & Linde, J. FungiFun2: a comprehensive online resource for systematic analysis of gene lists from fungal species. Bioinforma. (Oxf., Engl.) 31, 445–446 (2015).
Lee, M., Park, H., Han, K., Hong, S. & Yu, J. High molecular weight genomic DNA mini-prep for filamentous fungi. Fungal Genet. Biol. 104, 1–5 (2017).
Samson, R. A. et al. Phylogeny, identification and nomenclature of the genus Aspergillus. Stud. Mycol. 78, 141–173 (2014).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907 [q-bio.GN] (2012).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80 (2012).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 28, 3326–3328 (2012).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Robert, C. Edgar. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Pontecorvo, G., Roper, J. A., Chemmons, L. M., Macdonald, K. D. & Bufton, A. W. J. The Genetics of Aspergillus nidulans. Adv. Genet. 5, 141–238 (1953).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671 (2012).
Acknowledgements
We thank Annika Haagsman (Veterinary Medicine, Utrecht University, The Netherlands) for isolation of fungal plaques from dogs with SNA and her contributions and comments during the course of this research.
Author information
Authors and Affiliations
Contributions
I.V. and HdC designed the experiments; I.V., H.W., and HdC wrote the article; I.V., AHdR, C.T., J.B. performed the experiments; All authors read and approved the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Valdes, I.D., Hart de Ruijter, A.B.P., Torres, C.J. et al. The sino-nasal warzone: transcriptomic and genomic studies on sino-nasal aspergillosis in dogs. npj Biofilms Microbiomes 6, 51 (2020). https://doi.org/10.1038/s41522-020-00163-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-020-00163-7
- Springer Nature Limited