

Abstract
In eukaryotes, the origin recognition complex (ORC) faciliates the assembly of pre-replicative complex (pre-RC) at origin DNA for replication licensing. Here we show that the N-terminal intrinsically disordered region (IDR) of the yeast Orc2 subunit is crucial for this process. Removing a segment (residues 176-200) from Orc2-IDR or mutating a key isoleucine (194) significantly inhibits replication initiation across the genome. These Orc2-IDR mutants are capable of assembling the ORC-Cdc6-Cdt1-Mcm2-7 intermediate, which exhibits impaired ATP hydrolysis and fails to be convered into the subsequent Mcm2-7-ORC complex and pre-RC. These defects can be partially rescued by the Orc2-IDR peptide. Moreover, the phosphorylation of this Orc2-IDR region by S cyclin-dependent kinase blocks its binding to Mcm2-7 complex, causing a defective pre-RC assembly. Our findings provide important insights into the multifaceted roles of ORC in supporting origin licensing during the G1 phase and its regulation to restrict origin firing within the S phase.
Similar content being viewed by others


Introduction
Eukaryotic DNA replication begins at multiple replication origins scattered throughout the genome1,2. To ensure accurate genome duplication, each origin is activated only once per cell cycle. This precise control is achieved through the assembly of a pre-replicative complex (pre-RC) at the origin DNA during the G1 phase, followed by its activation when cells enter S phase3,4.
In the yeast Saccharomyces cerevisiae, the pre-RC assembly is facilitated by the origin recognition complex (ORC), which binds to replication origins known as autonomously replicating sequences (ARSs) containing an ARS consensus sequence (ACS)1. The ORC consists of six subunits, Orc1-65. While Orc1-5 each possesses an AAA + or AAA + -like domain and a winged helix domain (WHD), Orc6 bears little similarity to other ORC subunits and instead resembles a transcription factor II B (TFIIB). Together, these six subunits, along with Cdc6 and Cdt1, collaborate to assemble two copies of Mcm2-7 complexes into a head-to-head double hexamer (DH) at the origin DNA6,7,8,9,10,11,12,13. This process is also known as replication licensing or pre-RC assembly1. Once loaded, the MCM-DH remains inactive throughout the G1 phase. However, upon entering the S phase, Dbf4-dependent kinase (DDK) and S cyclin-dependent kinase (S-CDK) work in concert with multiple origin-firing factors to activate the MCM-DH, leading to the formation of two active replisomes that drive bidirectional DNA synthesis1,4,14,15,16,17.
The in vitro recapitulation of replication licensing in yeast has provided valuable insights into the process of pre-RC assembly1,18,19. It begins with the recognition of origin DNA by ORC, serving as a platform for subsequent assembly events5. Cdc6 is then recruited by ORC to facilitate the loading of the first Cdt1-Mcm2-7 heptamer into a transient ORC-Cdc6-Cdt1-Mcm2-7 (OCCM) intermediate on DNA20,21. Upon ATP hydrolysis, OCCM undergoes a structural reconfiguration, transforming into a Mcm2-7-ORC (MO) complex where ORC’s binding site is changed from ACS to a downstream B2 DNA element21,22. The MO complex serves as a foundation for directing the recruitment of the second Cdt1-Mcm2-7 heptamer in the proper orientation to fuse with the initially loaded Mcm2-7, forming a tightly coupled DH. Recent single-molecule studies have demonstrated that a single ORC can flip over the loaded Mcm2-7 single hexamer to capture its downstream binding site during MO formation22,23. To complete this process, ORC must remain on a stable tether to the first loaded Mcm2-7 after it is released from the ACS site and before it re-engages with the B2 DNA. The involvement of Orc6 in this process has been proposed due to its interaction with Mcm2 via its N-terminal domain (NTD)21. However, the exact mechanism by which OCCM drives this conversion and how ORC accomplishes its flipping remain poorly understood.
ORC’s activities at origin DNA are regulated by post-translational modifications1,24. The function of ORC in replication licensing is limited to the G1 phase due to the oscillating activities of CDKs throughout the cell cycle1. It is well established that S-CDKs target Orc2 and Orc6, resulting in inhibitory effects on ORC1. Recent studies have revealed that S-CDK phosphorylation of ORC does not prevent MCM recruitment to the origin DNA. However, it significantly suppresses the MO formation25. Interestingly, Orc6 phosphorylation allows its NTD to interact with the ORC-Cdc6 complex, specifically at a shared surface with the C-terminal WHD of Mcm726, slowing down the OCCM formation. Nevertheless, the molecular details of how S-CDK phosphorylation of Orc2 blocks pre-RC assembly remain unclear.
In this study, we mapped the N-terminal IDR of Orc2 and identified a short α-helix (SH) containing segment (176-200) that is essential for cell viability (Fig. 1 and Supplementary Fig. 1). Our further analyses indicate that this IDR region is required for MO formation through regulating ATP hydrolysis by OCCM to promote pre-RC assembly. Moreover, S-CDK targets Orc2-SH for inhibitory phosphorylation to block replication licensing. Our findings provide mechanistic insights into the essential role of a short linear motif embedded in Orc2-IDR in the regulation of DNA replication initiation.

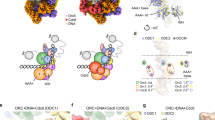
a Side view of the segmented cryo-EM density map (EMD-6941) of the yeast ORC in complex with origin DNA. WHD: winged helix domain; BP: basic patch. b Domain organization and DISOPRED analysis of Orc2. ℗ represents CDK consensus sites. DISOPRED scores around/above the cut-off value (0.5) are considered highly disordered regions. This indicates that the first 200 residues of Orc2 are highly flexible. c, d Upper panels: schematic illustration of ORC2-IDR mutant construction. Lower panels: the indicated Orc2-IDR mutants were tested for complementation of Orc2-1 depletion in GAL-orc2-1 cells.
Results
Orc2-IDR is essential for cell viability
In the yeast ORC-DNA structure, three NTDs from subunits Orc1, Orc2, and Orc6 exhibit high flexibility27. Among them, Orc1-NTD (1-355) contains a bromo-adjacent homology (BAH) domain that can bind to nucleosome core particles, assisting in origin selection and other chromatin-related functions28,29,30. The flexible Orc6-NTD (1-267) has also been implicated in MO formation21,22. However, the specific role of Orc2-NTD (1-235) in pre-RC assembly is largely unknown. Sequence analysis using DISOPRED predicts that Orc2-NTD (1-235) is an extended intrinsically disorder region (IDR), in agreement with its flexible feature in the ORC-DNA structure (Fig. 1a, b) and the AlphaFold predicted model (Supplementary Fig. 1) (https://alphafold.ebi.ac.uk/entry/P32833).
To investigate the role of Orc2-IDR, we generated yeast strains with the endogenous orc2-1 mutant gene under the control of the GAL promoter (GAL-orc2-1)31, expressing either wild-type (WT) or mutant version of ORC2 ectopically under its native promoter (Fig. 1c, d). The GAL-orc2-1 strain with an empty vector was used as a negative control. Notably, GAL-orc2-1 cells can readily induce an efficient depletion of Orc2-1 proteins31. When the GAL-orc2-1 cells were grown in a glucose-only medium (referred to as YPD), the expression of endogenous Orc2-1 proteins was suppressed, resulting in cell lethality. However, this lethality was rescued by the ectopic expression of Orc2 (WT) (Fig. 1c, d).
To facilitate the construction of orc2 mutants, we divided Orc2-IDR into three segments: segment1 (S1: 1–175), S2 (176–200, also referred to as SH), and S3 (201–235), based on the DISOPRED score (Fig. 1b, c). We observed that S1 was not required for the growth of GAL-orc2-1 cells on the YPD plate (Fig. 1c). However, the removal of S2/SH from Orc2 resulted in cell lethality (Fig. 1c), indicating that this short α-helix containing region is essential for cell proliferation. To identify the specific residue(s) that is/are crucial for cell viability, we introduced mutations in two groups of amino acids within the SH: (1) all positively charged amino acids were changed to alanine (PC-to-A), and (2) all hydrophobic amino acids were replaced with glutamic acid (HF-to-E) (Fig. 1d), aiming to disrupt any potential electrostatic or hydrophobic interactions mediated by Orc2-SH. Our results demonstrated that while the orc2-PC-to-A mutant supported cell growth, the orc2-HF-to-E mutant was lethal (Fig. 1d). Further mapping revealed that I194 was a key residue responsible for cell viability, as the orc2-I194E mutant cells displayed lethality on YPD plate (Fig. 1d). Interestingly, an orc2-I194V mutant, where I194 was replaced with a similar hydrophobic residue, valine (V), was able to sustain cell growth (Fig. 1d). These findings suggest that Orc2-SH plays a crucial role in cell cycle progression, likely through mediating hydrophobic interactions with its binding partner(s).
Orc2-IDR is required for DNA replication initiation on chromatin
To examine the requirement of Orc2-SH in DNA replication, we performed nocodazole followed by an alpha-factor block-and-release assay. GAL-orc2-1 cells were cultured in a galactose-containing medium and initially synchronized at the G2/M phase with nocodazole. The cells were subsequently released into YPD medium supplemented with alpha factor, with the GAL promotor switched off to deplete Orc2-1 (Fig. 2a). This ensured G1 synchrony before further release into fresh YPD medium. Cell samples were then collected and subjected to FACS analysis. Our analysis revealed a significant delay in S phase entry for the control strain carrying the empty vector (Fig. 2b), indicating that Orc2-1 depletion inhibits DNA replication initiation. This defect was largely rescued by the ectopic expression of ORC2 (WT), but not by orc2-ΔN200 or orc2-I194E mutant (referred to as SH mutant) (Fig. 2b). While the ORC2 (WT) cells were able to complete S phase and progress to the next cell cycle, both SH mutant cells exhibited a similar delay in S phase entry and prolonged S phase progression.

a Scheme for Orc2-1 depletion by glucose repression with GAL-orc2-1 strain. b Flow cytometry profiles of cells collected from nocodazole followed by alpha-factor block-and-release assays with the indicated strains after Orc2-1 depletion. c–e Plots showing relative DNA copy numbers in the sections of chromosome (chr) VIII (c), chr IX (d), and chr XVI (e) (see methods for details). Open circles indicate confirmed replication origins according to OriBD.
To assess the impact of the SH mutations on replication initiation across the yeast genome, we performed sync-seq assays using both WT and mutant cells. We compared their DNA copy number between replicating samples taken from the early S phase (30 min after G1 release) and non-replicating samples taken from the G1 phase. The whole-genome sequencing data from the relevant G1- and S-phase cells were used to generate replication profiles. As shown in Fig. 2c–e and Supplementary Fig. 2, DNA replication was initiated from early origins in the WT cells. In contrast, the majority of these initiation events were suppressed in the SH mutant cells. These results provide compelling evidence that Orc2-SH plays a crucial role in initiating DNA replication on chromatin. Notably, we also observed robust initiation events upon a few origins, such as ARS1614 and ARS1621, in the SH mutant cells (Fig. 2e and Supplementary Fig. 2), suggesting the existence of an Orc2-SH-independent pathway for replication initiation in yeast cells. However, this pathway is not sufficient to sustain bulk DNA replication across the genome, leading to cell lethality.
Pre-RC assembly mediated by Orc2-IDR
To determine the requirement of Orc2-IDR for replication licensing, we conducted chromatin-binding assays to examine the overall levels of pre-RC on G1 chromatin in the orc2-SH mutant cells after Orc2-1 depletion. G1 cells from nocodazole block and release were collected (Fig. 3a), and cell samples were processed to analyze the chromatin association of Orc3 and Mcm6. As shown in Fig. 3b, chromatin-bound ORC was absent after Orc2-1 depletion, resulting in a failure of Mcm2-7 loading in the control strain (NC). However, similar levels of Orc3 were observed on chromatin in both WT and SH mutant cells, suggesting that the association of ORC with chromatin is independent of Orc2-IDR. Interestingly, the chromatin-bound Mcm6 exhibited a significant reduction in the SH mutants compared to the WT strain (Fig. 3b). These results indicate that Orc2-SH is required for promoting pre-RC assembly in vivo.

a A brief flowchart of sample collection for chromatin binding assay. b Fractions of whole cell extract (WCE) and chromatin taken from the indicated strains were analyzed by SDS-PAGE and immunoblotting. WT: GAL-orc2-1 pRS405-ORC2-WT strain; NC (negative control): GAL-orc2-1 pRS405 strain; ΔN200: GAL-orc2-1 pRS405-orc2-ΔN200 strain; I194E: GAL-orc2-1 pRS405-orc2-I194E strain. c SDS-PAGE of the purified ORCs used in the in vitro MCM loading assays. d Schematic illustration of on-beads MCM loading assay. e, f Pre-RC complexes were formed in the presence of ATP, washed with low salt buffer (e) or high salt buffer (f), released from DNA with DNase I, analyzed by SDS-PAGE, and visualized by silver staining and western blot. g MCM loading was performed in the presence of ATPγS. h, i, Time-course analysis of MCM loading process by negative-stain electron microscopy. The proportions of MCM-DH (h) and OCCM (i) were calculated during the loading process in the presence of ATP. j Same as (i) but with the reaction performed in the presence of ATPγS. Similar results in (b, c), and (e–g) were obtained in two independent experiments. Error bars in h–j represent mean ± s.d. from three independent experiments. Source data are provided as a Source Data file.
To elucidate the detailed role of Orc2-IDR in replication licensing, we purified Orc2-SH mutant containing ORCs to examine their impact on MCM loading using in vitro pre-RC assembly assays (Fig. 3c, d). We also used Orc2 (WT) and Orc2-ΔN175 containing ORCs as controls for the reactions. In the presence of ATP, all ORCs supported the DNA binding of ORC and Mcm2-7; however, the reactions with the SH mutants (Orc2-ΔN200 and Orc2-I194E) were less efficient in driving MCM loading (Fig. 3e). In addition, salt-stable Mcm2-7 bound to DNA was obtained only with Orc2 (WT) and Orc2-ΔN175, but not with the SH mutants (Fig. 3f). In contrast, in the presence of ATPγS, the levels of ORC, Cdc6, Cdt1, and MCM on origin DNA were similar in the reactions with all tested ORCs (Fig. 3g). These results suggest that Orc2-SH is dispensable for OCCM formation but essential for pre-RC assembly.
To further characterize the role of Orc2-IDR in MCM loading, we performed a time-course assay to monitor MCM loading using electron microscopy (EM) imaging. As expected, efficient DH formation was observed in the reactions with Orc2 (WT) (Fig. 3h). However, the efficiency of DH formation was significantly reduced in the reactions with Orc2-SH mutants (Fig. 3h). In addition, during this assembly process, OCCM particles appeared at 2 minutes in the reactions with either WT or mutant ORCs (Fig. 3i), corroborating the conclusion that Orc2-IDR is not required for OCCM formation. Consistently, in the presence of ATPγS, the OCCM intermediates were formed on DNA at similar rates in the reactions with all tested ORCs (Fig. 3j). Interestingly, in the presence of ATP, the WT OCCM particles only lasted for a short period on DNA (Fig. 3i), eventually forming the end product MCM-DH (Fig. 3h). In contrast, the SH mutant containing OCCM particles did not disappear like the WT ones; instead, they gradually accumulated to a high level (Fig. 3i). Supporting this result, Cdt1 retention on DNA was also associated with the SH mutants in the pre-RC assays (Fig. 3e). These findings suggest that Orc2-IDR is essential for the conversion of OCCM into MCM-DH during pre-RC assembly.
Orc2-IDR modulates ATP hydrolysis for pre-RC assembly
The release of Cdt1 and the subsequent formation of MO require ATP hydrolysis by OCCM32. We hypothesized that Orc2-IDR can stimulate the ATPase activity of OCCM. To test this hypothesis, we employed an in vitro assay to measure the rate of ATP hydrolysis during pre-RC assembly. We observed robust ATP hydrolysis in the reactions with a complete set of pre-RC components, compared to reactions with ORC or Cdt1-Mcm2-7 alone (Fig. 4a). However, when the Orc2-WT was replaced with the SH mutants, the rates of ATP hydrolysis were reduced by almost half, similar to the level induced by Cdt1-Mcm2-7 alone (Fig. 4a). This result suggests that Orc2-SH regulates ATPase activities induced by pre-RC assembly.

a Analysis of ATPase activities with the indicated ORCs during pre-RC assembly. ORC-DNA or Cdt1-Mcm2-7only serve as controls. Statistical significance was evaluated using one-way ANOVA. *P = 0.0126. ns, not significant, P > 0.05. b The effect of SH peptide on the pre-RC-induced ATP hydrolysis with the indicated ORCs. P-values calculated with two-sided Welch’s t test; *P = 0.0226. ns, not significant, P > 0.05. c, d The addition of SH peptide can restore the defects of Orc2 mutants in MO formation (c) and pre-RC assembly (d). Similar results in (d) were obtained in two independent experiments. Error bars in (a–c) represent mean ± s.d. from three independent experiments. Source data are provided as a Source Data file.
Next, we asked whether the impaired ATP hydrolysis in the pre-RC reactions with the mutant ORC could be restored by the Orc2-SH peptide. To test this, we included a synthetic Orc2-SH peptide (residues 187–205) in the pre-RC assays. We found that this peptide significantly increased the rate of ATP hydrolysis in the reaction with the mutant ORC (Fig. 4b). Accordingly, Orc2-SH also partially restored the ability of the SH mutant to promote MO formation (Fig. 4c), resulting in the assembly of salt-stable MCM-DHs on DNA (Fig. 4d). These results highlight an important role of Orc2-SH in the OCCM-MO transition by regulating pre-RC assembly-induced ATP hydrolysis.
Orc2-SH binds to Mcm2-7 containing complexes
To determine whether Orc2-SH stimulates ATP hydrolysis by OCCM via Mcm2-7 binding, we conducted a GST pulldown assay to measure the ability of Orc2-NTD (residues 1–200) (referred to as Orc2N200) binding to MCM-containing complexes. Purified GST-Orc2N200 was immobilized on glutathione resin and incubated with either Cdt1-Mcm2-7 or OCCM (Fig. 5a, b and Supplementary Fig. 3a). We found that both Cdt1-Mcm2-7 and OCCM can be pulled down by GST-Orc2N200, but not by GST-Orc2N175 or GST alone (Fig. 5a, b), indicating a direct interaction between Orc2-SH(175-200) and Mcm2-7 containing complexes. The low pull-down efficiency may reflect a weak or transient interaction between them. In addition, the Orc2N200-I194E mutant maintained its binding affinity to Mcm2-7 containing complexes (Fig. 5a, b). These results suggest that Orc2-SH directly binds to Mcm2-7, enabling Orc2-I194 to transform OCCM into a state capable of robust ATP hydrolysis.

a, b GST-Orc2N200 but not GST-Orc2N175 can pull-down Cdt1-Mcm2-7 (a) and OCCM (b). c S-CDK phosphorylation disrupts the binding of Orc2N200 to Mcm2-7. The elutes from the GST pull-down assays were analyzed by SDS-PAGE and visualized by silver staining. The elutes from the GST pull-down assays (a–c) were analyzed by SDS-PAGE and visualized by silver staining. d Analysis of pre-RC assembly in the presence of S-CDK or phospho-mimic mutant S188D of Orc2-ΔN175. Pre-RC complexes were released from DNA with DNase I and analyzed by SDS-PAGE and silver staining. Clb5 refers to the regulatory subunit of S-CDK. Similar results were obtained in two independent experiments.
Inhibitory regulation of Orc2-SH binding to Mcm2-7 by S-CDK
S-CDK phosphorylation imposes an inhibitory effect on ORC to prevent origin licensing1. Intriguingly, all CDK phosphorylation sites on Orc2 are clustered in its flexible NTD, with S188 positioned within the SH region (Fig. 1b). We asked whether S-CDK phosphorylation at this site could disrupt Orc2-SH binding to Mcm2-7, resulting in a defective pre-RC assembly. To test this hypothesis, the binding affinity of Orc2N200 to Mcm2-7 was examined after S-CDK phosphorylation using an in vitro pulldown assay. Our results revealed that the phosphorylated Orc2N200, which displayed a noticeable shift in its gel migration upon S-CDK phosphorylation, failed to interact with Cdt1-Mcm2-7 (Fig. 5c and Supplementary Fig. 3b).
Next, to investigate the specific effect of S-CDK phosphorylation of Orc2-SH on pre-RC assembly, we performed in vitro MCM loading assays with Orc2ΔN175-containing ORC, in which the non-essential S1 region (1–175) was removed. Our results indicated that S-CDK treatment significantly inhibited pre-RC assembly (Fig. 5d). Moreover, we generated a phospho-mimic mutant of Orc2ΔN175, referred to as Orc2ΔN175-S188D, in which S188 was changed to D to mimic SH phosphorylation. The Orc2ΔN175-S188D phospho-mimic mutant exhibited a severe defect in DH formation, similar to that observed with the phosphorylated Orc2ΔN175-containing ORC (Fig. 5d). Thus, we conclude that Orc2-SH is one of the major targets for inhibitory phosphorylation by S-CDK, which prevents pre-RC assembly and DNA re-replication outside of the G1 phase during the cell cycle.
Discussion
The highly flexible motifs/domains or IDRs of macromolecules often play important roles in mediating protein-protein interactions or facilitating liquid-liquid phase separation (LLPS) to support their relevant functions in vivo33,34,35. In the case of the yeast ORC, three NTDs belonging to Orc1, Orc2, and Orc6 contain highly flexible regions, enabling ORC to coordinate the events leading to replication licensing on chromatin. The flexible NTD of Orc1 consists of a BAH domain and a long IDR with multiple basic patches (BPs). The conserved Orc1-BAH domain can bind to nucleosomes, allowing ORC to select sites for MCM loading within local chromatin structures28,29. Orc1-BP has been shown to contribute to the recognition of ACS by ORC in yeast27. In addition, metazoan Orc1-IDR has been implicated in regulating replication licensing by facilitating LLPS or serving as a platform to recruit other factors33,34,35.
In our study, we identified a crucial SH segment (175–200) embedded within Orc2-IDR that is essential for MO formation. Our results revealed that removing this region from Orc2 interrupts pre-RC assembly, leading to a stall at the OCCM stage. While the Orc2-SH mutants can support OCCM formation, the assembled OCCM appears to be relatively stable on DNA and accumulates to a higher level compared to Orc2 (WT). This suggests that the mutant OCCM loses its ability to transform into MO. Interestingly, we found that this blockage can be bypassed by adding the Orc2-SH peptide in our in vitro pre-RC assays. Our results indicate that this peptide enhances the rate of pre-RC induced ATP hydrolysis, promotes MO formation, and further rescues pre-RC assembly in the reactions with the SH mutant. As Orc2-SH can bind to Mcm2-7 complexes, we believe that this interaction reconfigures the Mcm2-7 ring into a state that is competent for efficient ATP hydrolysis. This is a critical step for OCCM to allow Cdt1 release, Mcm2-7 ring closure, and subsequent ORC flipping upon the loaded Mcm2-7 prior to MO formation. In the OCCM structure, Orc2-IDR is positioned adjacent to the CTDs of Mcm2-720 (Supplementary Fig. 4). Therefore, our study highlights the importance of Orc2-SH in modulating ATPase activities of Mcm2-7 to promote pre-RC assembly (Fig. 6). Notably, Orc2-I194E mutation does not affect the interaction between Orc2-SH and Mcm2-7, suggesting that the residue I194 plays a crucial role in remodeling Mcm2-7 ring following Orc2-SH binding. Further study is necessary to elucidate the structural basis of how Orc2-IDR impacts Mcm2-7, inducing conformational changes that trigger ATP hydrolysis by OCCM.

With the help of Cdc6, ORC recruits the first Cdt1-Mcm2-7 onto the origin DNA, resulting in the formation of ORC-Cdc6-Cdt1-Mcm2-7 (OCCM). Subsequently, Orc2-IDR binds to Mcm2-7, which triggers ATP hydrolysis by OCCM. This leads to the release of Cdt1 and Cdc6. Following this step, ORC switches its binding site from ACS to B2 by flipping upon the first loaded Mcm2-7. The resultant Mcm2-7-ORC (MO) complex acts as a platform for the recruitment of Cdc6 and the second Cdt1-Mcm2-7, ultimately forming the MCM-DH. Note that the ability of Orc2-IDR to promote MO formation can be hindered by S-CDK phosphorylation.
In the MO structure, the flexible Orc6-NTD (1-119) interacts with the Mcm2/5 gate-forming subunits21. This interaction has been proposed as an anchoring point for ORC to flip at a point after Orc2-IDR’s function in driving MO formation. Interestingly, it has been shown that the removal of Orc6-NTD reduces the efficiency of MO formation by half21, implying that other element(s) from ORC also contribute to this process. Given that Orc2-SH can interact with the Mcm2-7 containing complex, we speculate that this interaction could provide an additional binding site for ORC to secure its stable association with the MCM ring during its flipping. It is quite possible that Orc2-IDR, along with Orc6-NTD, escorts ORC to switch its binding site from ACS to B2 on the origin DNA.
To ensure that all replication origins fire no more than once per cell cycle, cells have evolved multiple redundant mechanisms to prevent DNA re-replication1. ORC phosphorylation by S-CDK serves as an effective way to inhibit pre-RC assembly. We found that the function of Orc2-SH in MCM loading is tightly regulated by S-CDK (Fig. 6). Phosphorylation of Orc2-SH alone has the same inhibitory effect as the full phosphorylation of both Orc2 and Orc6 to prevent replication licensing. This finding is consistent with recent single-molecule studies, which have demonstrated that phosphorylation of either Orc2 or Orc6 significantly inhibits MO formation and the subsequent DH formation25. It also explains the necessity of eliminating both Orc2 and Orc6 phosphorylation to induce DNA re-replication in G2/M cells.
Interestingly, replication initiation in yeast can occur through an Orc2-SH independent manner at certain origins, suggesting that there are two distinct strategies for pre-RC assembly on chromatin in yeast. The choice between these strategies is likely influenced by the involvement of Orc2-SH in driving MO formation. Recent in vitro studies have demonstrated that pre-RC assembly can be achieved in the absence of Orc2-NTD (1-236) on an artificial DNA that contains two ORC binding sites and a 90-bp insertion in between36. In line with this finding, our analysis indicates that both ARS1614 and ARS1621, which can support Orc2-SH insensitive origin firing (Fig. 2e), contain an additional ACS- or B2-like element and are located in regions with a wider nucleosome-depleted region (NDR) compared to ARS1 (Supplementary Fig. 5). It is likely that these special origins can facilitate pre-RC assembly through simultaneous or independent loading of two Mcm2-7 complexes on DNA as previously suggested37, without the involvement of MO formation driven by Orc2-SH. If this is true, it will have significant implications for understanding replication licensing in higher eukaryotes where no DNA consensus sequence has been identified for ORC binding to drive MCM loading. It is noteworthy that Orc2-SH is only conserved among yeast species but not in higher eukaryotes (Supplementary Fig. 6). Further investigation is required to understand the detailed mechanism regulating pre-RC assembly independent of Orc2-SH in yeast. This will offer valuable insights into how the Orc2-SH independent pathway has evolved as the dominant strategy for pre-RC assembly in metazoans.
Methods
Yeast strain construction
The open reading frame of the wild type ORC2, along with its upstream 400 bp and downstream 200 bp, was inserted into pRS405 at the BamHI and XhoI sites to generate pRS405-ORC2-WT. The orc2 mutant plasmids were derived from pRS405-ORC2-WT through mutagenesis. The pRS405-ORC2-related plasmids, linearized at the BstEII site, were integrated into GA168031 (GAL-orc2-1, a gift from Susan Gasser), respectively. All strains used for expressing and purifying mutant ORCs are derivatives of ySDORC6,19 (a gift from John Diffley). The relevant codon-optimized ORC2 genes were cloned into pRS405-GAL1-10-FLAG at the PspOMI and XhoI sites, respectively, and then integrated into ySDORC.
Protein purification
The wild type ORC was purified as previously described19,27. The mutant ORCs with FLAG-tagged Orc2 were purified using a two-step immunoprecipitation (IP) strategy. Yeast cells of each mutant (10 liters) were cultured in a YPR (raffinose) medium until reaching an OD600 of 2.0. The cells were then arrested in the G1 phase with 50 ng/ml alpha factor for 1.5 h, followed by adding galactose (final 2%) into the culture for an additional 3 h to induce ORC overexpression. After centrifugation at 13,000 × g for 5 min at 4 °C and washing with pre-cooled H2O, the cells were resuspended in lysis buffer [25 mM HEPES-KOH pH 7.4, 0.5 M KCl, 10% glycerol, 0.05% NP40S, 2 mM CaCl2, 1 mM DTT, 1 × protease inhibitor (Roche)], and flash-frozen in liquid nitrogen to make “popcorns”. The cell pellets were then lysed using a Freezer Mill (Spex Sample Prep). The supernatant was clarified by centrifugation at 38,900 × g for 30 min at 4 °C. Subsequently, 2 ml of pre-equilibrated Calmodulin-Sepharose beads (Cytiva, 17-0529-01) were added to the supernatant. The mixture was allowed to bind at 4 °C for 2 h with rotation. The beads were then washed with 150 ml of lysis buffer, and elution was performed three times using 2 ml of lysis buffer containing 1 mM EDTA and 2 mM EGTA. The elution fractions were combined for further FLAG IP with 2 ml of pre-equilibrated M2 FLAG-beads (Sigma, A2220) at 4 °C with rotation for 3 h. The beads were then washed with 20 ml of washing buffer [25 mM HEPES-KOH pH 7.4, 0.15 M KCl, 10% glycerol, 0.05% NP40S, 1 mM β-ME]. Elution was performed three times by adding 0.5 mg/ml 3 × FLAG peptide (GenScript). The elution fractions were pooled, concentrated, and loaded onto a Superdex 10/300 GL column (GE Healthcare) for size-exclusion chromatography. Peak fractions containing ORC were combined and snap-frozen in liquid nitrogen for storage at − 80 °C.
His-SUMO-Cdc6 was purified from E. coli. 2 liters of BL21-DE3 cells bearing pET-His-SUMO-Cdc6 were cultured until reaching OD600 of 0.6 at 37 °C. IPTG was then added to a final concentration of 0.5 mM, and induction was continued for 14 h at 16 °C. The cells were harvested by centrifugation at 13,000 × g for 5 minutes at 4 °C. The cells were resuspended in 50 ml of lysis buffer [50 mM K2HPO4/KH2PO4 pH 7.5, 5 mM MgCl2, 1% Triton, 1 mM β-ME, 2 mM ATP, 0.15 M KOAc, 1 × protease inhibitor], and lysed using high-pressure homogenizer (700 kPa, 3 min, 4 °C). The lysate was clarified by centrifugation at 38,900 × g for 30 min at 4 °C. 2 mL of pre-equilibrated Ni-beads (Takara, 635660) were added to the supernatant fraction, and binding was allowed for 0.5 hour at 4 °C. The beads were then washed with 100 ml of washing buffer [lysis buffer + 10 mM imidazole]. Elution was conducted with 6 ml of elution buffer [lysis buffer + 0.25 M imidazole]. The elution fraction from the Ni-beads was loaded onto a Superdex 200 increase 10/300 GL column for gel filtration chromatography. Peak fractions containing His-SUMO-Cdc6 were pooled, snap-frozen in liquid nitrogen, and stored at − 80 °C.
Cdt1-Mcm2-7 was purified from strain yJF3819,38 (a gift from John Diffley), following the previously described method38. Yeast cells were cultured in YPR medium at 30 °C until reaching an OD600 of 2.0. Subsequently, the cells were synchronized in G1 with 50 ng/ml α factor for 1.5 h. Overexpression of Cdt1 and Mcm2-7 was induced by adding galactose (final concentration: 2%) for 3 h. The cells were harvested by centrifugation, washed twice with ice-chilled water, and then resuspended in lysis buffer [25 mM HEPES-KOH pH 7.5, 0.1 M K-glutamate, 10% glycerol, 0.02% NP40S, 2 mM ATP, 1 mM DTT, and 1 × protease inhibitor]. The resuspended cells were rapidly frozen in liquid nitrogen and crushed using a Freezer Mill (Spex Sample Prep). The lysates were clarified by centrifugation at 38,900 × g for 30 min at 4 °C. Then, 2 ml of pre-equilibrated M2 FLAG-beads (Sigma, A2220) was added to the clarified fraction and incubated for 3 h at 4 °C with rotation. The beads were then washed with 150 ml of lysis buffer, and elution was performed by adding 2 ml of lysis buffer containing 0.5 mg/ml 3 × FLAG peptide (GenScript) three times. The elutes were combined and concentrated to a volume suitable for loading onto a 20–40% glycerol gradient [25 mM HEPES-KOH pH 7.5, 0.1 M K-glutamate, 20–40% glycerol, 0.02% NP40S, 1 mM ATP, 5 mM Mg(OAc)2, and 1 mM DTT] for ultracentrifugation for 14 h at 137,000 × g using a TLS-55 rotor (Beckman). The fractions containing the heptamer complex were pooled, frozen in liquid nitrogen, and stored for further use.
GST-Orc2-N200 and its mutant were purified from BL21-DE3 bearing relevant pGEX6p-1-ORC2 constructs. 2 liters of relevant cells were cultured to an OD600 of 0.6. IPTG was added to 0.5 mM, and induction was conducted at 16 °C for 14 h. The cells were collected and resuspended in lysis buffer [50 mM Tris-HCl pH 7.0, 300 mM NaCl, 1 mM DTT, 1 × protease inhibitor] and lysed with a homogenizer (700kPa, 3 min, 4 °C). 1.5 mL of pre-equilibrated glutathione beads (Cytiva, 17075601) were used for each binding reaction. The beads were then washed with 150 mL of lysis buffer. The proteins used in the GST pull-down assay were eluted with 5 mM glutathione. Fractions containing the targeted proteins were pooled and frozen in liquid nitrogen.
S-CDK was purified from strain yAE37 (a gift from John Diffley) using the method previously described19, with minor modifications. Yeast cells were cultured in YPR medium at 30 °C until the OD600 reached 2.0. Benomyl (Sigma, 381586) was added to a final concentration of 40 µg/ml to arrest cells at the G2/M phase before the addition of galactose (final 2%) to induce S-CDK overexpression for 4 h. The cells were harvested, washed twice with ice-chilled water, and resuspended in the lysis buffer (40 mM HEPES-KOH pH 7.6, 10% glycerol, 0.02% NP40S, 300 mM KOAc, 2 mM CaCl2) with 1 x protease inhibitor. The cell suspension was drop-wise frozen in liquid nitrogen and crushed using a Freezer Mill (Spex Sample Prep). Cell debris was removed by centrifugation (32,300 × g, 4 °C, 1 h). The supernatant was collected and incubated with 3 ml pre-equilibrated calmodulin affinity resin (Agilent Technologies, 214303-52) at 4 °C for 2 h. The beads were washed with lysis buffer, and S-CDK was eluted by incubating with 200 µg/ml TEV protease in digestion buffer (40 mM HEPES-KOH pH 7.6, 10% glycerol, 0.02% NP40S, 2 mM CaCl2) at 4 °C for 12 h. The supernatant was collected and separated by Superose 6 column (GE Healthcare) equilibrated with lysis buffer without CaCl2. Peak fractions were pooled, frozen in liquid nitrogen, and stored at − 80 °C.
FACS analysis
To arrest cells in the G1 phase, log-phase yeast cultures in YPG medium were treated with α-factor (10 μg/ml) at 30 °C for 2 h. The cultures pre-synchronized in the G1 phase were released into fresh YPD medium containing nocodazole (Cell Signaling, 2190, 10–15 μg/ml) for 3 h, which allowed for efficient G2/M arrest and Orc2-1 depletion. The cells were released into fresh YPD medium in the absence or presence of α-factor. The cells, after the second round of G1 synchronization, were further released into a fresh YPD medium. The relevant samples were taken at indicated intervals. Cells were then spun down, washed with H2O, and fixed with pre-cooled 70% ethanol overnight. The fixed yeast cells were washed with and resuspended in sodium citrate solution. RNase A (Thermo Scientific, EN0531) and Proteinase K (Thermo Scientific, EO0491) were added sequentially to remove RNAs and proteins. Then, cells were stained in PBS containing 0.5 mg/ml Propidium Iodide (Sigma, P4170) at 4 °C overnight. Flow cytometry was performed using BD FACSAriaIII flow cytometer and analyzed by FlowJo software.
Chromatin binding assay
200 ml of G1 cells were harvested as described in the above section for FACS analysis. The pellet was resuspended in pre-spheroplasting buffer [100 mM PIPES pH 9.4, 10 mM DTT, 1 mM PMSF, 2ug/ml pepstatinA] and incubated on ice for 10 min. The cells were then pelleted and resuspended in spheroplasting buffer [50 mM K2HPO4/KH2PO4 7.5, 0.6 M sorbitol, 10 mM DTT] containing 0.2 mg of lyticase for cell removal at 37 °C for 30 min. The spheroplasts were collected by centrifugation and washed with washing buffer [50 mM HEPES-KOH pH 7.5, 100 mM KCl, 2.5 mM MgCl2, 0.4 M sorbitol]. The spheroplasts were lysed in lysis buffer [50 mM HEPES-KOH pH 7.5, 100 mM KCl, 2.5 mM MgCl2, 0.5% TritonX-100, 1 × protease inhibitor] and incubated on ice for 30 min. The lysates were spun twice at 2400 × g for 1 min to remove under-lysed cells. The supernatant fractions were loaded onto an equal volume of lysis buffer containing 30% sucrose and centrifuged at 20,800 × g for 10 min at 4 °C. The insoluble chromatin fractions were then washed twice with the lysis buffer. The relevant samples were collected for SDS-PAGE and immunoblotting. For immunoblotting, the antibodies against Mcm6 (a gift from Karim Labib), SB3 (a gift from Bruce Stillman), and histone H2B (Abcam, ab1790) were applied at dilutions of 1:20000, 1:20000, and 1:2500, respectively.
Syn-seq, copy-number calculation, and nucleosome positioning
Syn-seq and copy-number calculations were carried out as previously described39. The Orc2-WT, Orc2-ΔN200, and Orc2-I194E cells were collected at 0 min and 30 min after release from G1 synchronization with Orc2-1 depletion. 1 μg of genomic DNA was used for library preparation and sequencing by BGI using the DNBseq platform, achieving a sequencing depth of approximately 75 reads per nucleotide. Trim Galore (Version 0.6.7) was used to remove adapters, and low-quality reads from the sequencing data, which were then mapped to the S288c reference genome (SGD, SacCer3) in 1-kb bins using the localMapper script. Duplicates were removed using the localMapper script. The Repliscope R package (Version 1.1.1) under R (Version 4.3.0) was utilized for further analysis39. All raw and processed sequencing data were deposited in the GEO database under the accession number GSE262693.
Previously published datasets of MNase-seq with G1 yeast cells were obtained from the Sequence Read Archive (SRA) with accession number SRP04131440. The MNase-seq data were first cleaned using Trim Galore (Version 0.6.7) and mapped to the S288c reference genome (SGD, SacCer3) using bowtie2 (Version 2.5.3)41. Duplicates were removed using Picard tools (Version 2.27.5, http://broadinstitute.github.io/picard). Nucleosome positioning around ARS in 1-bp bins was generated using deepTools (Version 3.5.4)42.
In vitro MCM loading assay
MCM loading assay was performed as previously described6,21 with modifications as detailed below. For the on-beads reaction, 2 pmol of ORC(or SH mutants), 2.5 pmol of Cdc6, and 5 pmol of Cdt1-Mcm2-7 were incubated with 2.5 pmol of a biotin-labeled ARS1 DNA (260 bp) immobilized to M280 Streptavidin beads (Invitrogen, 11205D) in pre-RC assembly buffer [25 mM HEPES-KOH pH 7.4, 150 mM K-glutamate, 0.02% NP40S, 5 mM Mg(OAc)2, 10% glycerol, 1 mM DTT, 3 mM ATP]. To assay OCCM formation, 1 mM ATPγS was used to replace ATP. The reactions were carried out at 30 °C for 30 min. For the rescue assay with Orc2-IDR peptide (TSPLKQIIMNNLKEYKDST, GenScript), 3 ng of the peptide was first incubated with 5 pmol of Cdt1-Mcm2-7 on ice for 30 mins before being added to the reaction. In the reaction with S-CDK, ORC and ARS1 DNA was first incubated on ice for 10 min, followed by the addition of 0.25 nM of S-CDK for further incubation at 30 °C for 10 min. Cdc6 and Cdt1-Mcm-2-7 were then included in the reaction. The beads were washed with either low salt buffer [25 mM HEPES-KOH pH 7.4, 300 mM K-glutamate, 0.02% NP40S, 5 mM Mg(OAc)2, 10% glycerol, 1 mM DTT, 3 mM ATP] or high salt buffer [25 mM HEPES-KOH pH 7.4, 600 mM NaCl, 0.02% NP40S, 5 mM Mg(OAc)2, 10% glycerol, 1 mM DTT, 3 mM ATP], respectively. DNA-bound proteins were eluted by digesting the DNA with 0.2 units of DNase I (NEB, M0303) at 37 °C for 30 min. For immunoblotting, the antibodies against Cdc6 (Santa Cruz, sc6317) and Cdt1 (a gift from Stephen P Bell) were applied at dilutions of 1:2000 and 1:10,000, respectively. The samples used for electron microscopy imaging were collected directly from the reactions with ARS1 DNA which was not immobilized to M280 beads.
To isolate OCCM, the assembled intermediates were loaded onto a 20%–40% glycerol gradient in buffer [25 mM HEPES-KOH pH 7.6, 150 mM K-glutamate, 5 mM Mg(OAc)2, 20–40% glycerol, 0.02% NP40S, 0.1 mM ATPγS] for ultracentrifugation at 137,000 × g, 14 h using a TLS-55 rotor (Beckman). Fractions containing OCCM were pooled and concentrated for GST pulldown assay in the presence of 0.1 mM of ATPγS.
To enrich MO, 25 nM of ORC mutants and 28 nM of Cdc6 were first incubated with 350 ng of ARS1 DNA (350 bp) in MO buffer (25 mM HEPES-KOH pH 7.6, 10 mM Mg(OAc)2, 200 mM KOAc, 1 mM DTT, 0.5 mM ATP) for 10 minutes (Eppendorf, 30 °C, 1250 rpm). Next, 8 nM of Mcm2-7-Cdt1 (or Cdt1-Mcm2-7 premixed with Orc2-SH peptide) was added and incubated for 4 min before EM analysis.
Negative stain electron microscopy
Negative-stain sample preparation was performed on 400-mesh copper grids with carbon film (PELCO). Grids were glow-discharged for 30 s at 45 mA (PELCO easiGlow) before a 3 μl sample was applied. After 1 min incubation, the residual sample was removed with filter paper. Grids were stained with 2% uranyl formate (Electron Microscopy Sciences, 22450) three times.
Data collection was carried out on a Talos120c microscope (Thermo Scientific) operating at 120 keV. Micrographs were taken with a Ceta CMOS camera at a nominal magnification of 57,000 (2.7 Å pixel size) within a − 0.5 to − 2.5 μm defocus range. Negative stain particles were processed with RELION 3.1. The CTF of each micrograph was estimated using CTFFIND 4.1, and particles were extracted and subjected to reference-free 2D classification.
To capture the process of pre-RC assembly, the MCM loading reaction was fixed at different time points (2, 10, 20 min) by applying 3 μl of the samples onto glow-discharged grids for staining with uranyl formate. For each sample, ~ 20 micrographs were collected to obtain sufficient particles for classification. We used EMAN2 v2.9 for the semi-automatic selection of particles with a box size of 210 Å, which were then classified by RELION 3.1. Following 2D classification, all classes showing MCM-containing particles were identified, including Cdt1/Mcm2-7/Mcm2-7, OCCM, MO, and MCM-DH. The total number (n) of the MCM-containing particles was calculated as nCdt1-Mcm2-7/Mcm2-7 + nOCCM + nMO + 2 × nMCM-DH. The particle numbers of each class were recorded to calculate their percentage over the total MCM.
The quantification of MO formation was carried out following a previous study21. The typical shape of the MO particles consists of one ORC coupled to a single MCM ring, which is tilted at an angle of about 90° offset from the central channel of the ORC. EM data was collected for MO formation after the pre-RC reactions for 4 min. Moreover, the Orc2-SH peptide was premixed with Cdt1-Mcm2-7 before adding them to the reactions for MO formation.
ATPase assay
To examine ATPase activities, the MCM loading reactions were performed in an ATPase buffer [25 mM HEPES-KOH pH 7.4, 150 mM KOAc, 5 mM Mg(OAc)2] with 500 pmol of ATP. One reaction with the buffer only was used as a control. After bead removal, the remaining ATP concentration was measured with an ATP Fluorometric Assay kit (Sigma, MAK190). Fluorescence quantification was conducted with BioTex Cytation 1 cell imaging multi-mode reader at a wavelength of Ex575/Em620. The standard deviation was generated from three independent experiments.
GST Pulldown assay
5 μg of GST, GST-O2N175, GST-O2N200, or GST-O2N200-I194E were first incubated with 50 μl of pre-equilibrate Glutathione-Sepharose beads (Cytiva, 17075601) in 500 μl of binding buffer 1 (BF1) [50 mM Tris-HCl pH7.0, 100 mM NaCl, 1 mM DTT] at 4 °C for 2 h. The bound beads were washed with 500 μl of BF1, followed by washing with 500 μl of binding buffer 2 (BF2) [50 mM HEPES-KOH pH 7.4, 100 mM K-glutamate, 8 mM MgCl2, 0.02% NP40S, 5 mM ATP, 1 mM EDTA, 1 mM DTT]. The beads were then resuspended with 500 μl of BF2 with the inclusion of 20 μg of Cdt1-Mcm2-7 heptamer or OCCM and incubated for 2 h. After washing the beads three times with 500 μl of BF2, elution was conducted with 100 μl of BF1 containing 5 mM glutathione. For the assays with OCCM, 0.1 mM ATPγS was used to replace ATP in all the buffers. For immunoblotting, the antibodies against Mcm2 (a gift from Bruce Stillman) and GST (Abcam, ab111947) were applied at dilutions of 1:10,000 and 1:1000, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw sequence data reported in this paper have been deposited in the GEO database under the accession number GSE262693. Source data are provided in this paper.
References
Bell, S. P. & Labib, K. Chromosome duplication in Saccharomyces cerevisiae. Genetics 203, 1027–1067 (2016).
Parker, M. W., Botchan, M. R. & Berger, J. M. Mechanisms and regulation of DNA replication initiation in eukaryotes. Crit. Rev. Biochem Mol. Biol. 52, 107–144 (2017).
Bleichert, F., Botchan, M. R. & Berger, J. M. Mechanisms for initiating cellular DNA replication. Science 355, https://doi.org/10.1126/science.aah6317 (2017).
Tanaka, S. & Araki, H. Helicase activation and establishment of replication forks at chromosomal origins of replication. Cold Spring Harb. Perspect. Biol. 5, a010371 (2013).
Hoggard, T. & Fox, C. A. In The Initiation of DNA Replication in Eukaryotes (ed Daniel L. Kaplan) 159-188 (Springer International Publishing, 2016).
Remus, D. et al. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 139, 719–730 (2009).
Evrin, C. et al. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 106, 20240–20245 (2009).
Li, N. et al. Structure of the eukaryotic MCM complex at 3.8 A. Nature 524, 186–191 (2015).
Abid Ali, F. et al. Cryo-EM structure of a licensed DNA replication origin. Nat. Commun. 8, 2241 (2017).
Noguchi, Y. et al. Cryo-EM structure of Mcm2-7 double hexamer on DNA suggests a lagging-strand DNA extrusion model. Proc. Natl. Acad. Sci. USA 114, E9529–E9538 (2017).
Azmi, I. F. et al. Nucleosomes influence multiple steps during replication initiation. Elife 6, https://doi.org/10.7554/eLife.22512 (2017).
Kurat, C. F., Yeeles, J. T. P., Patel, H., Early, A. & Diffley, J. F. X. Chromatin controls DNA replication origin selection, lagging-strand synthesis, and replication fork rates. Mol. Cell 65, 117–130 (2017).
Devbhandari, S., Jiang, J., Kumar, C., Whitehouse, I. & Remus, D. Chromatin constrains the initiation and elongation of DNA replication. Mol. Cell 65, 131–141 (2017).
Douglas, M. E., Ali, F. A., Costa, A. & Diffley, J. F. X. The mechanism of eukaryotic CMG helicase activation. Nature 555, 265–268 (2018).
Lewis, J. S. et al. Mechanism of replication origin melting nucleated by CMG helicase assembly. Nature 606, 1007–1014 (2022).
Suski, J. M. et al. CDC7-independent G1/S transition revealed by targeted protein degradation. Nature 605, 357–365 (2022).
Kang, S., Kang, M. S., Ryu, E. & Myung, K. Eukaryotic DNA replication: Orchestrated action of multi-subunit protein complexes. Mutat. Res. 809, 58–69 (2018).
Heller, R. C. et al. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146, 80–91 (2011).
Yeeles, J. T., Deegan, T. D., Janska, A., Early, A. & Diffley, J. F. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519, 431–435 (2015).
Yuan, Z. et al. Structural basis of Mcm2-7 replicative helicase loading by ORC-Cdc6 and Cdt1. Nat. Struct. Mol. Biol. 24, 316–324 (2017).
Miller, T. C. R., Locke, J., Greiwe, J. F., Diffley, J. F. X. & Costa, A. Mechanism of head-to-head MCM double-hexamer formation revealed by cryo-EM. Nature 575, 704–710 (2019).
Gupta, S., Friedman, L. J., Gelles, J. & Bell, S. P. A helicase-tethered ORC flip enables bidirectional helicase loading. Elife 10, https://doi.org/10.7554/eLife.74282 (2021).
Stillman, B. The remarkable gymnastics of ORC. Elife 11, https://doi.org/10.7554/eLife.76475 (2022).
Regan-Mochrie, G. et al. Yeast ORC sumoylation status fine-tunes origin licensing. Genes Dev. 36, 807–821 (2022).
Amasino, A. L., Gupta, S., Friedman, L. J., Gelles, J. & Bell, S. P. Regulation of replication origin licensing by ORC phosphorylation reveals a two-step mechanism for Mcm2-7 ring closing. Proc. Natl. Acad. Sci. USA 120, e2221484120 (2023).
Schmidt, J. M. et al. A mechanism of origin licensing control through autoinhibition of S. cerevisiae ORC.DNA.Cdc6. Nat. Commun. 13, 1059 (2022).
Li, N. et al. Structure of the origin recognition complex bound to DNA replication origin. Nature 559, 217–222 (2018).
Muller, P. et al. The conserved bromo-adjacent homology domain of yeast Orc1 functions in the selection of DNA replication origins within chromatin. Genes Dev. 24, 1418–1433 (2010).
Kuo, A. J. et al. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 484, 115–119 (2012).
Nguyen, V. Q., Co, C. & Li, J. J. Cyclin-dependent kinases prevent DNA re-replication through multiple mechanisms. Nature 411, 1068–1073 (2001).
Shimada, K. & Gasser, S. M. The origin recognition complex functions in sister-chromatid cohesion in Saccharomyces cerevisiae. Cell 128, 85–99 (2007).
Fernandez-Cid, A. et al. An ORC/Cdc6/MCM2-7 complex is formed in a multistep reaction to serve as a platform for MCM double-hexamer assembly. Mol. Cell 50, 577–588 (2013).
Hossain, M., Bhalla, K. & Stillman, B. Multiple, short protein binding motifs in ORC1 and CDC6 control the initiation of DNA replication. Mol. Cell 81, 1951–1969.e1956 (2021).
Parker, M. W. et al. A new class of disordered elements controls DNA replication through initiator self-assembly. Elife 8, https://doi.org/10.7554/eLife.48562 (2019).
Parker, M. W., Kao, J. A., Huang, A., Berger, J. M. & Botchan, M. R. Molecular determinants of phase separation for Drosophila DNA replication licensing factors. Elife 10, https://doi.org/10.7554/eLife.70535 (2021).
Lim, C. T. et al. Cell cycle regulation has shaped budding yeast replication origin structure and function. Preprint at bioRxiv https://doi.org/10.1101/2024.01.10.575016 (2024).
Coster, G. & Diffley, J. F. X. Bidirectional eukaryotic DNA replication is established by quasi-symmetrical helicase loading. Science 357, 314–318 (2017).
Frigola, J., Remus, D., Mehanna, A. & Diffley, J. F. ATPase-dependent quality control of DNA replication origin licensing. Nature 495, 339–343 (2013).
Batrakou, D. G., Muller, C. A., Wilson, R. H. C. & Nieduszynski, C. A. DNA copy-number measurement of genome replication dynamics by high-throughput sequencing: the sort-seq, sync-seq and MFA-seq family. Nat. Protoc. 15, 1255–1284 (2020).
Belsky, J. A., MacAlpine, H. K., Lubelsky, Y., Hartemink, A. J. & MacAlpine, D. M. Genome-wide chromatin footprinting reveals changes in replication origin architecture induced by pre-RC assembly. Genes Dev. 29, 212–224 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–165 (2016).
Acknowledgements
We thank Bruce Stillman, John Diffley, and Susan Gasser for yeast strains and antibodies, and members in our laboratories for discussion. This work was supported by the Research Grants Council (RGC) of Hong Kong (GRF17119022, GRF17109623, C7035-23GF, and Y.L.Z.).
Author information
Authors and Affiliations
Contributions
Y.L.Z conceived and supervised the project; Y.W. constructed strains; Y.W. and Q.Z. purified proteins and performed functional experiments; Y.L. and Y.W. conducted bioinformatic analyses; Y.L.Z., Y.W., Q.Z., Y.L., and W.L. analyzed the data, prepared the figures, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Matthew Parker and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Y., Zhang, Q., Lin, Y. et al. Replication licensing regulated by a short linear motif within an intrinsically disordered region of origin recognition complex. Nat Commun 15, 8039 (2024). https://doi.org/10.1038/s41467-024-52408-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52408-0
- Springer Nature Limited