

Abstract
Progressive supranuclear palsy (PSP), a rare Parkinsonian disorder, is characterized by problems with movement, balance, and cognition. PSP differs from Alzheimer’s disease (AD) and other diseases, displaying abnormal microtubule-associated protein tau by both neuronal and glial cell pathologies. Genetic contributors may mediate these differences; however, the genetics of PSP remain underexplored. Here we conduct the largest genome-wide association study (GWAS) of PSP which includes 2779 cases (2595 neuropathologically-confirmed) and 5584 controls and identify six independent PSP susceptibility loci with genome-wide significant (P < 5 × 10−8) associations, including five known (MAPT, MOBP, STX6, RUNX2, SLCO1A2) and one novel locus (C4A). Integration with cell type-specific epigenomic annotations reveal an oligodendrocytic signature that might distinguish PSP from AD and Parkinson’s disease in subsequent studies. Candidate PSP risk gene prioritization using expression quantitative trait loci (eQTLs) identifies oligodendrocyte-specific effects on gene expression in half of the genome-wide significant loci, and an association with C4A expression in brain tissue, which may be driven by increased C4A copy number. Finally, histological studies demonstrate tau aggregates in oligodendrocytes that colocalize with C4 (complement) deposition. Integrating GWAS with functional studies, epigenomic and eQTL analyses, we identify potential causal roles for variation in MOBP, STX6, RUNX2, SLCO1A2, and C4A in PSP pathogenesis.
Similar content being viewed by others


Introduction
Tau proteinopathies (“tauopathies”), characterized by abnormal aggregates composed of the microtubule-associated protein tau inclusions, are a class of neurodegenerative diseases in aged individuals with varying yet overlapping clinical features including dementia, movement disorder, motor neuron disease, and psychiatric changes1,2. Among these is progressive supranuclear palsy (PSP; MIM #601104), a rare, late-onset neurodegenerative disease characterized by impaired movement, with symptoms including slowed movement (bradykinesia), loss of balance, frequent falls, and difficulty with eye movement (vertical supranuclear gaze palsy), as well as cognitive decline. Though an uncommon cause of dementia compared to AD, PSP is estimated to affect 5–17 per 100,000 persons in the US, making it the second leading cause of Parkinsonism after PD, and autopsy studies have found PSP pathology in 2–6% of individuals with no PSP neurological diagnosis prior to death, suggesting that it is more prevalent than appreciated in living individuals3,4,5. Given the sharing of common tau pathology across multiple neurodegenerative diseases (i.e., AD, corticobasal degeneration, chronic traumatic encephalopathy, and others) insights into the pathogenesis of PSP may yield potential therapeutic targets for a multitude of related diseases.
Key insights into PSP have come from decades of genetic studies which have demonstrated the disorder to be almost entirely sporadic disease, however careful clinical evaluation has revealed a tendency for family clustering6,7. While the chromosome 17q21.31 H1/H2 haplotype, an approximately 900 kbp inversion polymorphism encompassing the gene encoding the tau protein MAPT, remains the strongest known genetic risk factor for PSP (OR ≈ 5.5), loci containing variants with more modest effects have also been identified. These include variants associated with genome-wide significance (GWS; P < 5 × 10−8) in or near genes STX6, EIF2AK3, and MOBP8. Subsequent work identified a variant, rs2242367, in an intron of the chromosome 12q12 gene SLC2A13, approximately ~200 kbp upstream of the established PD/parkinsonism gene LRRK2 as a locus influencing PSP disease duration9. Additional novel PSP risk loci were identified at chromosomes 6p12.1 (near RUNX2) and 12p12.1 (near SLCO1A2), as well as a unique 17q21.31 association in MAPT-adjacent gene KANSL110,11. These loci help resolve only a portion of PSP heritability, and much of the genetic risk remains unexplained10.
In this work, given that PSP is an archetypical primary tauopathy, advancing our understanding of the genetic architecture of the disorder requires large and robustly characterized cohorts of diseases2,12, which have the potential to provide important insight into a number of diseases. Prior genome-wide association studies (GWAS) of PSP have been limited by relatively modest sample sizes, suboptimal control groups, and a paucity of downstream analyses to nominate causal genes surrounding or within the significant risk loci8,10,11. Here we perform the largest genetic association study of PSP to date, including 2779 cases (of which 2595 were autopsy-confirmed, building upon the 1069 autopsy confirmed cases from stage 1 of work from 20118) and 5584 age-matched, non-demented, autopsy-confirmed controls derived from the Alzheimer Disease Genetics Consortium13. We perform functional follow-up on each identified locus using a battery of annotation tools to pinpoint candidate causal genes and perform additional validation using molecular and histological approaches in human postmortem brain tissue. Taken together, our findings identify novel PSP genetic risk architecture and provide new functional insight into the genetic and molecular mechanisms driving tau proteostasis in PSP.
Results
Dataset collection and quality control
The cohort consisted of 2779 PSP cases and 5584 age-matched controls of which a majority were neuropathologically confirmed representing the largest PSP GWAS to date (Table 1). On average, the age-at-death of the controls was approximately ten years older than the cases and there were proportionally more females in the control population (60%) than in the cases (45%). To harmonize genotype data across multiple genotyping platforms and account for separate ascertainment of some sets of cases and controls, we constructed and implemented a genotype harmonization pipeline after quality control and prior to imputation (Supplementary Fig. 1). After imputation of individual case-control sets to the TOPMed-r2 reference panel, we filtered down to an overlapping set of 7,230,420 common (minor allele frequency (MAF) > 0.01), high quality (imputation R2 > 0.8) SNPs.
Genome-wide association study
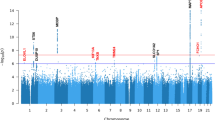
We observed six genome-wide significant loci (GWS; P < 5 × 10−8), of which one was novel on 6p21.32 near TNXB at SNP rs369580 (OR [95% CI]: 1.43 [1.28, 1.60]; P = 8.11 × 10−10) (Table 2). Our results confirm previously identified signals in loci containing MAPT, STX6, MOBP, RUNX2, and SLCO1A2. We observed a signal below the GWS threshold corresponding to the locus containing EIF2AK3 (P = 3.63 × 10−5) which was reported in previous studies10 (Fig. 1a). Genome-wide associations demonstrated modest genomic inflation with λ = 1.074 (Supplementary Fig. 2). As a sensitivity analysis, we repeated the analysis excluding the 125 non-autopsy confirmed subjects and did not observe any major differences (Supplementary Fig. 3). Additionally, we examined signals identified in prior neurodegenerative studies GWAS and found 22 PD SNPs and 13 AD SNP with modest association signals (0.05 > P > 0.0005, Supplementary Data 1, 2). Conditional analysis using the lead SNP in each locus did not reveal any secondary associations at any locus (Supplementary Figs. 4–8). The addition of the MAPT haplotype status as a covariate weakened the association observed in the 17q21.31 loci (P = 2.28 × 10−17 vs. P = 1.94 × 10−110 with MAPT adjustment) but did not influence the other five associations (Supplementary Fig. 9). We then performed stratified LD-score regression (S-LDSC)14,15, a method to estimate SNP heritability enrichment in sets of variants grouped by genomic features, using epigenomic annotations from four major CNS cell types assigning variants to promoters and enhancers16. We compared our S-LDSC results to recent GWAS conducted in Alzheimer’s disease (AD) and Parkinson’s disease (PD)17,18,19. As previously shown, AD heritability was enriched within microglial enhancers, whereas PD heritability was enriched in neuronal and oligodendrocyte promoters. In PSP, we observed a nominally significant enrichment in oligodendrocyte enhancer sequences (P = 0.027) (Fig. 1b). We then performed functional fine-mapping with PolyFun-FINEMAP, which estimated independent sets of variants (credible sets) at four loci, excluding the HLA-adjacent 6p21.32 (TNXB) and MAPT loci due to LD complexity20,21. At each locus we observed between one and three credible sets each containing 1–3 SNPs with a posterior inclusion probability (PIP) > 0.1 (Fig. 1c). We then overlapped the fine-mapped SNPs with the same epigenomic annotations as before, identifying several loci that contain fine-mapped SNPs overlapping CNS cell type epigenomic annotations. We also compared our fine-mapped SNPs to significant SNPs found in a recent massively parallel reporter assay (MPRA) using a previous PSP GWAS22. Only the 3p22.1 locus contained previously tested SNPs.

a Manhattan plot for PSP genome-wide association study (n = 2779 cases versus n = 5584 controls) identified six genome-wide significant (P < 5 × 10−8) loci indicated by the dotted line containing approximately 218 genes around a 2-Mb window from the lead SNP. b LD-score regression using cell type-specific regulatory elements (dotted line indicates p < 0.05) derived from ChIP-seq10 demonstrates a unique cell-specific profile in PSP when compared to Alzheimer’s disease (AD) and Parkinson’s disease (PD)18,19. c Fine-mapping of the four amenable loci identified different numbers of causal SNPs and credible sets, some of which overlapped with cell type-specific regulatory elements and previously identified regulatory SNPs from an MPRA22. Abbreviations: Oligo, oligodendrocytes; MPRA, massively parallel reporter assay.
Colocalization analysis and gen prioritization
We then sought to identify specific causal genes at each locus by incorporating expression quantitative trait loci (eQTLs) from bulk brain expression data from the Genotype Tissue Expression (GTEx) project and expression data from sorted CNS cell types from single nucleus RNA-seq (snRNA-seq)23 (Fig. 2). We performed colocalization tests estimating the probability that the same single causal variant is associated with both disease risk and with gene expression by comparing all matching SNPs within each GWAS locus (±1Mbp) to those tested in each eQTL. This approach prioritized STX6 and RUNX2 as the likely causal genes in the 1p25.3 and 6p21.1 loci respectively as they had a high posterior probability (PP4) across multiple brain regions. In the complex HLA-adjacent 6p21.32 locus, BTNL2 colocalized in only three brain regions. Cell type-specific eQTLs identified STX6 and RUNX2 in oligodendrocytes at PP4 > 0.8, whereas eQTLs for MOBP were found in both oligodendrocytes and excitatory neurons23. No gene was prioritized by eQTLs in the 12p12.1 locus. Additionally, we ran a transcriptome-wide association study (TWAS) analysis using genetically-predicted expression models24,25 from two cohorts of dorsolateral prefrontal cortex: the CommonMind Consortium, and the Accelerating Medicines Partnership in Alzheimer’s Disease (AMP-AD) Project26,27. By identifying shared associations between genetically-predicted gene expression and our PSP GWAS, we identified increased cortical expression of STX6, RUNX2 and MOBP were associated with increased risk of PSP (Bonferroni-adjusted P < 0.05; Supplementary Fig. 10). TWAS prioritized multiple genes in the 6p21.32 and 17q21.32 loci, including C4A, C4B, and MAPT. In summary, three of the six GWAS loci have evidence of acting through gene expression in oligodendrocytes. Guided by these cell type-specific colocalizations, we examined our loci more closely.

Datasets were GTEx (bulk RNA-seq) and Bryois et al (single nucleus RNA-seq) eQTLs across brain regions and purified cell types analyzed with COLOC. a All genes and loci shown with PP4 > 0.5 in at least one eQTL dataset. Point sizes are scaled to the magnitude of posterior probability of colocalization (PP4). *PP4 ≥ 0.75. OPC, Oligodendrocyte precursor cell. b Directions of effect on gene expression for STX6, MOBP, and RUNX2 relative to the risk allele (increased in frequency in PSP cases) of the lead GWAS SNP. Error bars denote standard error of the eQTL association.
1p25.3 - STX6. Colocalization of 1p25.3 prioritized STX6 in multiple brain regions and with oligodendrocyte-specific eQTLs. The risk allele of the lead GWAS SNP rs1044595-C is associated with increased expression of STX6 in bulk brain samples and in purified oligodendrocytes (Fig. 2b). Fine-mapping of 1p25.3 identified two credible sets each containing 2 SNPs with a PIP > 0.1. These SNPs overlapped with oligodendrocyte enhancer sequences as identified by cell type-specific ChIP-seq (Fig. 3a). The first credible set included the lead GWAS SNP rs1044595 as well as a second SNP rs3789362, in high LD (R2 = 0.96, 1000 Genomes European superpopulation). rs2789362 overlaps with an oligodendrocyte-specific enhancer within an intron of STX6. Taken together, this suggests that rs3789362-A increases STX6 expression by modifying an oligodendrocyte-specific enhancer sequence.

Functional annotations from cell type-specific regulatory regions (enhancers and promoters) and cell type-specific DNA interactome anchors from proximity ligation-assisted ChIP-Seq (PLAC-seq)16 are shown in the loci containing STX6 (a) and RUNX2 (b). SNPs are colored by their LD with the lead GWAS SNP in Europeans (1000 Genomes European superpopulation).
3p22.1 - MOBP. As a well-established marker gene for oligodendrocytes, we expected to see strong colocalization between the 3p22.1 locus and MOBP in bulk brain samples and oligodendrocytes specifically. The risk allele of the lead GWAS SNP rs631312-G is associated with increased MOBP expression in oligodendrocytes (Supplementary Fig. 11). We observed three single-variant credible sets in 3p22.1, two of which overlapped with a region at the transcription start site of the MOBP gene containing ChIP-seq regions defined as both promoters and enhancers in oligodendrocytes and densely connected by proximity ligation-assisted ChIP-seq (PLAC-seq) contacts (Supplementary Fig. 11)16. Taken together, this suggests that variants within 3p22.1 alter MOBP expression in oligodendrocytes specifically.
6p21.1 - RUNX2. Colocalization of 6p21.1 prioritized RUNX2 in multiple brain regions as well as in oligodendrocytes specifically, with the risk allele of the lead GWAS SNP rs12197948-A associated with increased levels of RUNX2 expression in all datasets (Fig. 2b). Fine-mapping identified a single credible set containing 3 SNPs with a PIP > 0.1. Two of the SNPs (rs12197948 and rs4714854, R2 = 0.99, 1000 Genomes European superpopulation) sit within the third intron of RUNX2, which contains an annotated enhancer in both microglia and oligodendrocytes (Fig. 3b). Although rs4714854 overlaps a microglia enhancer peak, it is also very close to the oligodendrocyte enhancer peak. Additionally, PLAC-Seq in microglia identified several contacts between the microglia enhancer region and the RUNX2 promoter. Therefore, although colocalization suggests oligodendrocytes to be the causal cell-type, we cannot rule out that the GWAS association at 6p21.1 may also affect RUNX2 expression in microglia through a shared enhancer sequence.
6p21.32 - C4A. Although we observed colocalization with bulk brain eQTLs for BTNL2, we reasoned that the known LD complexity in the locus may obscure genuine colocalizations. As an alternative approach we applied INFERNO, which uses LD pruning at each GWAS locus to construct a set of independent SNP sets, which can then be tested for eQTL colocalization separately, which is the main difference COLOC28 and INFERNO29,30. In the 6p21.32 locus, INFERNO identified three sets of SNPs (Fig. 4a). Each SNP set was then colocalized with all nearby genes (Fig. 4b) using eQTLs from 13 GTEx brain regions (v7). We observed multiple genes in the locus to be colocalized, with the largest number of colocalizations (PP4 > 0.9) in all 3 SNP sets being with eQTLs for the gene C4A (Fig. 4c). Comparing each P-value for association with PSP (PGWAS) with the eQTL association in GTEx Frontal Cortex (PeQTL), we observed that while including all SNPs from the locus resulted in a minimal probability of colocalization (PP4 = 2 × 10−5; Fig. 4d), using the individual sets of SNPs resulted in a much higher colocalization for all three sets with C4A eQTLs (PP4 > 0.8; Fig. 4e). The risk allele of the lead GWAS SNP rs2523524-G was associated with increased C4A expression (Supplementary Fig. 12). Given the known C4A copy number variation in this locus we hypothesized that variability in C4A copy number could explain the observed signal. To test this hypothesis, we generated imputed C4A and C4B copy number values and ran logistic regression for case control status based on alterations in the copy number of each gene and observed that C4A copy number was suggestively associated with PSP status (P = 1.58 × 10−6), whereas C4B copy number was not (P = 0.14). Additionally, we ran our association study including either C4A or C4B copy number status as a covariate and observed that the inclusion of only C4A copy number status weakened the signal such that genome-wide statistical significance was no longer observed in this locus (Fig. 5a–c). We therefore nominated C4A as the most likely causal gene at this locus.

a LD pruning selects three sets of credible SNPs in the locus which surround several genes (b). Colocalization of each SNP set with expression quantitative trait loci in multiple brain regions (GTEx v7) (c) shows that an eQTL for C4A is present in the majority of tested brain regions, more than any other gene, with a high posterior probability of colocalization (PP4) > 0.9. d Comparing the relationship between the P-values found in GWAS (PGWAS) and the C4A eQTL (PeQTL). e Using all SNPs in the locus resulted in a low PP4 with C4A eQTLs in the frontal cortex (PP4 < 0.01), whereas the three sets of SNPs chosen by INFERNO all show high probability of colocalization (PP4 > 0.8).

a Despite losing 14 cases due to the inability to impute C4A and C4B copy number, a genome-wide significant association is still observed on 6p21.32. b The addition of imputed C4B copy number to the association model still produces a robust genome-wide significant association, whereas adding imputed C4A copy number (c) results in a non-statistically significant association, suggesting that imputed C4A copy number is influencing the signal.
Differential gene expression in frontal cortex and cerebellum
Given the genetic, eQTL, and fine-mapping evidence suggesting that multiple genes contained in the GWS-associated loci are potentially implicated in PSP, we leveraged a previously generated bulk RNA-seq dataset to identify regionally specific changes in gene expression in the frontal cortex and cerebellum in patients with PSP compared to non-neurological disease controls31,32. After re-analyzing the raw data to include relevant covariates, we focused on 16 candidate genes identified in the significant GWAS loci and available in the bulk RNA-seq dataset (C4A, CYP21A1P, FLOT1, HLA DPB1, HLADMB, KIAA1614, MOBP, MSH5, PLA2G7, RPSA, RUNX2, SLCO1A2, STX6, SUPT3H, VILL, ZNF621). In the frontal cortex, we observed significantly increased expression of STX6 and FLOT1, and significantly decreased expression of PLA2G7, MOBP, MSH5, HLA-DPB1, HLA-DMB, and SLCO1A2 in PSP versus controls (P < 0.0025, Fig. 6a). In the cerebellum, the same significantly decreased expression pattern was observed in MOBP, MSH5, and SLCO1A2, but no significant differences were observed in HLA-DPB1, HLA-DMB, and FLOT1 (P > 0.0025, Fig. 6b). The data suggest regionally specific changes of multiple genes identified in loci identified from the GWAS data which may have downstream effects on disease relevant protein expression, however these differences may be attributed to a difference in cell composition between cases and controls and further single-cell analysis studies are warranted.

Bulk RNA-seq from PSP (n = 84 cortices, 83 cerebella) and control (n = 77 cortices, 77 cerebella) human post-mortem brain tissue showing significant differences in (a) six neocortical genes and (b) five cerebellar. The expression differences shown are normalized read counts. Sixteen total candidate genes were present in the dataset. Bulk RNA-seq fold change values shown only if the differential expression P < 0.0025. Box plots show the median, the first and third quartiles of the distribution, with whiskers extending to 1.5 times the interquartile range to the end of each figure legend.
Immunohistochemical and biochemical analysis
Given the novel genetic association observed in the 6p21.32 locus and the downstream computational evidence nominating C4A as a candidate gene, we examined tissues from both PSP cases and controls biochemically and immunohistochemically to see if there was any cell type-specific pathology relevant to the C4A signal. As expected, multiplex immunohistochemical staining of controls (n = 10) showed little hyperphosphorylated tau (p-tau, AT8) pathology and a minimal amount of C4A protein signal in the frontal cortex alongside positively stained healthy oligodendrocytes (OLIG2) (Fig. 7a, Supplementary Fig. 13). In PSP cases (n = 10), we observed a strong immunohistochemical p-tau signal in neurons and tufted astrocytes, a hallmark pathology of the disorder, as well as a marked increase in C4A protein expression in the axons in association with p-tau positive oligodendrocytic coiled bodies (Fig. 7b, Supplementary Fig. 13). Image analysis revealed significantly more C4A staining in PSP than controls (P = 0.0001, Fig. 7c) Furthermore, these axonal profiles were abnormal, being significantly shorter in PSP than controls (P = 0.001, Fig. 7d). Finally, quantitative immunoblots of the C4A alpha chain showed significantly higher levels in PSP (n = 6) compared to controls (n = 7, P = 0.008, Fig. 7e, Supplementary Fig. 14). Together, these findings provide histological and biochemical evidence of C4 abnormalities in association with tau pathology in PSP brains.

a Triple immunohistochemical stains of C4A (magenta), hyperphosphorylated tau (AT8, green), and OLIG2 (brown) show limited C4A and hyperphosphorylated tau staining in controls (b). Staining in PSP demonstrates colocalization of C4A and hyperphosphorylated tau in an OLIG2 positive coiled body (blue and brown). Tufted astrocyte cell bodies (green) and neurofibrillary tangles (not shown) are negative for C4A in PSP. The average number of C4A positive axons per subject (c) was significantly higher in PSP than controls (n = 10 cases and controls, frontal cortex, 5 regions of interest per subject, 1538 observations found in PSP, 124 in controls). Quantification of C4A positive axons revealed significantly shorter (d) axonal length in PSP when compared to controls. e Quantitative immunoblots of the C4A alpha chain showed significantly higher levels in PSP compared to controls (n = 7 cases, 6 controls). f C4A gene expression in whole blood from patients with PSP was significantly higher than controls (n = 51 cases, 281 controls). The scale bar is 50 micrometers.
C4A expression in whole blood
As we observed elevated C4A protein in PSP oligodendrocytes postmortem, we reasoned that C4A mRNA may be elevated in living patient blood samples. We re-analyzed a publicly available RNA microarray dataset generated from whole blood from non-neurological controls (n = 281) and clinically diagnosed PSP cases (n = 51)33. We observed C4A mRNA to be upregulated in blood from PSP patients compared to controls (P = 0.02, Fig. 7f, Supplementary Fig. 15).
Discussion
We have summarized our ensemble of downstream genetic analyses in a single table (Table 3) and assigned a gene priority score based on methods detailed elsewhere34. In the 1q25.2 locus, we nominate STX6 as the causal gene as it shows eQTL colocalization in bulk brain specifically in regions known to be vulnerable to pathology in PSP as well as in oligodendrocytes; the fine-mapped SNPs overlap an oligodendrocyte-specific enhancer region; and STX6 gene expression is upregulated in the cerebellum of PSP brains. In the 3p22.1 locus, we nominate MOBP as the causal gene, although it does not show eQTL colocalization in bulk brain and others have suggested its role in SLC25A38/Apoptosin gene locus 70 kbp away, we did observe a signal in oligodendrocyte single cell data as well as downregulation of expression in PSP brains in addition to fine mapping oligodendrocytic enhancers and promoters as well as an observed signal in the MPRA data22,35. In the 6p21.1 locus, we nominate RUNX2 based on similar observations however there is some discrepancy over the cell type-specificity given the signal mapped to microglial enhancers but also had an oligodendrocytic eQTL signal. We did not observe associations in the loci overlapping EIF2AK3 and LRRK2 which have been previously observed, albeit LRRK2 was found to be associated with PSP survival not susceptibility8,9. In the novel 6p21.32 locus reported here, nomination of a causal gene was challenging given the limited fine mapping and eQTL interactions reported. Thus, we turned to biochemical and immunohistochemical validation which strongly supports C4A’s role in complement activation and neuroinflammation in PSP and thus calls for further exploration of the mechanistic role of this gene in PSP. Lastly, in the 12p12.1 locus we nominate SLCO1A2 given the evidence provided for its significant downregulation of expression in PSP brains in multiple regions.
Tau proteinopathies, especially primary tauopathies such as PSP that arise independently of amyloid-β, hold significant clinical and scientific importance due to their prevalence and potential to provide novel mechanistic insights into neurodegeneration2,12,36. Furthermore, understanding abnormalities in tau dysfunction offers promise in advancing our knowledge of neurodegenerative diseases more broadly37. Tau-related neurodegeneration has been proposed to occur through various mechanisms, including apoptosis, excitotoxicity, oxidative stress, inflammation, mitochondrial dysfunction, prion-like propagation, and protein aggregation. However, the connection between these mechanisms and genetic drivers remains poorly understood38. Furthermore, the vulnerability of distinct cell populations in tauopathies remains incompletely characterized. Human GWAS continues to be an invaluable tool in elucidating causal mechanisms in neurodegenerative diseases39. Notably, increasingly large genetic studies of Alzheimer disease (AD) have continued to enable the discovery of new risk loci, including those related to neuroimmune mechanisms and other functions39. Genetic studies of the primary tauopathies, including PSP, have remained small despite their potential to highlight non-amyloid-driven mechanisms that are becoming increasingly relevant given the growing emphasis on combination therapy in AD40. This study of PSP, the prototype non-AD primary tauopathy, that includes 8363 total subjects in a genome-wide study, sheds new light on these candidate mechanisms.
Our study uncovered a novel genetic signal at C4A which encodes the acidic form of complement factor 4, part of the classical activation pathway. This finding was further supported by histological, biochemical, and blood biomarker analyses. Critically, co-localization of C4A protein with abnormal tau species in oligodendrocytes further supports that innate immune function plays a causal role in driving this pathological interaction in PSP. A significant locus in the HLA region near C4A has also been identified in a GWAS of ALS, which is hypothesized to be a distal (dying-back) axonopathy, suggesting the possibility that axon-myelin interactions might contribute to and link these disorders41,42. Furthermore, there is a genetic link between copy number variation in C4A and schizophrenia and functional studies have shown overproduction of this protein promotes excessive synaptic loss and behavioral changes in mice43,44,45. To this point, we also observed this link when we ran our association study with imputed C4A copy number as a covariate resulting in reduction of our primary signal in 6p21.32 below the genome-wide threshold. Additionally, using a CRISPR deletion assay targeting a SNP contained within 6p21.32 in the HLA region (previously found in a GWAS of AD22) reduced C4A expression in iPSC-derived astrocytes providing more evidence for the role of C4A in neurodegeneration. Similar to what we observed in a whole blood dataset, others have observed that differences in complement protein levels in the cerebrospinal fluid across various neurodegenerative diseases46,47,48. Lastly, although it has been hypothesized for some time that complement activation is involved in neurodegeneration and this has been shown in murine models, our histopathological evidence in human post-mortem tissue shows marked morphological features in oligodendrocytes with p-tau pathology, demonstrating a link back to the identified novel genetic loci45,49. In summary, although the genetic signals (e.g., lead SNP) differ in these genetic studies compared to the PSP genetics presented here, these findings underscore the importance of exploring the role of innate immune interactions and oligodendrocyte pathology in the pathogenesis of multiple neurodegenerative conditions.
Despite the insights gained from this study, several limitations should be considered when interpreting the findings. Although most cases were autopsy-confirmed to assure correct classification, this limited our overall sample size, compared to using clinically diagnosed, or even proxy cases. To this point, the relatively limited availability of genetically- and phenotypically-characterized PSP cases, the global majority of which are incorporated into this the largest study of PSP to-date, still limited this study to the observation of only one novel locus. GWAS typically requires very large sample sizes to achieve sufficient statistical power, and inadequate sample sizes can result in false-negative and false-positive findings, potentially missing true genetic associations. While we were able to provide biochemical and histological evidence prioritizing C4A at the 6p21.32 locus, this gene resides in the HLA region where complex structural genomic rearrangements complicate identification of causal variants. This also limits our ability to nominate genes on 17q21.31, thus there is a critical need for advanced computational tools and long-read sequencing in these loci. Follow-up studies, such as functional genomic analyses, model organism experiments, stratification based on potential comorbid pathological features, and a replication cohort are necessary to elucidate how the identified variants affect biological processes related to neurodegeneration.
We explored the genetic risk for PSP, confirming previous signals while also identifying one novel association. Among the confirmed genetic signals, MAPT remains the strongest, consistent with the well-established role of the MAPT haplotypes in tau proteinopathies50. We also confirmed the association with myelin-associated oligodendrocytic basic protein (MOBP) and assigned this signal to gene expression in oligodendrocytes, in line with its role in synthesis and maintenance of myelin. MOBP is also a candidate risk gene in amyotrophic lateral sclerosis (ALS) and a previous colocalization analysis has shown that the same causal SNPs are found in PSP, ALS, and corticobasal degeneration8,41,51,52. STX6 encodes syntaxin 6, a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) that localizes to endosomal transport vesicles and has a critical role in intracellular trafficking. Intriguingly, recent studies have implicated STX6 in regulation of immune function, but it is also highly expressed in other cells including oligodendrocytes, which we also observed here in this study53,54,55. However, our investigation did not support the involvement of the EIF2A locus identified in previous PSP GWAS, even though parallel human tissue research has implicated the integrated stress response in tauopathy56,57, although this is controversial58. Thus, additional validation is warranted. We observed a signal in RUNX2, which encodes runt-related transcription factor-2, which had previously been identified but not replicated until this study10,11. Although we observed an association in oligodendrocytes, RUNX2 is highly expressed in microglia and may play a role in regulation of phagocytosis59,60,61. Finally, we also replicated the signal at the SLCO1A2 locus, which encodes the solute carrier organic anion transporter family 1A2 protein, which is also highly expressed in human oligodendrocytes, although we did not observe a colocalizing eQTL, suggesting that the locus may act through an alternative molecular mechanism62. SLCO1A2 has been linked to beta-amyloid burden in AD suggesting a generalized role in brain homeostasis amongst tau proteinopathies63. Taken together, these findings highlight the potential significance of immune-related mechanisms in PSP and for the first time in the field, we have made use of cell type-specific data to nominate increased gene expression specifically in oligodendrocytes as the mechanism behind 3 out of 6 risk loci.
In summary, this study identified six independent susceptibility loci including a novel locus at 6p21.32 associated with PSP, a neurodegenerative disease characterized by movement, cognitive, and behavioral impairments. Through computational analyses and functional fine-mapping, several candidate genes were nominated, including MOBP, STX6, RUNX2, SLCO1A2, and C4A. Additionally, this work revealed a unique oligodendrocyte signature that could distinguish PSP from other neurodegenerative diseases. Further investigation of the identified susceptibility loci and their functional consequences, as well as the examination of cell specific pathologies, provides new insights into the genetic and molecular mechanisms underlying PSP. These findings contribute to our understanding of tau proteostasis and may have implications for related tauopathies.
Methods
Datasets
The cohort includes 8703 cases and controls (4850 women, 3853 men) with an average age of 66.5 in the cases and 72.8 in the controls. For a majority of cases included in the study, inclusion criteria were a neuropathological diagnosis of PSP (n = 2654, with the exception of a small number of cases, both living and deceased, that only had a neurological diagnosis (n = 125). PSP subjects with comorbid pathological features of other neurodegenerative disorders were not excluded from the study including AD-like features, Lewy bodies, and TDP-43 as prevalence of these comorbid features has been previously demostrated64. The controls had no clinical evidence of cognitive impairment or a movement disorder (n = 5584) and neuropathologically could only have age-related pathological changes. A full list of the institutions where the material was collected can be found in Supplementary Data 3 and it should be noted a majority of the samples included here were contained in previous studies8,10,11. Tissue was obtained from donors who had provided written informed consent for research use either directly or via their next of kin. Research with de-identified autopsy material does not meet the federal regulatory definition of human subject research as defined in 45 CFR part 46 and is otherwise exempt. However, HIPAA requirements still apply. Thus, all material was de-identified. For the living subjects, the study was reviewed and approved by the institutional board (IRB#11-001142) at University of California, Los Angeles.
Genotyping and quality control
PSP cases and controls were genotyped at three different institutions (University of Pennsylvania, Icahn School of Medicine at Mount Sinai, and the University of California Los Angeles) on three genotyping platforms (Illumina Human660W, Illumina OmniExpress 2.5, and Illumina Global Screening Array) in 10 total batches (Supplementary Fig. 1). DNA was isolated from the subjects’ using an automated robot (Kingfisher, Thermofisher Scientific), or manually using phenol chloroform extraction8,11,65. The cases and controls were genotyped at each of the respective institutions, merged, and harmonized to contain the same variants and single nucleotide polymorphism (SNP) and sample level quality control (QC, detailed below) was performed followed by imputation. The process was repeated by combining the data from the three centers and the overlapping variants were again harmonized. PLINK v1.9 was used to perform quality control. SNP exclusion criteria included MAF < 2%, genotyping call-rate filter less than 98%, and Hardy–Weinberg threshold of P < 10−6. Individuals with discordant sex, non-European ancestry, genotyping failure of >10%, or excess relatedness (\(\hat{\pi }\) > 0.4) were excluded. A principal components analysis (PCA) was performed to identify population substructure using EIGENSTRAT v6.1.466,67 and the 1000 Genomes reference panel. Samples were excluded if they were greater than six standard deviations away from the European population cluster. Population substructure was rechecked and plotted and overlayed on 1000 genomes (Supplementary Fig. 16). The entire quality control pipeline, scree plot, and indicators of which cases and controls were excluded are described in Supplementary Figs. 17, 18.
TOPMed imputation and post-processing
Each dataset was imputed on the Trans-Omics for Precision Medicine (TOPMed) Imputation Server using the multi-ancestry release 2 (r2) reference panel which includes data on from 97,256 participants with 308,107,085 SNPs observed on 194,512 haplotypes68,69. Phasing was performed using EAGLE with subsequent imputation using Minimac470,71. Imputed variants were filtered using a conservative quality threshold, R2 ≥ 0.8, to assure high quality of variants, and additional filtering on variants overlapping all genotype sets with MAF > 0.01 was performed prior to analysis.
Association analysis
Single-variant genome-wide association analyses was performed jointly on all imputed datasets using a score-based logistic regression under an additive model with covariate adjustment for sex, the first three PC eigenvectors for population substructure, and indicator variables for genotyping platform to mitigate potential batch effects. All association analyses were performed using the program SNPTEST72. After analysis, variants with regression coefficient of |β | >5 and any erroneous estimates (negative standard errors or P-values equal to 0 or 1) were excluded from further analysis as these values are likely indicative of an asymptotically effect. Conditional analysis was performed by conditioning the association on the lead SNP in the locus using PLINK and the dose dependent analysis of the MAPT sub haplotype (i.e. number of H1 alleles) was performed by adding this variable as a covariate into the main model.
External expression datasets
Expression quantitative trait locus (eQTL) full summary statistics for bulk RNA-seq from 13 human brain regions from the GTEx consortium v8 and v773,74 were downloaded from the GTEx web portal. Donor numbers ranged from 114 (substantia nigra) to 209 (cerebellum). eQTL summary statistics for 8 cortical cell types from single nucleus RNA-seq of 196 donors were downloaded from Zenodo23. GWAS summary statistics for Alzheimer's disease and Parkinson's disease were downloaded from their respective repositories17,18. The PSP bulk RNA sequencing data was downloaded from “The Mayo clinic RNAseq study” and the whole blood data was downloaded from the Gene Expression Omnibus portal from a study entitled “Systems-level analysis of peripheral blood gene expression in dementia patients reveals an innate immune response shared across multiple disorders”31. All summary statistics were coordinate sorted and indexed with Tabix to allow random access75.
Fine-mapping
For each locus, we gathered all SNPs within 2-Mbp windows (±1 Mbp flanking the lead GWAS SNP) and filtered out SNPs with a MAF < 0.001. We focused on common variants to maximize the relevance of these results to a larger proportion of the PSP population. LD correlation matrices (in units of r) were acquired for each locus from the UK Biobank (UKB) reference panel, pre-calculated by Weissbrod et al.21. Any SNPs that could not be identified within the LD reference were necessarily removed from subsequent analyses. Statistical fine-mapping was performed on each locus separately with FINEMAP20. Functional fine-mapping was performed using PolyFun + FINEMAP, both of which compute SNP-wise heritability-derived prior probabilities using an L2-regularized extension of stratified-linkage disequilibrium (LD) Score (S-LDSC) regression21. For PolyFun + FINEMAP, we used the default UK Biobank baseline model composed of 187 binarized epigenomic and genic annotations76. In all subsequent analyses presented here, SNPs that fall within the MAPT locus and HLA region/C4A locus were excluded due to the particularly complex LD structure77. PolyFun + FINEMAP provides a 1) posterior probability (PP) that each SNP is causal, on a scale from 0 to 1, and 2) credible sets (CS) of SNPs that have been identified as having a high PP of being causal, which we have set at a threshold of PP ≥ 0.95. PolyFun+FINEMAP meets the following criteria: 1) can take into account LD and 2) can operate using only summary statistics. For FINEMAP, we set the maximum number of causal SNPs to five.
Cell type-specific epigenomic annotations:
For all downstream fine-mapping analyses, we used functional annotations from cell type-specific ChIP-seq annotations of regulatory regions (enhancers and promoters) and cell type-specific DNA interactome anchors from proximity ligation-assisted ChIP-Seq (PLAC-seq)16. These same epigenomic datasets are used for both the fine-mapping summary overlap plot and each locus plot and consist of the following cell types: neurons, oligodendrocytes, microglia, and astrocytes. For the fine-mapping summary plot, we also compare the overlap between fine-mapped PSP GWAS SNPs and significant SNPs identified by the HEK293T cell-line MPRA for Alzheimer’s Disease (AD) and PSP22. Active promoters and enhancers were defined as follows. H3K4me3 and H3K27ac ChIP-seq data were collected for each purified cell type. Active promoters were defined as the intersection between H3K4me3 peaks and H3K27ac peaks that were within 2 kb of the nearest transcription start site (TSS). Active enhancers were defined as H3K27ac peaks that were not within H3K4me3 peaks.
LD-score regression
Stratified LD score regression (S-LDSC) was applied to determine whether specific brain cell type annotations were enriched for heritability of progressive supranuclear palsy14,15,16. Binary annotations were created using active promoters and enhancers, as well as the 1000 Genomes Phase 3 panel of common variants that was used in the LDSC baseline annotation model (annotation = 1 if the common variant falls in a promoter/enhancer peak in a particular cell type, annotation = 0 if not)78. The cell type-specific enhancer and promoter peak sets were then tested for enrichment of heritability while controlling for the full baseline model.
Colocalization and gene prioritization
Two independent pipelines were applied to the GWAS summary statistics to prioritize genes flanking and within the significant loci. We first used the COLOC package to test whether SNPs from different disease GWAS colocalized with expression QTLs from bulk RNA-seq or single-nucleus RNA-seq28. For each genome-wide significant locus in the GWAS we extracted the nominal summary statistics of association for all SNPs within 1 Mbp either side of the lead SNP (2Mbp-wide region total). In each QTL dataset we then extracted all nominal associations for all SNP-gene pairs within that range and tested for colocalization between the GWAS locus and each gene. Where MAF was missing, we used reference values from the 1000 Genomes (Phase 3) European superpopulations. Colocalization was performed by comparing the P-value distributions between matching sets of SNPs. To reduce false positives caused by long-range LD contamination, we removed the MAPT and HLA regions from consideration and restricted locus-gene colocalizations to GWAS-eQTL SNP pairs where the distance between their respective top SNPs was ≤500 kbp or the two lead SNPs were in modest LD (r2 > 0.1), taken from the 1000 Genomes (Phase 3) European superpopulations using the LDLinkR package79.
Analyses were then performed in GRCh37/hg19 using the INFERNO and SparkINFERNO pipelines30,80. Data was converted between genome references using LiftOver and all SNPs were preserved. LD-based pruning was run using the 1000 Genome EUR reference genotype panel on all GWS variants (P < 5 × 10−8, n = 3016) using r2 < 0.7 and 500 kb window. This resulted in 108 independent signals (loci). We defined loci to include variants in LD with the tag variant (r2 ≥ 0.7) restricting to the variants that are at most 1 Mbp away and no more than 1,000 variants between the tag variant and leftmost and rightmost variant in LD with the tag variant. Next, we performed colocalization on each of the 108 loci, against each of the eQTLs from the GTEx v7 dataset28,73. In both approaches, we used the posterior probability for colocalization between GWAS and eQTL signals (coloc_PP.H4.abf) at the locus level, as a ranking for causality of each gene at each locus.
Transcriptome-wide association study
The GWAS summary stats were first converted to z-scores using munge_sumstats.py from the LDSC toolkit. Panels of pre-computed TWAS weights from dorsolateral prefrontal cortex samples as part of the CommonMind Consortium (n = 452) and the AMP-AD project (n = 888) were downloaded from their respective sources. We used an existing 1000 Genomes European LD reference mapped to the hg19 build. TWAS estimates cis-SNP heritability (all SNPs 1Mbp from gene) for each gene then imputes expression in the GWAS to identify associations between gene expression and disease risk. Each gene was given a z-score and P-value. P-values were adjusted for multiple testing within each panel using the Bonferroni method. Genes were called significant at an adjusted P < 0.05.
Imputation of C4A and C4B copy number
C4 alleles from the genotypes were computed using the HapMap3 CEU reference panel using a protocol generated by Sekar et al.43 Briefly, VCF files were generated from chromosome six, and imputation was run using BEAGLE81. The results were compiled into a table containing C4A and C4B copy on each subject in the study, except for 16 cases which the program was unable to compute copy number status. Long and short isoforms were not considered in the model. Plink v. 1.90 was run using the same covariates as the main analysis with the addition of the imputed copy numbers for both genes and run separately. Additionally, logistic regression of case control status was run in R comparing C4A and C4B copy number using the same covariates as the primary association study. Validation of the imputation was performed using droplet PCR (n = 4, each sample with a unique number of C4A copies, run in duplicate) and was found to be within the previously reported accuracy (0.70 < r2 < 1.00, Supplementary Data 4)43.
Differential gene expression
Raw RNA-seq data from PSP and control postmortem brain was processed using the RAPiD-nf pipeline developed as part of the CommonMind consortium. RAPiD-nf is a pipeline in the NextFlow framework and uses Trimmomatic (version 0.36), STAR (version 2.7a), FASTQC (version 0.11.8), featureCounts (version 1.3.1), and Picard (version 2.20.0) for pre-processing and quality control. RSEM (1.3.1) was used for gene expression estimation82,83,84,85. After processing, 84 cortical samples and 83 cerebellar samples from the PSP cases were included and 77 cortical and cerebellar samples were used from controls. Principal component analysis on the normalized RNA-seq matrix was performed to identify outliers based on clustering. The RNA-seq matrix was normalized using trimmed mean of M values and transformed using the limma::voom() function and lowly expressed genes removed86. Covariates were selected to minimize gene expression differences based on technical variables. Clinical and technical variables from Picard were combined and correlated using variancePartition87. Variables that contributed the most to variance in gene expression and had the least overlap with one another were included. Final variables included as covariates were RNA integrity number (RIN), mean insert size, age at death, and biological sex. After normalization and covariate adjustment, differential gene expression (DGE) analysis was performed on 16 genes contained within a 2Mbp-wide region flanking each lead SNP using the limma package to compare gene expression of PSP cases and controls88. Limma calculated log2-fold change, t-statistics, and P-values for each gene. Because we looked specifically at 16 genes contained within five significant loci, a P < 0.05/16 = 0.0025 was considered differentially expressed based on a Bonferroni correcting for multiple comparisons.
Immunohistochemistry
Human brain tissues were fixed in 10% formalin, embedded in paraffin, and cut to a thickness of 6 micrometers (n = 10 for controls vs. n = 10 for PSP cases). Slides were baked and deparaffinized in EZ prep at 72 °C for 8 min, then pretreated with Heat Induced Epitope Retrieval (HIER) in Tris-EDTA buffer pH 7.8 at 95 °C for 64 min in standard cell condition solution one (CC1) using a Ventana Discovery ULTRA (Roche Indianapolis IN). Blocking was then performed in an inhibitor solution for 12 min at room temperature. Incubation was then performed with primary antibody oligodendrocyte transcription factor (OLIG2, pre-diluted by the manufacturer) for 40 min at room temperature. A secondary antibody, OmniMap anti-rabbit horseradish peroxidase (HRP), was added for 12 min followed by the addition of 3,3′-Diaminobenzidine (DAB) CM / H2O2 CM with an 8-min incubation time, and Copper CM was added and incubated for 5 min. Next, a denaturation cycle was then run at 95 °C for 8 min followed by incubation with primary antibody C4A (1:700) for 32 min at room temperature and then with OmniMap secondary anti-Rabbit HRP antibody for 12 min followed by purple / H2O2 incubation at 28 min to enhance the bright field color. A denaturation cycle was then run at 95 °C for 8 min. A final incubation with a third primary antibody against hyperphosphorylated tau (AT8, 1:1500) was run for 32 min at room temperature and OmniMap anti-Mouse HRP secondary antibody was added for 12 min, followed by GREEN HRP / H2O2 incubation for 16 min and another incubation Green Activator for 16 min to enhance visualization. Lastly, a counterstain with hematoxylin was added for 4 min, and then a post counterstain Bluing Reagent was added for 4 min. A detailed description of the reagents used, and their catalog number can be found in Supplementary Data 5.
Image analysis
Five regions containing marked C4a pathology in the white matter on all cases and controls were imaged on a Nikon Eclipse Ci (Nikon Melville, NY) at 20x magnification. The NeuronJ package contained within FIJI v.2.13.1 was used to assess cellular features of complement-activation quantitatively for both the length of the feature and the total number of features89,90.
Biochemical analysis
Western blots were performed using fresh-frozen brain tissues from the prefrontal cortex (n = 7 for PSP cases, 6 for controls). Samples were homogenized with a glass-Teflon homogenizer at 500 rpm in 10 volumes (wt/vol) of ice-cold Pierce RIPA buffer (Thermo Fisher Scientific, Waltham, MA) containing Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA), incubated on ice for 30 min, centrifuged at 16,000 g for 15 min, and then supernatants were collected. For each sample, 30 μg of proteins were boiled in Laemmli sample buffer (Bio-Rad, Hercules, CA) for 5 min, run on 10% PROTEAN TGX Precast Gels (Bio-Rad, Hercules, CA), blotted to nitrocellulose membranes, and stained with C4a antisera (ab170942, 1:1000; Abcam, Waltham, MA). Horseradish peroxidase-labeled secondary anti-rabbit antibody (1:20,000; Vector Labs, Burlingame, CA) was detected by Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific). To quantify and standardize protein levels without reliance on specific housekeeping proteins, total protein was detected with Amido Black (Sigma-Aldrich, St. Louis, MO). Chemiluminescence was measured in a ChemiDoc Imaging System (Bio-Rad, Hercules, CA), and relative optical densities were determined by using AlphaEaseFC software, version 4.0.1 (Alpha Innotech, San Jose, CA), normalized to total protein loaded.
Statistical analysis
All non-GWAS were performed in R v4.0 and plotted using ggplot2 v3.4.2. For non-normally distributed data a Wilcox test was used to test for significance, and a two-way ANOVA was used for normally distributed data.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The genotype summary statistics in this study have been deposited in the NIAGADS database under accession code NG00169. GWAS summary statistics with only P-values are open access and available to download here: https://dss.niagads.org/open-access-data-portal/#NG00169. As access to full summary statistics is controlled due to the presence of identifiable information, data can be accessed by selecting “Apply for Access” on the main summary statistics page: https://dss.niagads.org/datasets/ng00067/ng00169/. An NIH eRA Commons ID is required for application. Please allow two weeks for a response to the request. The raw genotype data are also restricted access as these data contain identifiable information, but requests for these data can be made by emailing adamnaj@pennmedicine.upenn.edu and kurt.farrell@mssm.edu. Please allow four weeks for a response to the request. Data is available for general research use according to the following data access and attribution requirements: https://www.niagads.org/data/request/data-request-instructions. We anticipate the individual-level genotypes will be available on NIAGADS under restricted access in 6–12 months. The additional data generated in this study are provided in the Supplementary Information/Source Data file. The publicly available data used here can be found in the following repositories: GTEx web portal, https://gtexportal.org/home/datasets eQTL single cell data, https://zenodo.org/record/5543735 AD GWAS summary statistics, https://www.niagads.org/datasets/ng00075 PD GWAS summary statistics, https://drive.google.com/drive/folders/10bGj6HfAXgl-JslpI9ZJIL_JIgZyktxn Mayo Clinic RNAseq Study, https://adknowledgeportal.synapse.org/Explore/Studies/DetailsPage/StudyDetails?Study=syn5550404. Whole blood microarray data, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140830. Brain PLAC-seq, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001373.v2.p2. Picard https://github.com/broadinstitute/picard/releases. C4 imputation panel, https://github.com/freeseek/imputec4. 1000 Genomes reference panel, https://www.internationalgenome.org. All other data supporting the findings described in this manuscript are available in the article and its Supplementary Information files. Please see legends for these files for details. Source data are provided with this paper.
Code availability
All software used in this study is publicly available at the URLs or references cited. The specific parameters and code used in this paper can be found in our GitHub repository at https://github.com/jackhump/PSP_GWAS and is permanently referenced with the https://doi.org/10.5281/zenodo.12668541.
References
Kovacs, G. G., Ghetti, B. & Goedert, M. Classification of diseases with accumulation of Tau protein. Neuropathol. Appl. Neurobiol. 48, e12792 (2022).
Stamelou, M. et al. Evolving concepts in progressive supranuclear palsy and other 4-repeat tauopathies. Nat. Rev. Neurol. 17, 601–620 (2021).
Nath, U. et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain 124, 1438–1449 (2001).
Lubarsky, M. & Juncos, J. L. Progressive supranuclear palsy: a current review. Neurologist 14, 79–88 (2008).
Evidente, V. G. H. et al. Neuropathological findings of PSP in the elderly without clinical PSP: Possible incidental PSP? Parkinsonism Relat. D. 17, 365–371 (2011).
Donker Kaat, L. et al. Familial aggregation of parkinsonism in progressive supranuclear palsy. Neurology 73, 98–105 (2009).
Baker, K. B. & Montgomery, E. B. Jr. Performance on the PD test battery by relatives of patients with progressive supranuclear palsy. Neurology 56, 25–30 (2001).
Hoglinger, G. U. et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 43, 699–705 (2011).
Jabbari, E. et al. Genetic determinants of survival in progressive supranuclear palsy: a genome-wide association study. Lancet Neurol. 20, 107–116 (2021).
Chen, J. A. et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol. Neurodegener. 13, 41 (2018).
Sanchez-Contreras, M. Y. et al. Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol. Neurodegener. 13, 37 (2018).
Shoeibi, A., Olfati, N. & Litvan, I. Frontrunner in Translation: Progressive Supranuclear Palsy. Front Neurol. 10, 1125 (2019).
Naj, A. C. et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43, 436–441 (2011).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Nott, A. et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019).
Lambert, J. C., Ramirez, A., Grenier-Boley, B. & Bellenguez, C. Step by step: towards a better understanding of the genetic architecture of Alzheimer’s disease. Mol. Psychiatry https://doi.org/10.1038/s41380-023-02076-1 (2023).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Benner, C. et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016).
Weissbrod, O. et al. Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat. Genet. 52, 1355–1363 (2020).
Cooper, Y. A. et al. Functional regulatory variants implicate distinct transcriptional networks in dementia. Science 377, eabi8654 (2022).
Bryois, J. et al. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat. Neurosci. 25, 1104–1112 (2022).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 47, 1091–1098 (2015).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Raj, T. et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 50, 1584–1592 (2018).
Gockley, J. et al. Multi-tissue neocortical transcriptome-wide association study implicates 8 genes across 6 genomic loci in Alzheimer’s disease. Genome Med. 13, 76 (2021).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Amlie-Wolf, A. et al. Using INFERNO to Infer the Molecular Mechanisms Underlying Noncoding Genetic Associations. Methods Mol. Biol. 2254, 73–91 (2021).
Kuksa, P. P. et al. SparkINFERNO: a scalable high-throughput pipeline for inferring molecular mechanisms of non-coding genetic variants. Bioinformatics 36, 3879–3881 (2020).
Allen, M. et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci. Data 3, 160089 (2016).
Ressler, H. W. et al. MAPT haplotype-associated transcriptomic changes in progressive supranuclear palsy. Acta Neuropathol. Commun. 12, 135 (2024).
Nachun, D. et al. Systems-level analysis of peripheral blood gene expression in dementia patients reveals an innate immune response shared across multiple disorders. bioRxiv, 2019.2012.2013.875112 https://doi.org/10.1101/2019.12.13.875112 (2019).
Fritsche, L. G. et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 48, 134–143 (2016).
Zhao, Y. et al. Appoptosin-Mediated Caspase Cleavage of Tau Contributes to Progressive Supranuclear Palsy Pathogenesis. Neuron 87, 963–975 (2015).
Silva, M. C. & Haggarty, S. J. Tauopathies: Deciphering Disease Mechanisms to Develop Effective Therapies. Int. J. Mol. Sci. 21 https://doi.org/10.3390/ijms21238948 (2020).
Wareham, L. K. et al. Solving neurodegeneration: common mechanisms and strategies for new treatments. Mol. Neurodegener. 17, 23 (2022).
Tacik, P., Sanchez-Contreras, M., Rademakers, R., Dickson, D. W. & Wszolek, Z. K. Genetic Disorders with Tau Pathology: A Review of the Literature and Report of Two Patients with Tauopathy and Positive Family Histories. Neurodegener. Dis. 16, 12–21 (2016).
Andrews, S. J. et al. The complex genetic architecture of Alzheimer’s disease: novel insights and future directions. EBioMedicine 90, 104511 (2023).
Salloway, S. P. et al. Advancing combination therapy for Alzheimer’s disease. Alzheimers Dement (NY) 6, e12073 (2020).
van Rheenen, W. et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 53, 1636–1648 (2021).
Moloney, E. B., de Winter, F. & Verhaagen, J. ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front Neurosci. 8, 252 (2014).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Yilmaz, M. et al. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci. 24, 214–224 (2021).
Zhou, J., Fonseca, M. I., Pisalyaput, K. & Tenner, A. J. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. J. Neurochem 106, 2080–2092 (2008).
Yamada, T., Moroo, I., Koguchi, Y., Asahina, M. & Hirayama, K. Increased concentration of C4d complement protein in the cerebrospinal fluids in progressive supranuclear palsy. Acta Neurol. Scand. 89, 42–46 (1994).
Tsuboi, Y. & Yamada, T. Increased concentration of C4d complement protein in CSF in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 57, 859–861 (1994).
Khosousi, S. et al. Complement system changes in blood in Parkinson’s disease and progressive Supranuclear Palsy/Corticobasal Syndrome. Parkinsonism Relat. Disord. 108, 105313 (2023).
Davies, C. & Spires-Jones, T. L. Complementing Tau: New Data Show that the Complement System Is Involved in Degeneration in Tauopathies. Neuron 100, 1267–1269 (2018).
Gallo, D., Ruiz, A. & Sanchez-Juan, P. Genetic Architecture of Primary Tauopathies. Neuroscience 518, 27–37 (2023).
van Rheenen, W. et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048 (2016).
Kouri, N. et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat. Commun. 6, 7247 (2015).
Allen, M. et al. Divergent brain gene expression patterns associate with distinct cell-specific tau neuropathology traits in progressive supranuclear palsy. Acta Neuropathol. 136, 709–727 (2018).
Stow, J. L., Manderson, A. P. & Murray, R. Z. SNAREing immunity: the role of SNAREs in the immune system. Nat. Rev. Immunol. 6, 919–929 (2006).
Ferrari, R. et al. Assessment of common variability and expression quantitative trait loci for genome-wide associations for progressive supranuclear palsy. Neurobiol. Aging 35, 1514 e1511–1512 (2014).
Nijholt, D. A., van Haastert, E. S., Rozemuller, A. J., Scheper, W. & Hoozemans, J. J. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol. 226, 693–702 (2012).
Verheijen, B. M. et al. Activation of the Unfolded Protein Response and Proteostasis Disturbance in Parkinsonism-Dementia of Guam. J. Neuropathol. Exp. Neurol. 79, 34–45 (2020).
Pitera, A. P. et al. Molecular Investigation of the Unfolded Protein Response in Select Human Tauopathies. J. Alzheimers Dis. Rep. 5, 855–869 (2021).
Nakazato, R. et al. Constitutive and functional expression of runt-related transcription factor-2 by microglial cells. Neurochem Int. 74, 24–35 (2014).
Nakazato, R. et al. Upregulation of Runt-Related Transcription Factor-2 Through CCAAT Enhancer Binding Protein-beta Signaling Pathway in Microglial BV-2 Cells Exposed to ATP. J. Cell Physiol. 230, 2510–2521 (2015).
Bronckers, A. L., Sasaguri, K. & Engelse, M. A. Transcription and immunolocalization of Runx2/Cbfa1/Pebp2alphaA in developing rodent and human craniofacial tissues: further evidence suggesting osteoclasts phagocytose osteocytes. Microsc Res. Tech. 61, 540–548 (2003).
Brown, A. L. et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137 (2022).
Roostaei, T. et al. Genome-wide interaction study of brain beta-amyloid burden and cognitive impairment in Alzheimer’s disease. Mol. Psychiatry 22, 287–295 (2017).
Jecmenica Lukic, M. et al. Copathology in Progressive Supranuclear Palsy: Does It Matter? Mov. Disord. 35, 984–993 (2020).
Farrell, K. et al. Genome-wide association study and functional validation implicates JADE1 in tauopathy. Acta Neuropathol. 143, 33–53 (2022).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Loh, P. R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Fuchsberger, C., Abecasis, G. R. & Hinds, D. A. minimac2: faster genotype imputation. Bioinformatics 31, 782–784 (2015).
Marchini, J., Howie, B., Myers, S., McVean, G. & Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39, 906–913 (2007).
Consortium, G. T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Consortium, G. T. et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Gazal, S. et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 49, 1421–1427 (2017).
Anderson, J. E. & Willan, A. R. Estimating the size of family practice populations. Quadratic Odds Estimation. Med. Care 26, 1228–1233 (1988).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Myers, T. A., Chanock, S. J. & Machiela, M. J. LDlinkR: An R Package for Rapidly Calculating Linkage Disequilibrium Statistics in Diverse Populations. Front Genet. 11, 157 (2020).
Amlie-Wolf, A. et al. INFERNO: inferring the molecular mechanisms of noncoding genetic variants. Nucleic Acids Res. 46, 8740–8753 (2018).
Browning, B. L., Zhou, Y. & Browning, S. R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 103, 338–348 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 12, 323 (2011).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Hoffman, G. E. & Schadt, E. E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinforma. 17, 483 (2016).
McCarthy, D. J. & Smyth, G. K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 25, 765–771 (2009).
Meijering, E. et al. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytom. A 58, 167–176 (2004).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Acknowledgements
Crary/Farrell Labs: [R01 AG054008, R01 NS095252, R01 AG060961, R01 NS086736, and R01 AG062348 P30 AG066514 to J.F.C. K01 AG070326 and CurePSP 685-2023-06-Pathway to K.F.], the Rainwater Charitable Foundation / Tau Consortium, Karen Strauss Cook Research Scholar Award, Stuart Katz & Dr. Jane Martin. Penn/Lee/Naj/Wang/Schellenberg Labs: [P01 AG017586, U54 NS100693, and UG3 NS104095; RF1 AG074328-01, and P30 AG072979; CurePSP Consortium; Controls were drawn from the ADGC (U01 AG032984, RC2 AG036528), and included samples from the National Cell Repository for Alzheimer’s Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the National Institute on Aging (NIA). We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible; Control data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24-AG041689); additional salary and analytical support were provided by NIA grants R01 AG054060 and RF1 AG061351] to A.N., W.P.L., H.W., and G.S. Raj/Humphrey/Ravi: [R56-AG055824, U01-AG068880, U54-NS123743 to J.H., A.R., and T.R.]. Goate Lab: [Rainwater Charitable Foundation, NS123746 to A.G.]. UCLA/Geschwind lab: [K08AG065519 to T.C, 3UH3NS104095, Larry L Hillblom Foundation, Tau Consortium to D.G.]. Ross/Dickson: U54 NS100693, P50 AG016574, CurePSP Foundation, Mayo Foundation to D.W.D., and O.A.R. Hardy lab: The Dolby Foundation to J.H. Höglinger Lab: Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198), DFG (HO2402/18-1 MSAomics), the German Federal Ministry of Education and Research (BMBF, 01KU1403A EpiPD; 01EK1605A HitTau); Niedersächsisches Ministerium für Wissenschaft und Kunst / VolkswagenStiftung (Niedersächsisches Vorab), Petermax-Müller Foundation (Etiology and Therapy of Synucleinopathies and Tauopathies) to G.U.H. Walker/Nirenberg: Department of Veterans Affairs, CX002342 to R.H.W. and M.J.N. This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai and supported by the Clinical and Translational Science Awards (CTSA) grant UL1TR004419 from the National Center for Advancing Translational Sciences. Research reported in this paper was supported by the Office of Research Infrastructure of the National Institutes of Health under award number S10OD026880 and S10OD030463. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additionally, the authors would like to acknowledge the Neuropathology brain bank and research CoRE at Mount Sinai. The authors would like to acknowledge the following tissue repositories for providing the materials necessary to conduct the study: University of Louisville, Australian Brain Bank Network and Flinders University, Barcelona Biobanc and The University of Barcelona, Brain-Net Germany and Neurobiobank Munich, Emory University, Harvard Brain Tissue Resource Center, McLean Brain Bank, Indiana University School of Medicine, Johns Hopkins University, London brain bank, Los Angeles Veterans Association hospital brain bank, Ludwig-Maximilians-Universität München, German Center for Neurodegenerative Diseases (DZNE), Madrid (Universidad Autónoma de Madrid Spain), Massachusetts General Institute for Neurodegenerative Disease, Mayo Clinic Jacksonville, Netherlands Brain Bank and Erasmus University, New York Brain Bank, Columbia University, University of Paris, Southern Texas University, Sun Health Research Institute, University College London Queen Square Institute of Neurology Queen Square Brain Bank for Neurological Disorders, University of California San Diego, University of California San Francisco Memory and Aging Center, University of Antwerp, University of Michigan, University of Navarra, University of Saskatchewan, University of Southern California, University of Toronto, University of Washington, University of Würzburg, Victorian Brain Bank, Boston University, Emory University, Netherlands Brain Bank and Erasmus University, Oregon Health Sciences University, University of Pittsburgh, University of Miami, University of Washington, University of California Irvine and the NIH Neurobiobank. The authors would like to express their gratitude to the donors and their families which made this work possible.
Author information
Authors and Affiliations
Consortia
Contributions
J.F.C, A.N., G.S., A.G., D.H.G. and K.F. conceived the study. K.F, A.N., J. Humphrey, and J.F.C wrote the manuscript. K.F., A.N., Y.Z., S.K., J. Humphrey, and J.F.C performed the computational genetic association study. J. Humphrey, K.F., T.C., Y.Z., Y.Y.L., P.P.K., V.P., A.R., N.H., S.K., and A.E.R. performed the downstream computational data analysis. D.H.G., G.S., W.P.L., A.G., L.S.W., T.R., T.C., G.C., G.U.H., H.R.M., J.F.C., Y.Y.L., and J. Hardy, consulted on the statistical methods. A.B.K., O.V., L.B.C., H.R., K.W., C.D.S., T.D.C., M.M.K., H.W., G.C., G.U.H, U.M., L.I.G., R.A., S.K., H.R.M., T.R., T.T.W., Z.J., K.Y.M, R.R., D.W.D., O.A.R., G.S., D.H.G., M.A.I., PSP Genetics Study Group., R.H.W., M.J.N., J.F.C., A.N., K.F., performed samples selection and confirmation of diagnosis form respective brain bank. S.H.K. performed western blot analysis. T.D.C. C.D.S., K.W., M.M.K., D.D., A.C., B.B. performed immunohistochemical preparations and downstream analysis. T.C., A.E.R., G.C., T.R., G. U. H., T.T.W., O.A.R., L.S.W., A.G., G.S., D.W.D., D.H.G, J.F.C, and A.N. provided advice on interpreting the results. K.F., R.A., S.K. completed the reporting summary. J.F.C., A.N., and K.F., oversaw the study, provided direction and resources. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
There are no competing interests to the work published here, but in full transparency the following authors wish to disclose their industry relations. A.M.G. is an SAB member for Genentech and Muna Therapeutics. H.M. consultants for Roche, Aprinoia, AI Therapeutics, and Amylyx and is a co-applicant on a patent application PCT/GB2012/052140. L.G. consults for AI Therapeutics, Amylyx, Apellis, Aprinoia, Ferrer, Mitochon, Mitsubishi Tanabe, P3Lab, Roche, Springer, Switch, UCB, and Woolsey G.C. is currently employed by Regeneron. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiong-Jian Luo, Artur Schuh, Jin-Tai Yu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Farrell, K., Humphrey, J., Chang, T. et al. Genetic, transcriptomic, histological, and biochemical analysis of progressive supranuclear palsy implicates glial activation and novel risk genes. Nat Commun 15, 7880 (2024). https://doi.org/10.1038/s41467-024-52025-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52025-x
- Springer Nature Limited