

Abstract
For accurate mitotic cell division, replicated chromatin must be assembled into chromosomes and faithfully segregated into daughter cells. While protein factors like condensin play key roles in this process, it is unclear how chromosome assembly proceeds as molecular events of nucleosomes in living cells and how condensins act on nucleosomes to organize chromosomes. To approach these questions, we investigate nucleosome behavior during mitosis of living human cells using single-nucleosome tracking, combined with rapid-protein depletion technology and computational modeling. Our results show that local nucleosome motion becomes increasingly constrained during mitotic chromosome assembly, which is functionally distinct from condensed apoptotic chromatin. Condensins act as molecular crosslinkers, locally constraining nucleosomes to organize chromosomes. Additionally, nucleosome-nucleosome interactions via histone tails constrain and compact whole chromosomes. Our findings elucidate the physical nature of the chromosome assembly process during mitosis.
Similar content being viewed by others

Introduction
Genome information copied by DNA replication must be faithfully transmitted into two daughter cells during mitotic cell division. To secure this transmission process, replicated DNA is assembled into mitotic chromosomes. In terms of local organization, the 10 nm-fiber, in which a long negatively charged DNA is wrapped around positively charged core histones, seems somewhat irregularly folded without the regular 30-nm fibers1,2,3,4,5,6. Several players have been identified and extensively investigated regarding how higher-order organization is able to form the chromosome shape. These include condensins, topoisomerase IIα, KIF4α, and Ki-67, which are all involved in chromosome organization7,8,9,10,11,12,13,14,15,16,17,18,19.
Two conserved structural maintenance of chromosome (SMC) protein complexes, condensin I and condensin II20,21, are critical protein factors in vertebrates. They share SMC2 and SMC4 core proteins but have distinct sets of non-SMC regulatory proteins: CAP-H, CAP-D2, and CAP-G for condensin I and CAP-H2, CAP-D3, and CAP-G2 for condensin II21. Condensins are also found in the classical “chromosome scaffold” in histone-depleted chromosomes, which retains the overall size and shape of the chromosomes22,23,24. In vitro reconstituted chromosomes are swollen with the depletion of condensins8,20,21. Condensins are assumed to actively form and stabilize DNA loops25,26. Condensin II is primarily involved in establishing the mitotic chromosome axis, while condensin I mediates axis shortening and regulates the size of chromatin loops7,25,26. Condensin I is more abundant than condensin II during mitosis21,24,25. Condensin II binds more stably to chromosomes (residence time >5 min) than condensin I (∼2 min) and is confined more centrally to the axis while condensin I occupies a wider area25,27,28. Recent in vitro and in silico studies proposed that condensin is a molecular motor and can actively push out DNA to make a loop, called loop extrusion18,29,30,31. Another model is diffusion capture in which condensin stabilizes interactions between binding sites in chromatin to make a loop32,33,34.
A recent chromosome conformation capture (Hi-C) study created a fine contact probability map of genomic DNA and, together with subsequent computational modeling, suggested condensin II forms large and condensin I forms small loop structures26. This finding agrees with the quantitative estimation by super-resolution imaging25. Loop shortening by the binding of additional condensin I in anaphase seems to be coupled with a maximum chromosome compaction10,25,35. Surprisingly, these short loop sizes are comparable to those of DNA loops from the classical radial loop/chromosome scaffold model (30–90 kb)22.
If condensin loop formation is crucial to chromosome assembly, then the removal of condensins should result in chromosomal decompaction. However, the overall chromosome volume following depletion of both condensins I and II remains largely unchanged, while chromosome shape becomes abnormal36,37. To explain this observation, a global chromosome compaction force would be required, in addition to that exerted by condensins. This force might be direct interactions between nucleosomes via histone tails. Indeed, acetylation of histone tails, which can weaken nucleosome-nucleosome interactions, significantly decreases during mitosis37,38,39,40,41,42. Chromatin in interphase and mitosis becomes decompacted and more mobile when treated with the histone deacetylase (HDAC) inhibitor trichostatin A (TSA)37,43,44,45,46, which increases the acetylation of histone H3 and H4 tails and suppresses nucleosome–nucleosome interactions. Hyperacetylation of histone tails induces a segregation defect in mitotic chromosomes39. In addition, a transient increase in free Mg2+ observed during mitosis may also contribute to this global compaction47. Nucleosomes have a net negative charge and repulse each other. An increase in free Mg2+ decreases the repulsion and compacts chromosomes in vitro47,48,49.
Because of its highly charged nature, chromatin/chromosome structure varies greatly depending on the surrounding environment (e.g., cations, molecular crowding)50,51 or by fixation with chemical crosslinking52. To investigate the physical nature of the mitotic chromosome assembly process in living cells, genome-wide single-nucleosome imaging and tracking is a promising method45,53,54,55,56,57,58,59. Local nucleosome motions obtained through single-nucleosome imaging provide structural information about chromatin in living cells. Local nucleosome motions can also facilitate chromatin accessibility54,60,61. While transcription is almost silenced in mitotic chromosomes62, large molecules such as condensins and topoisomerase IIα should move around inside the compact chromosome54,63.
In this study, using single-nucleosome imaging combined with rapid protein depletion technology and coarse-grained Brownian dynamics modeling, we reveal nucleosome motion in mitotic chromosomes, how it varies over time, and what might underlie the observed effects. Local nucleosome motions are increasingly constrained during mitotic chromosome assembly. Two condensins locally constrain chromatin fibers around the axis to organize chromosomes. Nucleosome-nucleosome interactions via histone tails drive global compaction during mitosis, in which extensive histone tail deacetylation occurs. The obtained findings enhance our understanding of the physical nature of mitotic chromosome assembly in living human cells.
Results
Single-nucleosome imaging/tracking in mitotic HeLa cells
We performed single-nucleosome imaging to measure local chromatin motion in mitotic chromosomes and to ascertain how they are organized and assembled in living human cells. Histone H2B was tagged with HaloTag (H2B-Halo), to which a HaloTag ligand dye (e.g., TMR (Tetramethylrhodamine), JF549, or JF64664) can specifically bind in living cells. Tagged H2B was stably expressed in HeLa cells (Fig. 1a). The H2B-Halo was incorporated into nucleosomes throughout the chromosomes, including euchromatic and heterochromatic regions (Fig. 1b, c). Presumably, this occurred by histone replacement on a scale of hours65. Stepwise-salt washing of nuclei isolated from the established HeLa cells expressing H2B-Halo previously confirmed that expressed H2B-Halo behaved similarly to endogenous H2B60. FRAP (fluorescence recovery after photobleaching) analysis showed about 90% of H2B-Halo bound very stably to mitotic chromosomes (Supplementary Fig. 1a, b). These results suggest that the H2B-Halo molecules were appropriately incorporated into the nucleosomes within the cell. For single-nucleosome imaging, H2B-Halo was sparsely labeled with a very low concentration of TMR, JF549, or JF646 (Fig. 1d) to produce a relatively small number (~100–200 nucleosomes per frame (50 ms) in a nucleus or ~50 nucleosomes per frame (50 ms) in a mitotic cell) of fluorescent nucleosomes, leading to a stochastic labeling of nucleosomes genome-wide. We used oblique illumination microscopy, which enabled the illumination of a thin area within a single cell while reducing the background noise (green line in Fig. 1e45,57,66). Clear, well-separated dots were detected (Fig. 1f) with a single-step photobleaching profile (Supplementary Fig. 1c), suggesting that each dot represents a single fluorescent H2B-Halo molecule in a single nucleosome.

a Western blot of H2B-Halo expression in HeLa cells using the anti-HaloTag and anti-H2B antibody. Lane 1, parental HeLa cells; lane 2, H2B-Halo expressed HeLa cells. b HeLa cells expressing H2B-Halo labeled with HaloTag TMR ligand (center). (Left) DAPI-stained DNA. (Right) Merged image (DNA, cyan; H2B-Halo, magenta). Representative images of interphase nuclei (top) and mitotic chromosome spreads (bottom). Note that the TMR pattern is similar to DNA staining on interphase nuclei. c Enlarged image of the mitotic chromosomes in b (marked with the dot rectangle) showing a more uniform TMR labeling than DAPI, as depicted in the line plot below (DNA, cyan; TMR, magenta). A.U. means arbitrary unit. d A small fraction of H2B-Halo in mitotic chromosomes was sparsely labeled with the HaloTag fluorescent ligand (red). The labeled nucleosome movements can be tracked at super-resolution. e Scheme of oblique illumination microscopy. The illumination laser (green) excites fluorescent nucleosomes within a thin optical layer (red) of the mitotic cells to reduce background. f Single-nucleosome (H2B-Halo-TMR) images of interphase chromatin (left) or metaphase chromosomes (right) in a live HeLa cell after background subtraction. Also see Supplementary movies 1 and 2. g Two representative trajectories of nucleosome of interphase chromatin (left) or metaphase chromosomes (right). h MSD plot (±SD among cells) of TMR-labeled nucleosomes in live interphase (black, n = 20 cells), metaphase (magenta, n = 17 cells), apoptotic (blue, n = 19 cells), and FA-fixed (gray, n = 13 cells) HeLa cells from 0.05 to 0.5 s. MSD exponents from Supplementary Fig. 1f and asymmetry coefficient (AC) values from Supplementary Fig. 2b are also shown. The Kolmogorov-Smirnov test (two-sided) was used. ***, P < 0.0005, for interphase versus metaphase (P = 6.0 × 10−8), metaphase versus apoptosis (P = 4.2 × 10−9), and apoptosis versus FA-fixed (P = 1.2 × 10−4). i Schematic for angle-distribution analysis60,74. j Schematic for AC. See Methods for details. AC shows deviation from a homogeneous distribution and is negative for angular distributions, where pulling back force is dominant.
Local chromatin motion is more constrained in mitotic chromosomes than in interphase chromatin
We recorded the TMR/JF549-nucleosome dots observed in interphase chromatin and mitotic chromosomes at 50 ms/frame (~450 frames, 22.5 s in total) in living cells (Supplementary movies 1 and 2). The individual dots were fitted with a 2D Gaussian function to estimate the position of the nucleosome67,68,69. The motions of individual nucleosomes were tracked using u-track software (Fig. 1g)70. The position determination accuracy was 15.5 nm (Supplementary Fig. 1d). Notably, we tracked only the signals of H2B-Halo-TMR incorporated into nucleosomes (Supplementary movies 1 and 2) since free histones moved too fast to be detected as dots and tracked under our imaging conditions. The effects of cellular movements were likely to be negligible on our time scale. From the nucleosome tracking data (Fig. 1g), we calculated the mean square displacement (MSD), which shows the spatial extent of random motion over a certain period71. In both interphase nuclei and mitotic chromosomes, the plot of calculated MSDs appeared sub-diffusive (black and magenta lines, respectively, Fig. 1h), which agrees with those reported in previous work60,72,73. Formaldehyde (FA) fixation of chromatin in the cells severely suppressed motions of fluorescent nucleosomes (grey line, Fig. 1h). The MSD plot shows that local nucleosome motions in mitotic chromosomes are much more constrained than in interphase chromatin (Fig. 1h).
In addition, we examined apoptotic cells, whose chromatin is also highly condensed (Supplementary Fig. 1e). Apoptotic chromatin had an even lower nucleosome motion than mitotic cells (Fig. 1h), suggesting that the compaction profiles of mitotic and apoptotic chromatin appear distinct, although both are highly condensed (discussed later). Furthermore, the mitotic MSD exponent in the time-range up to 0.5 s (0.37, Fig. 1h; Supplementary Fig. 1f) was lower than that of interphase (MSD exponent, 0.47), indicating that nucleosomes in mitotic chromosomes were less mobile than interphase chromatin.
We quantitatively investigated the moving angle distribution of individual nucleosomes (Fig. 1i, j)60,74 and found that nucleosomes in mitotic chromosomes were more constrained than those of interphase chromatin. Compared to the Brownian motion of a control particle (Supplementary Fig. 2a), biased nucleosome moving angles toward 180° (Supplementary Fig. 2b) suggest that nucleosomes are often pulled back to their original position (Fig. 1i). Note, nucleosomes in mitotic chromosomes had a more biased distribution than in interphase chromatin (Supplementary Fig. 2b). To quantify this, we introduced the asymmetry coefficient (AC) (Fig. 1j), which is calculated as the ratio between the frequency of forward angles (between −30° and 30°) and the backward angles (150°–210°). More negative AC values indicate higher constraining60,74. The AC value (−1.598, Fig. 1h; Supplementary Fig. 2b) of mitotic chromosomes was lower than the interphase one (−1.185), suggesting nucleosomes are more constrained in mitotic chromosomes.
To support our findings in HeLa cells, we examined other cell lines, i.e., human HCT116 cells, human RPE-1 cells75, and Indian Muntjac DM cells, which express H2B-Halo (Fig. 2a–c). HCT116 and RPE-1 cells have relatively normal numbers of chromosomes. DM cells are derived from deer fibroblast cells and have very large mitotic chromosomes54,76,77. All three cell lines showed a similar tendency (Fig. 2d–f) for local nucleosome motion in mitotic chromosomes, which was much more constrained than in interphase nuclei. We also confirmed the constrained motion of mitotic chromatin using a well-defined specific genomic locus in living HT-1080 diploid human cells, which have a tet operator (tetO) array integrated into a low gene density region of chromosome 5 (Fig. 2g, h)78. These results indicate that constrained local chromatin motion is a general feature of mitotic chromosomes.

a–c Western blots of H2B-Halo in lysates of HCT116, RPE-1, and Indian Muntjac DM cells (lane 2) using an anti-HaloTag antibody and a control anti-histone H2B antibody. Lane 1 is for their parental cells. d MSD plot (±SD among cells) of TMR-labeled interphase (black, n = 21 cells) and metaphase (magenta, n = 21 cells) nucleosomes in living HCT116 cells from 0.05 to 0.5 s. ***, P ≤ 0.0005, for interphase versus metaphase (P = 4.4 × 10−4). e MSD plot (±SD among cells) of TMR-labeled interphase (black, n = 18 cells) and metaphase (magenta, n = 15 cells) nucleosomes in living RPE1 cells from 0.05 to 0.5 s. ***, P < 0.0005, for interphase versus metaphase (P = 6.4 × 10−8). f MSD plot (±SD among cells) of TMR-labeled interphase (black, n = 17 cells) and metaphase (magenta, n = 20 cells) nucleosomes in living Indian Muntjac DM cells from 0.05 to 0.5 s. ***, P < 0.0005, for interphase versus metaphase (P = 4.7 × 10−9). Note that mitotic chromosomes in all three cell lines have more constrained nucleosome motions. g (left) Schematic depicting the location of tetO repeats (×250) in a gene-poor region of human chromosome 5. (right) TetR-4×mCherry foci images of living HT-1080 G160 and mitotic cells after background subtraction. Foci are indicated by yellow arrowheads. h MSD plots (±SD among cells) of TetR-4×mCherry foci motion in G1 phase (black, n = 21 cells, data reproduced from ref. 60) and metaphase (magenta, n = 9 cells) HT-1080 cells from 0.05 to 0.5 s. **, P < 0.005, for G1 versus metaphase (P = 5.0 × 10−3). For (d), (e), (f), and (h), the Kolmogorov-Smirnov test (two-sided) was used to determine P values.
How do the constraints emerge during the chromosome assembly process?
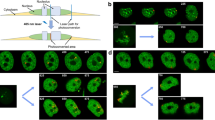
We investigated the chromosome assembly process at various mitotic stages in HCT116 cells. Note that nucleosome motions in the G1, S, and G2 phases of HCT116 cells were similar60. G2 phase cells synchronized by the CDK1 inhibitor RO-330679 were released, and we monitored the nucleosomes in G2 phase, prophase, prometaphase, metaphase, anaphase, and telophase cells (Fig. 3a). Single-nucleosome imaging/analysis revealed that chromatin was increasingly more constrained as assembly progressed from prophase to anaphase (Fig. 3b, c). Interestingly, chromosomes in prophase are individualized (Fig. 3a) but have a similar nucleosome motion as those in interphase (Fig. 3b, c), suggesting that chromosome assembly/individualization does not automatically imply reduced nucleosome motion. Maximal nucleosome constraining in anaphase is consistent with previous reports on maximal chromosome compaction in anaphase10,35. Once the decompaction started at telophase (Fig. 3a), the constraints were reduced (Figs. 3b, c). MSD exponents and AC values during the mitotic process showed similar tendencies (Fig. 3b; Supplementary Fig. 2c, d). We wondered whether whole chromosome motion, especially chromosome oscillation during metaphase80 or chromosome transmission during anaphase81,82, could affect the result or not. However, local nucleosome motion in such a short time (~50 nm/50 ms) was much larger than both types of chromosome movements (~1 nm/50 ms80,81,82), suggesting that the effects were negligible.

a Fluorescent images of FA-fixed HCT116 cells in different cell cycle stages. From left to right: G2, prophase, prometaphase, metaphase, anaphase, and telophase. DNA was stained with DAPI. b MSD plot (±SD among cells) of nucleosomes during the chromosome assembly process of HCT116 cells. Cells were released from G2, which had been arrested with RO-3306107. Nucleosomes in G2, prophase, prometaphase, metaphase, anaphase, and telophase were monitored. MSD plot (±SD among cells) of TMR-labeled nucleosomes in living HCT116 cells in G2 (black, n = 22 cells), prophase (pale blue, n = 13 cells), prometaphase (pale purple, n = 23 cells), metaphase (pale magenta, n = 17 cells), anaphase (magenta, n = 16 cells), and telophase (blue, n = 14 cells) from 0.05 to 0.5 s. The Kolmogorov-Smirnov test (two-sided) was used to determine P values. ***, P ≤ 0.0005 for G2 phase versus metaphase (P = 2.1 × 10−4), G2 phase versus anaphase (P = 7.0 × 10−6), and G2 phase versus telophase (P = 2.2 × 10−4). MSD exponents from Supplementary Fig. 2c and asymmetry coefficient (AC) values from Supplementary Fig. 2d are also shown in the brackets. c MSD values at 0.5 sec in each cell stage from Fig. 3b were plotted with distributions. Whiskers and bounds of boxes show maxima, minima, and first/third quantiles. Gray and black bars show their mean and median values, respectively. Note that nucleosomes are increasingly more constrained with the progression of chromosome assembly. The Kolmogorov-Smirnov test is described in Fig. 3b.
Condensins I and II constrain chromatin in mitotic chromosomes
What constrains chromatin during the assembly process? Because condensins I and II are key players for mitotic chromosome assembly in vertebrates (Fig. 4a)8,20,21 and locate to a chromosomal axis (upper row, Fig. 4b)23,27, we examined the involvement of condensins in the constraining process. First, we performed rapid depletion of both condensins in metaphase chromosomes using the auxin-inducible degron (AID) system in human HCT116 cells (Supplementary Fig. 3a)83,84. Here endogenous SMC2 (an essential subunit of both condensins) was tagged with mAID and mClover to monitor the depletion efficiency of condensins in human HCT116 cells (Supplementary Fig. 3b). H2B-Halo was stably expressed in the cell. Two hours after the addition of IAA (indole-3-acetic acid, auxin), SMC2 was almost completely depleted in the observed mitotic cells (Fig. 4b, c). The resultant chromosomes showed abnormal shapes while still keeping a compacted state (Fig. 4b), which agrees with the previous report17. Rapid depletion of condensins I and II drastically increased nucleosome motion to the interphase level (Fig. 4d; Supplementary Fig. 3c, d; Supplementary movies 3 and 4). On the other hand, rapid-depletion of RAD21 (a cohesin subunit) in mitotic chromosomes did not significantly increase local nucleosome motion (Supplementary Fig. 4a, b), presumably because cohesin mostly dissociates from chromatin during mitosis85, while it constrains chromatin during interphase86,87,88,89,90. During the assembly process, increasing amounts of condensins bind chromatin fibers, presumably to generate loops (Supplementary Fig. 4c, d) as proposed by refs. 25,26,34 and constrain chromatin motion.

a Diagram of condensins I and II, which share the same SMC ATPase subunits (SMC2 and SMC4) and have distinct sets of non-SMC regulatory proteins (CAP-H, -D2, and -G for condensin I, and CAP-H2, -D3, and -G2 for condensin II). b Fluorescent images of FA-fixed HCT116 cells depleted (KD) of SMC2 (condensins I and II). Top, control HCT116 cells (DMSO vehicle only). Bottom, SMC2-mClover depleted HCT116 cells with IAA treatment. From left to right, DNA (DAPI), SMC2-mClover, and merged. c Box plots with dots of the SMC2-mClover signal intensity. A.U. means arbitrary unit. Whiskers, bounds of box and gray bars show maxima, minima, first/third quantiles, and median values. Median values of control and SMC2 KD are 1.6 × 103 (n = 13 cells) and 1.5 × 102 (n = 10 cells), respectively. ***P < 0.0001 (P = 1.7 × 10−6) by Wilcoxon rank sum test (two-sided). d MSD plot (±SD among cells) of TMR-labeled interphase (Interphase, gray, n = 13 cells) and metaphase nucleosomes (Metaphase, black, n = 13 cells) in living HCT116 cells without or with condensins I and II depletion (SMC2 KD, magenta, n = 10 cells) from 0.05 to 0.5 s. The Kolmogorov-Smirnov test (two-sided) was used to determine P values. ***, P ≤ 0.0005, for with versus without SMC2 KD (P = 1.3 × 10−4), not significant (N.S.) for interphase versus metaphase with condensins KD (P = 0.44). MSD exponents from Supplementary Fig. 3c and AC values from Supplementary Fig. 3d are also shown in brackets.
Condensin I plays a major role in constraining chromatin
To pursue the distinct functional roles of the two condensins, we first depleted condensin I alone, which presumably forms small loops in chromosomes (Supplementary Fig. 4c, d)25,26. We used HCT116 cells expressing H2B-Halo, whose endogenous CAP-H was tagged with mAID and mClover (Supplementary Fig. 5a)17. After the rapid depletion (KD) of CAP-H in mitotic cells (Supplementary Fig. 5b, c), the chromosome shape slightly changed and local chromatin motion significantly increased (Supplementary Fig. 5b, d), but the increase was not as large as that in SMC2-depleted cells (Supplementary Fig. 5d, also see Fig. 4d). Furthermore, condensin II-specific depletion was performed in mitotic HCT116 cells expressing H2B-Halo, whose endogenous CAP-H2 was conjugated with mAID and mClover (Supplementary Fig. 5a, e, f)17(for more details, see Methods). Nevertheless, the local nucleosome dynamics did not significantly change (Supplementary Fig. 5g). Our result is reasonable because the tethered domains of condensins I and II are smaller or larger, respectively (Supplementary Fig. 4d). Our results suggest that condensin I is a major contributor for restraining chromatin, while we cannot completely exclude the possibility that residual condensin II contributed to the effect. Our results also support the previously proposed model (Supplementary Fig. 4d) that condensin II forms large DNA loops, which are in turn subdivided by condensin I into smaller, nested DNA loops25,26. On the other hand, the fact that double depletion of condensins I and II had a larger effect than single depletion suggests that condensin II also contributes to constraining nucleosome motions but that its contribution is usually masked by the greater contribution condensin I makes.
Computational modeling reconstructs chromatin-constraining by condensin
To further understand how condensins I and II constrain chromatin in mitotic chromosomes, we reconstructed the mitotic chromosome environment based on a previously published coarse-grained Brownian dynamics modeling of chromatin fibers and condensins I and II molecules (Fig. 5a, b)91. In this modeling based on previous reports25,26, activity of condensins, consecutive loop formation with DNA by condensins I and II29 was considered to shape chromosomes91. Motions of randomly or specifically identified points on the simulated chromatin fibers were tracked and analyzed (Fig. 5a; Supplementary movie 5). These simple models might not completely reflect the chromatin state in live mitotic cells, but they are still valuable for providing qualitative information on nucleosome behaviors in mitotic chromosomes. With an optimal set of parameters, the modeling result revealed that local chromatin motion was constrained in the axis regions where condensins I and II are enriched, while peripheral chromatin that is almost free from condensins was more mobile (Supplementary Fig. 6).

a, b Coarse-grained Brownian dynamics modeling of mitotic chromosome based on previous work91 and models (Supplementary Fig. 4d)25,26. Chromatin beads, condensin I, and condensin II are represented by blue, red, and green spheres, respectively. Also see Supplementary movie 5. Simulated mitotic chromosome without condensin I (c) or condensin II (d), respectively. Also see Supplementary movies 6 and 7. e Calculated MSDs (±SE among 10 chromosomes) of randomly identified points on the chromatin fibers of simulated chromosomes under the indicated conditions. Simulated mitotic chromosome without condensins (f) and that in a cylinder with restraint, where global constraining was provided (g). Also, see Supplementary movies 8 and 9.
Computational modeling and experimental data anticipate an additional global constraining factor(s)
Notably, this model recapitulates the chromosome morphologies with condensin I or condensin II depletion, which were experimentally and theoretically shown previously7,26,92. Removal of condensin I resulted in a slightly thicker chromosome shape (Fig. 5c) and condensin II removal caused a thinner chromosome shape (Fig. 5d).
However, once either of the condensins were removed from the simulated chromosomes, MSD values increased much more than their experimental data (Fig. 5e; Supplementary movies 6 and 7). This tendency became more prominent when both condensins were removed (Fig. 5f; Supplementary movie 8). Furthermore, the volume of a simulated chromosome increased after the removal of condensins (Fig. 5f; Supplementary movie 8), which is inconsistent with the report using 3D EM36: The total mitotic chromosome volume was similar to that in control cells after rapidly depleting both condensins36. These points suggest that a global constraining factor, in addition to condensins, was needed to better recapitulate the experimental results. Indeed, we obtained a suppression of the drastic increase in local motion, which is closer to the experimental result in the living cell, when cylindrical spaces were included with the simulated chromosomes (Fig. 5g), and both condensins were depleted (red line, Fig. 5e; Supplementary movie 9).
There is another indication that suggests the existence of an additional constraining factor(s) in mitotic chromosomes, besides this computational model finding. How can the chromosome periphery be compacted without condensins because the periphery seems to normally lack condensin (Fig. 4b; Supplementary Fig. 4c)? Furthermore, although nucleosome motion increased to the interphase level after rapid SMC2 depletion (Fig. 4d), the increased MSD values did not increase as high as the MSD values for interphase chromatin depleted of cohesin, which constrains interphase chromatin86,87,88,89. Then, what other constraining factor(s) is present?
Ki-67 does not contribute to additional constraining in mitotic chromosomes
Ki-67 localizes to the chromosome surface (upper, Supplementary Fig. 7a)17,93 and may be another constraining factor. Ki-67 seems to act as a surfactant to disperse mitotic chromosomes16 and be another important player in chromosome organization17,93. When Ki-67 and SMC2 were rapidly depleted from HCT116 simultaneously (Supplementary Fig. 7a–c), the chromosomes became abnormal, like sticky rice (lower, Supplementary Fig. 7a)17. Their nucleosome motion was like SMC2-depleted chromosomes from 0.05 to 0.5 s, while their motion may be more constrained on a longer time scale (Supplementary Fig. 7d). These data show that Ki-67 is not an additional constraining factor for mitotic chromosomes.
Nucleosome-nucleosome interactions globally constrain chromatin in mitotic chromosomes
Another potential constraining factor involves nucleosome-nucleosome interactions via histone tails. Schneider et al37. recently observed that treatment with the HDAC inhibitor TSA94 slightly decompacted mitotic chromosomes (upper, Fig. 6a), presumably because hyperacetylation of histone tails by TSA treatment lead to the loss of some positive charges in those histone tails and a loss of nucleosome–nucleosome contacts43,44,45,46. The decompaction effect by TSA became drastic in the absence of condensins (lower, Fig. 6a)37. To test the involvement of nucleosome-nucleosome interactions in the global constraining process, we treated cells with 5 µM TSA and examined nucleosome motions in mitotic chromosomes of HCT116 cells. This drug treatment increased histone H3 and H4 tail acetylation (left, Fig. 6b). The resultant nucleosome motion was upregulated to a level similar to SMC2-depleted chromosomes (Fig. 6c; Supplementary movie 10), presumably by inhibiting nucleosome–nucleosome interactions.

a Illustration of the effects caused by trichostatin A (TSA) and condensin depletion (KD)37. Chromosome cross-section view (top left). TSA treatment induces histone tail hyperacetylation and weakens nucleosome interactions (top right). Abnormal chromosome after condensin depletion (bottom left). TSA treatment of condensin-depleted cells results in greatly decompacted hyperacetylated chromatin, which is diffusely distributed throughout the cytoplasm (bottom right). b (left) Western blot of tail acetylated H3 and H4 from untreated (lane 1), SMC2 KD (2), 4 h TSA-treated (3) and 4 h TSA-treated SMC2 KD (4) HCT116 cells. H2B was used as a control. (right) Box plots (whiskers, maxima/minima; bounds of box, first/third quantiles) of SMC2-mClover signal. A.U. means arbitrary unit. Gray and black bars show mean and median values. Data from n = 27 (control), n = 20 (SMC2 KD), n = 24 (TSA), n = 16 (SMC2 KD + TSA) cells are shown. ***P < 0.0001 by Wilcoxon rank sum test (two-sided). c MSD plot (±SD among cells) of JF549-labeled nucleosomes in the mitotic HCT116 cells with condensins I and II depletion (SMC2 KD, blue, n = 20), condensins I and II depletion +5 µM TSA treatment (4 h) (SMC2 KD + 5 µM TSA, purple, n = 16), 5 µM TSA treatment (5 µM TSA, magenta, n = 24), and untreated control (control, black, n = 27). The Kolmogorov-Smirnov test (two-sided) was used. *** P ≤ 0.0005 for control versus SMC2 KD + 5 µM TSA (P = 7.5 × 10−12), control versus SMC2 KD (P = 2.2 × 10−8), control versus 5 µM TSA (P = 2.4 × 10−7). Not significant (N.S.) for SMC2 KD versus 5 µM TSA (P = 0.11). MSD exponents and AC values from Supplementary Fig. 8 are also shown. d Fluorescent images of FA-fixed HCT116 cells with SMC2-mAID-mClover expressing H2B-Halo; H2B-Halo-JF549 (top), SMC2-mClover (middle), and merged images (bottom). From left to right, untreated control, condensins I and II depleted cell with IAA addition, TSA-treated cell, and TSA-treated cell with condensins I and II depletion.
When combined with the rapid depletion of SMC2 (right, Fig. 6b), 5 µM TSA treatment substantially decompacted mitotic chromosomes in HCT116 cells (most right, Fig. 6d). Mitotic chromosomes became so swollen that individual chromosomes were difficult to distinguish, which is consistent with the previous report37. The nucleosome motions increased more drastically than either of those with only TSA treatment or SMC2 depletion (Fig. 6c; Supplementary Fig. 8a, b; Supplementary movie 11). MSD values of mitotic nucleosomes treated with TSA and depleted of SMC2 were even higher than interphase nucleosomes where interphase constraining factors (i.e., cohesin and transcription machinery) were depleted87. Treatment of TSA at a tenfold lower concentration (0.5 µM) also similarly affected mitotic nucleosome motion to that of 5 µM TSA (Supplementary Fig. 8d; also see Supplementary Fig. 8a, b, e). Taken together, these results indicate that nucleosome-nucleosome interactions constrain whole mitotic chromosomes, which contributes to their faithful segregation during mitosis.
Consistently, when we incorporated nucleosome-nucleosome interactions (chromatin beads attraction) into the previous computational model (Fig. 5), MSD plots of the modeling became closer to the experimental results (Fig. 7; Supplementary movies 12–15). The mean values of chromosome axis length, chromosome width, and volume under various conditions were also calculated. Our findings suggest that the cylindrical constraint, which the modeling in Fig. 5g introduced, corresponds to nucleosome–nucleosome interactions as a global constraint. We also found the modeling did not completely recapitulate the experimental results, especially the effects of either condensin I- or condensin II-depletion, suggesting that further refinements of condensins’ role(s) and bead attraction in the model and/or consideration of additional factor(s) would be required.

a Simulated mitotic chromosome with chromatin bead-bead attractions. Also, see Supplementary movie 12. The calculated mean values (±SD among chromosomes) of chromosome axis length, chromosome width, and volume are 0.5613 ± 0.0322 µm, 0.3436 ± 0.0495 µm, and 0.0521 ± 0.0165 µm3, respectively. b Simulated mitotic chromosome without condensin I. Also, see Supplementary movie 13. The calculated mean values (±SD among chromosomes) of axis length, chromosome width, and volume are 0.3993 ± 0.0210 µm, 0.4327 ± 0.0471 µm, and 0.0587 ± 0.0143 µm3, respectively. c Simulated mitotic chromosome without condensin II. Also, see Supplementary movie 14. The calculated mean values (±SD among chromosomes) of axis length, chromosome width, and volume are 1.3132 ± 0.0917 µm, 0.2798 ± 0.0333 µm, and 0.0808 ± 0.0221 µm3, respectively. d Simulated mitotic chromosome without condensins. Also, see Supplementary movie 15. The calculated mean values (±SD among chromosomes) of axis length, chromosome width, and volume are NA, 0.6050 ± 0.0701 µm, and 0.1160 ± 0.0403 µm3, respectively. e Calculated MSDs (±SE among 10 chromosomes) and AC values of randomly identified points on the chromatin fibers of simulated chromosomes under the indicated conditions.
Condensins act locally to constrain nucleosomes
Finally, we investigated how differentially condensins and nucleosome-nucleosome interactions constrain chromatin. While nucleosome-nucleosome interactions work as a global constraint, how do condensins work? Do condensins locally act on nucleosomes and constrain them, or globally, such as a condensin-generated torsional stress95? To pursue this issue, we examined TSA-treated chromosomes to visualize the actions of condensins more clearly because their nucleosome-nucleosome interactions were weakened by TSA (upper, Fig. 6a). While TSA-treated chromosomes were slightly decompacted, especially at the chromosome periphery (Fig. 6a, d), the signal intensity of condensins did not seem to change (Fig. 6b right and 6d)37. We wondered if nucleosome motion was more constrained in the axis of the TSA-treated chromosome, where both condensins are enriched (upper row, Fig. 4b; Fig. 6d)27. To test this possibility, we defined the SMC2-mClover regions in the imaging data as axes (Fig. 8a), analyzed nucleosome motions inside and outside each axis, and compared them. Interestingly, the nucleosomes outside the axis (periphery), which seemed relatively free from condensins, had higher mobility than the condensin-rich axis region (Fig. 8b, c; Supplementary Fig. 9a, b), consistent with the modeling result (Supplementary Fig. 6). Note that the nucleosome motion in the condensin-rich axis region did not change even upon TSA-treatment (Figs. 8b, c; Supplementary Fig. 9a, b). On the other hand, chromosomes without TSA treatment showed no significant difference in nucleosome motions between the axis and periphery (Fig. 8b; Supplementary Fig. 9a, b), though this might be due to our resolution limitation in the measurement of untreated mitotic chromosomes. These data suggest that condensins locally act on chromatin where they bind and constrain it (Fig. 8c), rather than globally through a condensin-generated torsional stress95, while altering transcription by TSA treatment might also affect condensin-dependent chromosome architecture. Our findings also indicate condensins are sufficient to constrain nucleosomes near the chromosome axis, and nucleosome–nucleosome interactions (histone deacetylation) are required to constrain nucleosomes outside the axis. This notion is further supported by the data that condensin I-specific depletion for TSA-treated chromosomes caused additional increases in nucleosome motion (right, Supplementary Fig. 9c).

a Upper, cartoon depiction of nucleosomes inside or outside the chromosomal axis. Image 1, enhanced SMC2-mClover region, which corresponds to the axis; image 2, extracted axis region (yellow dashed line) and nucleosome dots. b MSD plot (±SD among cells) of TMR-labeled nucleosomes inside (axis, solid blue, n = 14) and outside (periphery, solid magenta, n = 14) the SMC2-mClover enriched axis regions of mitotic chromosomes in TSA-treated HCT116 cells (0.05 to 0.5 s). The MSD data without TSA treatment are shown as dashed lines [axis, blue (n = 18); periphery, magenta (n = 18)], which almost overlap the inside axis of TSA-treated chromosomes. ***, P ≤ 0.0005, Kolmogorov–Smirnov test (two-sided) for TSA peripheral versus TSA axis regions (P = 1.9 × 10−5). N.S. for TSA axis regions versus non-TSA treated axis (P = 0.33) or peripheral (P = 0.15) regions. MSD exponents from Supplementary Fig. 9a and AC values from Supplementary Fig. 9b are also shown in the brackets. c Nucleosomes around the periphery (those mostly free from condensins) in the TSA-treated chromosomes, whose nucleosome–nucleosomes interactions were weakened, were less constrained and had higher mobility than those around the axis.
Discussion
In this study, single-nucleosome imaging and tracking combined with the AID rapid protein depletion system83,84 and computational modeling91 revealed how local chromatin behavior spatially and temporally changes during the chromosome assembly process. The nucleosome motion measurements presented, which were performed under a range of conditions, contain important information about the molecular behavior of a mitotic chromosome. While local nucleosome motion, which seems to be driven by thermal fluctuation, remains steady during interphase60, this motion becomes increasingly more constrained during the chromosome assembly process. Condensins I and II create loops and thereby constrain chromatin fibers (Fig. 9a). Deacetylation of histone tails causes chromatin to become sticky, facilitating nucleosome-nucleosome interactions. Consistently, highly decompacted chromosomes, which were depleted of condensins and had hyperacetylated histone tails by TSA treatment, showed an MSD exponent (0.49, Fig. 6c) closer to that of a Rouse polymer (0.5), whose motion is driven by thermal fluctuations. A Rouse polymer is an ideal chain consisting of beads connected by harmonic springs73,96.

a Condensins I and II seem to act as an ATP-dependent molecular crosslinkers, presumably to make loops like those in Supplementary Fig. 4d25,26 and constrain nucleosomes, together with nucleosome-nucleosome interactions. Without condensins, chromosomes still compact but the motion of their nucleosomes increases (left). b Cross-sectional view of protein infiltration into mitotic chromosomes. Proteins such as condensin and topoisomerase IIα can move around a mitotic chromosome with nucleosome fluctuations.
Local chromatin behavior provides new insights into understanding the physical nature of chromosome organization and the assembly process during mitosis. Condensin-depleted chromosomes were still highly compacted but their individual nucleosomes were more mobile than non-depleted ones (Fig. 4d). On the other hand, nucleosomes in condensed apoptotic chromatin were significantly constrained compared to those in mitotic chromatin (Fig. 1h; Supplementary Fig. 1e). This finding is consistent with previous work using fluorescence correlation spectroscopy (FCS), which showed apoptotic chromatin was difficult to access54. While normal mitotic, SMC2 depleted, and apoptotic chromatin are all highly compact, local chromatin behavior reveals that their properties seem physically and functionally different (i.e., rather gel-like, liquid-like, and solid-like, respectively). Mitotic chromosome assembly is a highly continuous and dynamic process in which many protein factors, including condensins and topoisomerase IIα, are involved7,8,9,10,11,12,13,14,15,16,17,18,19. Mitotic cells need to retain appropriate accessibility for the protein factors to penetrate their compact chromatin for different functions (Fig. 9b)54,63 while ensuring the faithful segregation of their mitotic chromosomes during cell division. This property differs from irreversibly aggregated nucleosomes in dying apoptotic cells, which prevent the release of DNA fragments from those cells. The mitotic chromosome seems to behave like a functional hydrogel, as suggested by Schneider et al.37.
How do the condensin complexes assemble mitotic chromosomes inside living human cells? We expected condensins to promote nucleosome motion by altering the axis length and loop size in chromosomes as the cell progressed from prometaphase to anaphase. However, nucleosome motion was increasingly constrained instead (Figs. 3b, c). We found that condensins locally constrain nucleosome fibers around a chromosome axis (Fig. 8b, c). It is unlikely that the peripheral regions are compacted by the global torsional stress of chromatin loops generated by condensins around this axis, as discussed in ref. 95. Consistently, computational modeling implies that condensin activities for loop formations contribute to constraining chromatin at the axis of mitotic chromosomes (Supplementary Fig. 6). These findings suggest a structural role for condensins in mitotic chromosome organization, which agrees with the classical chromosome scaffolding model22. Since more and more condensins slow local nucleosome motions during the assembly process, condensins in the chromosome axis behave like ATP-dependent molecular crosslinkers, which generate a loop (Fig. 9a)25,26,32,33,34. Our computational modeling suggests that this crosslinking activity of condensins contributes to constraining chromatin in mitotic chromosomes (Supplementary Fig. 10; Supplementary movies 16 and 17). Gerguri et al. found that loop extrusion leaves chromatin mobility largely unaltered, while a diffusion capture mechanism would be expected to reduce chromatin mobility33. On the other hand, if loop extrusion occurs at a slower time scale than the nucleosome’s diffusional motion, this condensin-mediated crosslinking may be compatible with condensin-mediated loop extrusion18,29,30. Our data cannot distinguish between the mechanisms of how DNA loops are formed by condensin (loop extrusion vs. capture). Further investigation is needed. It is also interesting that chromosomes are still individualized in the absence of both condensins (Fig. 4b). In addition to nucleosome-nucleosome interactions, can cohesin97 or depletion attraction (entropic force)51,98 contribute to this process? This is an interesting issue to be explored.
Recent studies suggest euchromatin also forms condensed chromatin domains, which are composed of collapsing chromatin loops59,99,100. Histone tails of nucleosomes on the domain surface are often acetylated for transcription activation and repulse each other because of their loss of nucleosome-nucleosome interactions. During mitosis, histone tails are deacetylated37,38,39,40,41, free Mg2+ levels rise, and macromolecular crowding increases47,51. These mitotic events may induce close interactions between domains.
In conclusion, we have measured nucleosome motion quantitatively at a high spatiotemporal resolution in mitotic chromosomes, which had yet to be conducted. We have captured the process of mitotic chromosome assembly as molecular events of nucleosomes in living human cells, revealing condensin-mediated crosslinking and nucleosome-nucleosome interactions for chromosome assembly. Our findings suggest that there are two potential ways nucleosome motion could be constrained: (i) by crosslinking two segments together (e.g., in a loop) without changing chromatin state or (ii) by sticking nucleosomes together. It would be an intriguing future issue to investigate how these two effects might interplay and contribute to the mitotic chromosome assembly process, keeping genome integrity during cell division. Our quantitative data also provides critical physical parameters that can be used for multiscale computational modeling (e.g., refs. 101,102), which will advance our mechanistic understanding of chromatin/chromosomes. Future investigations that combine single-condensin analysis and multiscale computational modeling will clarify how condensin mechanically generates a loop that constrains nucleosomes in mitotic chromosomes of living cells.
Methods
DNA construction of PB-EF1α-H2B-HaloTag-IRES-Puro plasmid
The PB-EF1α -MCS-IRES-Puro plasmid was constructed based on PB-EF1α-MCS-IRES-Neo (PB533A-2, SBI) and PB-CMV-MCS-EF1α-Puro (PB510B-1, SBI). PB-EF1α-MCS-IRES-Neo was digested with NcoI and SalI and blunted as a vector. Puro sequence was amplified from PB-CMV-MCS-EF1α-Puro with PCR with the addition of a SalI site using the following primer pair: 5′-GCGTCCGGAATGACCGAGTACAAGCCCAC-3′ and 5′-ACGCGTCGACTCAGGCACCGGGCTTGCGGGT-3′. The fragment was inserted into the vector. The PB-EF1α-H2B-HaloTag-IRES-Puro plasmid was constructed based on PB-EF1α-MCS-IRES-Puro and pPB-CAG-H2B-HaloTag. pPB-CAG-H2B-HaloTag digested with XhoI and HpaI was blunted and inserted into PB-EF1α-MCS-IRES-Puro that was digested with EcoRI and blunted.
Cell lines and establishment of stable cell lines
HeLa S3103, RPE-1 (CRL-4000; ATCC), and HT-1080 cells with tetO/TetR-mCherry (a clone of TT75, TT165, a gift from T. Tanaka at University of Dundee, UK)78 cells were cultured at 37 °C with 5% CO2 in Dulbecco’s Modified Eagle’s medium (DMEM) (D5796- 500 ML; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) (FB-1061/ 500; Biosera). HCT116 cells (CCL-247; ATCC) were cultured at 37 °C with 5% CO2 in McCoy’s 5A medium (SH30200.01; HyClone), supplemented with 10% FBS. Indian Muntjac cells (DM cells) were generously gifted to us from Dr. H. Kimura and Dr. P. Cook (Tokyo Tech and Oxford University, respectively)54,76. These cells were cultured at 37 °C with 5% CO2 in DMEM supplemented with 15% FBS.
The transposon system was used to stably express H2B-Halo in the RPE-1 and DM cell lines. The constructed plasmids pPB-CAG-IB-H2B-HaloTag45 and pPB-EF1α-H2B-HaloTag-IRES-puro were cotransfected with pCMV-hyPBase (provided from the Sanger Institute with a materials transfer agreement) to RPE-1 and DM cells with the Effectene Transfection Reagent kit (301425; QIAGEN), respectively. Transfected cells were then selected with 10 μg/ml blasticidin S (029–18701; Wako) for RPE-1 cells or 2 µg/ml puromycin (P8833-25MG; Sigma-Aldrich) for DM cells. For HeLa cells to stably express H2B-Halo, the Flp-In system (V602020, Invitrogen) was used60. pFRT-bla was first transfected into HeLa cells to integrate it into the genome using the Effectene Transfection Reagent kit. Cells that contained the flippase recognition target (FRT) site were selected using blasticidin S (5 µg/ml) and used for isolation of stable transformants. The isolation procedure using the Flp-In system was performed as previously described103. pEF1-H2B-Halo-FRT45 was transfected into HeLa cells that harbored an FRT site. Transformants were selected using hygromycin B (200 µg/ml) (10687-010, Invitrogen).
The HCT116 Tet-OsTIR1 SMC2-mAID-mClover line was generated as previously described83. For this purpose, we designed a CRISPR plasmid targeting the C-terminus cording region of the SMC2 gene (5′-ACCACCCAAAGGAGCACATG-3′) using pX330-U6-Chimeric_BB-CBh-hSpCas9 (#42230; Addgene).
Cell lines using the AID system were established based on the following cell lines: HCT116 expressing Tet-OsTIR1 and SMC2-mAID-mClover, HCT116 expressing CMV-OsTIR1 and CAP-H-mAID-mCherry17, HCT116 expressing CMV-OsTIR1 and CAP-H2-mAID-mCherry17, HCT116 expressing CMV-OsTIR1(F74G) and RAD21-mAID-mClover84, HCT116 expressing CMV-OsTIR1(F74G) and SMC2-mAID-mClover84, HCT116 expressing Tet-OsTIR1 and Ki-67-mAID-mClover and SMC2-mAID-mCherry17. The transposon system was used to stably express H2B-Halo in the above cell lines. The constructed plasmid pPB-CAG-IB-H2B-HaloTag75 was cotransfected with pCMV-hyPBase to the cells with the Effectene Transfection Reagent kit. Transfected cells were then selected with 10 μg/ml blasticidin S.
H2B-HaloTag expression and localization in the cell
To check the expression levels of our stable cell lines (Fig. 1a; Fig. 2a–c) and increased histone acetylation levels in TSA-treated cells (Fig. 6b), the cells were lysed in Laemmli sample buffer supplemented with 10% 2-mercaptoethanol (133-1457; Wako) and incubated at 95 °C for 5 min to denature the proteins104. The cell lysates, equivalent to ~1 × 105 cells per well, were subjected to SDS–polyacrylamide gel electrophoresis (PAGE) (14%) and subsequent Western blotting. For Western blotting, the fractionated proteins in the gel were transferred to a polyvinylidene difluoride membrane (PVDF) (IPVH00010, Millipore) by a semidry blotter (BE-320, Biocraft) or (Blotting System, ATTO). After blocking with 3% or 10% skim milk (190-12865; Wako), the membrane-bound proteins were probed by the anti-H2B rabbit (1:10,000; ab1790, Abcam) or anti-HaloTag mouse (1:1000; G9211, Promega) antibodies. To examine the histone acetylation level, the rabbit anti–acetyl histone H4 (06-866; Millipore), rabbit anti–acetyl histone H3 (06-599; Millipore), and rabbit anti-H2B (ab1790; Abcam) were used as primary antibodies. The secondary antibody used was either the anti-rabbit (1:10,000; 170-6515, Bio-Rad) or anti-mouse (1:5000; 170-6516, Bio-Rad) horse-radish peroxidase (HRP) conjugated goat antibody. Chemiluminescence reactions were used (WBKLS0100, Millipore) and detected by EZ-Capture MG (AE-9300H-CSP, ATTO).
To test the localizations of H2B-Halo/RAD21-mClover and morphology of the chromosomes (Fig. 1b; Fig. 6d; Supplementary Fig. 4a), H2B-HaloTag expressing cells were grown on poly-L-lysine–coated (P1524-500MG, Sigma-Aldrich) coverslips (C018001, Matsunami) and labeled with 5 nM HaloTag TMR ligand (G8251, Promega) for 1 h. The cells were fixed with 1.85% formaldehyde (FA) (064-00406, Wako) at room temperature for 15 min, permeabilized with 0.5% Triton X-100 (T-9284, Sigma-Aldrich) for 5 min, and stained with 4′,6-diamidino- 2-phenylindole (DAPI) (0.5 µg/ml) (10236276001, Roche) for 5 min, followed by PPDI (20 mM Hepes (pH 7.4), 1 mM MgCl2, 100 mM KCl, 78% glycerol, and paraphenylene diamine (1 mg/ml) (695106-1G, Sigma-Aldrich)) mounting. Single optical sectioning images were acquired using FV3000 confocal laser microscopy (Olympus) equipped with oil immersion 60× objectives (UPLXAPO, NA 1.42, Olympus) or silicon oil immersion 60× objectives (UPLSAPO, NA 1.30, Olympus) at room temperature. The microscopy was operated by FV31S-SW software (Olympus). Images were processed with the deconvolution software CellSens (Olympus) to enhance contrast by removing the haze.
Chromosome spreads
HeLa cells were grown on a 60 mm dish (430166, CORNING) and were arrested at G2 phase by 9 µM RO3306 (217699, Signa-Aldrich) treatment for 16 h. The G2-cells were labeled with 1 µM HaloTag TMR ligand for 1 h. The cells were released from G2 by removing RO3306 and treated with APC inhibitors for 1 hr to arrest the cells at metaphase. The cells were incubated in the hypotonic solution (2 times dilution of medium) at room temperature for 10 min, and then the mitotic cells were collected by mitotic shake-off. The cells were cytocentrifuged for 10 min at 1800 rpm with medium acceleration on the poly-L-lysine–coated coverslip using Cytospin 4 (Thermo Fisher Scientific). The cells were fixed with FA and labeled with DAPI followed by PPDI mounting as described above. Single optical sectioning images were recorded using FV3000 confocal laser microscopy equipped with oil immersion 60× objectives at room temperature. ImageJ (https://imagej.net/ij/index.html) was used to measure the intensity of TMR and DAPI along the chromosome. The TMR intensity around a centromere was matched with DAPI intensity around a centromere.
Fluorescence recovery after photobleaching (FRAP) of H2B-Halo-JF549
Photobleaching and time-lapse imaging (Supplementary Fig. 1a, b) were carried out under cell culture conditions (37 °C, 5% CO2, and humidity) on a FLUOVIEW FV1000 confocal laser scanning microscope (OLYMPUS) as described in ref. 59. Chromatin was labeled with 50 nM JF646-HaloTag ligand for 20 min one day before observation. Photobleaching and time-lapse imaging were performed using an inverted confocal microscope Olympus FV1000D (Evident; Japan) with a 20 mW 635-nm laser diode (NTT Electronics, Japan) for imaging and a 15 mW 559-nm laser diode (Evident) for photobleaching. H2B-Halo-JF646 in living cells was excited by the 635-nm laser through an objective lens (a 60× UPlan SApo S2 Silicon objective, NA 1.30; Evident) and detected at 650–750 nm. For FRAP, one and a half of z scanning was performed with 8% 635-nm laser power, followed by a single bleach pulse (100% 559-nm laser power) of 10 s using a spot of about 5 µm in diameter with a tornado mode of scanning. Z-stack images were taken every 1.0 μm in the z direction, 10 sections in total at 1-min intervals for 30 min in total. Similar z-sections were picked out visually. Before normalizing intensity, xy-drifts of cells were corrected by Fiji Plugin StackReg. Fluorescence intensity of the photobleached region was normalized by calculating the ratio of the photobleached region to the nonbleached region after subtracting background noise.
Fluorescent images of chromosome morphology in fixed cells
HCT116 cells were grown on poly-L-lysine-coated coverslips. The cells were fixed with 1.85% FA on coverslips at room temperature for 15 min, permeabilized with 0.5% Triton X-100 for 15 min, and stained with DAPI for 5 min, followed by PPDI mounting. For Fig. 3a, z-stack images with 0.3 μm thickness were acquired with a DeltaVision system (Applied Precision, Issaquah, WA) with a UPlanApo 60 × 1.40 NA objective lens (Olympus) and 1.6× magnification. The z-stack images were deconvolved with softWoRx (Applied Precision) and presented as maximum intensity projections of four sections. For Fig. 4b, HCT116 cells were grown on poly-L-lysine–coated coverslips to deplete SMC2 as described in the drug treatment section, but treatment using the APC inhibitors was omitted.
For Supplementary Figs. 5b, e, and 7a, cells were seeded on coverslips treated with 10 μg/ml fibronectin (Wako, Osaka, Japan) one day before the experiments. Cells were treated with 2 mM thymidine for 16 h, released in a thymidine-free medium for 6–8 h, treated with 10 μM RO-3306 (Tocris, Minneapolis, MN) for 3 h (to cause an arrest at the G2/M boundary), and then released in medium containing 10 μM S-Trityl-L-cysteine (STLC) (Tokyo Chemical Industry, Tokyo, Japan) to arrest the cells in mitosis. Cells were mock-treated (with DMSO) or treated with 0.5 mM IAA (Tokyo Chemical Industry) during the incubation periods with RO-3306 and STLC. For Ki-67-mACl/SMC2-mACh (#35), additional incubation with 2 μg/ml doxycycline (DOX) (MP Biomedicals, Santa Ana, CA) was included to induce the expression of OsTIR1 after release from the thymidine block. Cells were fixed with 3.7% formaldehyde in PBS at room temperature for 10 min 1 h after removing RO-3306. DNA was counterstained with 0.5 μg/ml Hoechst 33342. Z-stack images with 0.5 μm thickness were acquired with a DeltaVision Core system with a UplanApo 60 × 1.40 NA objective lens. Z-stack images were deconvolved with softWoRx and presented as maximum-intensity projections.
Fluorescent imaging of live cells
For Supplementary Fig. 1e, HeLa cells grown on poly-L-lysine–coated coverslips were labeled with 5 nM HaloTag TMR ligand for 2 h at 37 °C in 5% CO2. Twenty min before observation, the cells were labeled with 1 µM calcein-AM (JX028, Dojindo) mixed with pre-warmed 0.04% Pluronic F-127 (P6866, Molecular Probes) for 10–15 min at 37 °C in 5% CO2. The cells were washed with HBSS buffer (H1387; Sigma-Aldrich) and then observed for 1 h. Optical sectioning images of the cells were recorded with 0.52-µm thickness in the z direction, 18 to 32 sections in total using FV1000D confocal laser microscopy (Olympus) equipped with water immersion 60× objectives (UPLSAPO, NA 1.20, Olympus) at 37 °C in 5% CO2. Images were acquired using FV10-ASW software (v4.2, Olympus). ImageJ was used to project z-stacks to cover each whole chromosome.
HaloTag labeling for single nucleosome imaging
Cells stably expressing H2B-Halo were cultured on poly-L-lysine coated glass-based dishes (3970-035, Iwaki). H2B-Halo molecules were fluorescently labeled with 50-100 pM HaloTag TMR ligand (G8251, Promega), Janelia Fluor HaloTag 549 ligand (GA1110, Promega), or Janelia Fluor HaloTag 646 ligand (GA1120, Promega) according to the manufacturer’s instructions and observed in living cells cultured in the proper medium supplemented with 10% FBS without phenol red.
Drug treatments
Cells were arrested in G2 and their proteins were depleted using the AID system. First, cells were treated with 9 µM RO3306 and 1 μg/ml doxycycline (631311; Clontech) for 16 h. Then, only RO3306 was removed and cells were washed with medium, or HBSS, 3–5 times to release them from the G2 phase. Cells were incubated in medium containing doxycycline (1 μg/ml) for 15–30 min at 37 °C in 5% CO2. APC inhibitors, 50 µM Apcin (I-444-05M, R&D Systems) and 20 µM proTAME (I-440-01M, R&D Systems), were added to the medium to arrest the cells in metaphase with or without 0.5 mM IAA (3-indoleacetic acid (auxin), 19119-61, Nacalai Tesque). Cells were incubated with IAA for 1–3 h at 37 °C in 5% CO2 to allow IAA to degrade mAID-tagged target proteins. For the AID2 system84, 1 µM 5Ph-IAA was used without doxycycline pretreatment. The cells in metaphase were observed within 1 h. Instead of APC inhibitors, cells expressing H2B-Halo were treated with 0.1 µg/mL nocodazole (140-08531, Wako) in culture medium at 37 °C in 5% CO2 for 1 hr and then mitotic cells were observed within 1 h. To induce apoptosis, cells were treated with 20 μM VM26 (Teniposide T3109, Tokyo Chemical Industry Co., Ltd.) for 18–47 hrs or with 40 J/m2 UVC irradiation in 200 µL medium without phenol red using ICX-2000 (Thermo Fisher Scientific).
To monitor depletion efficiency, mean intensity values of mClover/mCherry signals in the chromosome regions excited by a 488 nm (10%)/561 nm (20%) laser were measured. Mean signal intensity values in regions outside chromosomes were used as background. The mean intensities after background subtraction were shown as mean intensity plots.
The AID system was used to deplete proteins and its depletion efficiencies for HCT116/CAP-H-mACh (mAID-mCherry) (Supplementary Fig. 5a), HCT116/CAP-H2-mACh (mAID-mCherry) (Supplementary Fig. 5a), and HCT116/Ki-67-mACl (mAID-mClover) + SMC2-mACh (mAID-mCherry) (Supplementary Fig. 7b) was verified by western blotting associated cell lysates as described in ref. 17. The denatured protein samples were electrophoretically separated on a SuperSep Ace 5–20% gradient gel (Wako, Osaka, Japan) and blotted onto an Immobilon-P membrane (Merck Millipore, Billerica, MA). The following antibodies were used as primary antibodies at the indicated dilutions or concentrations: mouse anti-β-actin (1:5000, AC-15; Sigma-Aldrich, St Louis, MO), rabbit anti-NCAPH/CAP-H (1:1000, 11515-1; ProteinTech, Rosemont, IL), rabbit anti-CAP-H2 (1 μg/ml, AfR205-4L21), rabbit anti-Ki-67 (1:1000, sc-15402; Santa Cruz Biotechnology, Dallas, TX), rabbit-OsTIR1 (1:100083), and rabbit anti-SMC2 (1:1000, ab10412; Abcam, Cambridge, UK) antibodies. The following antibodies were used as secondary antibodies at the indicated dilutions: goat anti-mouse-IgG conjugated to horseradish peroxidase (HRP) (1:3000, 170-6516; Bio-Rad, Hercules, CA) and goat anti-rabbit-IgG conjugated to HRP (1:3000, 170- 6515; Bio-Rad) antibodies. Protein bands were visualized by chemiluminescence using Immobilon Western (Merck Millipore).
Western blots of HCT116/SMC2-mAID (Supplementary Fig. 3b) were used to monitor the depletion efficiency. Cells were lysed with RIPA buffer. Protein extracts were collected after centrifugation, and the same volume of 2 × SDS sample buffer was added. After denaturing at 95 °C for 5 min, proteins were separated on a 7.5% TGX Stain-Free gel (BioRad) and subsequently transferred to a nitrocellulose membrane (Cytiva, #10600003). The membrane was treated with 1% skim milk in TBS-T for 15 min and subsequently incubated with primary antibody at 4 °C overnight. After washing with TBS-T, the membrane was incubated with the secondary antibody for 1 h at room temperature. Proteins were detected by ChemiDoc Touch MP imaging system (BioRad). The following antibodies were used. Primary antibodies: anti-OsTIR1 (1:1000, MBL, #PD048), anti-mAID (1:1000, MBL, #M214-3), anti-SMC2 (1:1000, Bethyl, #A300-058A-T), and anti-GAPDH conjugated with rhodamine (1:5000, Bio-Rad, 12004167). Secondary antibodies used were: anti-rabbit DyLight 800 (1:5000, Thermo Fisher Scientific, #SA5-35571) and anti-mouse IgG StarBright Blue 700 (1:2500, Bio-Rad, #12004158).
Single nucleosome imaging microscopy
Single nucleosomes were observed using an inverted Nikon Eclipse Ti microscope with a 100 mW Sapphire 561-nm laser (Sapphire-561-100CW CDRH, Coherent) and the scientific complementary metal-oxide semiconductor (sCMOS) ORCA-Flash 4.0 v2 camera (C11440-22CU, Hamamatsu Photonics). Fluorescently labeled H2B-Halo-TMR nucleosomes in living cells were excited by the 561-nm laser through an objective lens (100× Apo TIRF, NA 1.49; Nikon) and detected at 575-625 nm. An oblique illumination system with a TIRF unit (TI-TIRF-E, Nikon) was used to excite H2B-Halo-TMR molecules within a limited thin area in the cell nucleus and reduce the background noise. Six hundred sequential image frames were acquired using NIS elements software (AR v4.50.00 64 bit, Nikon) at a frame rate of 50 ms under continuous illumination. The frame rate and exposure time of the laser were controlled via a NI-DAQ board (BNC-2110, National Instruments). To maintain cell culture conditions (37 °C, 5% CO2, and humidity) under the microscope, a live cell chamber with a digital gas mixer (INUBG2TW-TIZW-D15N, Tokai Hit) and a warming box (TIZHB, Tokai Hit) were used.
Single nucleosome tracking analysis
The methods for image processing, single molecule tracking, and single-nucleosome movement analysis were described previously45,60,75. Sequential images were converted to 8-bit grayscale and the background noise signals were subtracted with Fiji software105. The nuclear regions in the images were extracted. Following this step, the centroid of each fluorescent dot in each image was determined and its trajectory was tracked with u-track (MATLAB package)70. The point spread function (PSF) sigma value was set as 2.0 to improve tracking quality. Sigma values sensitively affect AC value results, leading to a different value in the G2 phase from the previous report60. The individual nucleosome positions were mapped using R software (65 nm/pixel). For single-nucleosome movement analysis, the displacement and mean square displacement (MSD) of the fluorescent dots were calculated based on their trajectories. The originally calculated MSDs were in 2D. To obtain three-dimensional values, the 2D values were multiplied by 1.5 (4 Dt ->6 Dt). Histograms of the displacements were prepared using KaleidaGraph (v4.0, Synergy Software).
Angle-distribution analysis and extraction of chromosome axis regions
For the tracked consecutive points {(x0, y0), (x1, y1), …, (xn, yn), …} of a single particle on the xy plane, we converted the data into a set of displacement vectors, ∆rn = (xn + 1 – xn, yn + 1 – yn)t, where t was the transposed matrix. Then, we calculated the angle between two vectors ∆rn and ∆rn + 1. We carried out this procedure for all the points of each trajectory in our experiments and simulations. Last, we plotted the normalized polar histogram by our Python program. The angle distribution was normalized by 2π, and the values correspond to the probability density. Colors indicate the angles. The asymmetry coefficient (AC) was calculated as the logarithm to the base of 2 of the ratio between the frequencies of forward (FWD) angles (−30° to +30°) and backward (BWD) angles (150–210°).
To extract an axis region of a chromosome, an SMC2-mClover image was acquired using oblique illumination based on the single nucleosome imaging microscopy described above with a 100 mW Sapphire 561-nm laser (Sapphire-488-100CW CDRH, Coherent). The SMC2-mClover signal was binarized and extracted as the axis region of the chromosome with ImageJ. The detected single nucleosomes located inside and outside the axis region were categorized based on the centroids of their trajectories (i.e., time average of xy coordinates). The categorized trajectories were used in subsequent analyses.
Computational modeling
We employed coarse-grained molecular dynamics (MD) simulations with a Langevin thermostat based on a model from a previous study91. Chromosomes were considered as flexible polymer chains composed of \(N\) spherical monomers with a diameter of \(\sigma=20{{\rm{nm}}}\), each corresponding to about 10 nucleosomes (i.e., several kilobases of DNA). The natural length of the springs between monomers was set to be the same; a simulated chain of \(N\)= 5000 monomers corresponding to several tens of mega-bases of DNA, which was close to the size of the shortest arm of a human chromosome. The spring between monomers was modeled as a spring without excluded volume (phantom spring). Phantom springs can pass through other chains, which is mediated by the strand-passing activity of topoisomerase IIα. The actual frequency of strand passing events was small because of the excluded volume of the monomers connected by the springs. Thus, chromosomes were described as the potentials \({U}_{{chrom}}={U}_{{excl}}+{U}_{{spr}}\), where \({U}_{{excl}}\) and \({U}_{{spr}}\) represented by the volume exclusion among monomers and spring interactions between neighboring monomers in the chain, respectively. The excluded volume interaction \({U}_{{excl}}\) between monomers was modeled by a Weeks-Chandler-Andersen (WCA) potential, which corresponds to the repulsive part of the Lennard-Jones potential:
for \({r}_{i,\, j} < ^{6} {\sqrt{2}\sigma}\) and 0 elsewhere, where \({r}_{i,\, j}\) denotes the distance between the centers of the \(i\)-th and \(j\)-th monomers. To avoid numerical instability, we introduced a cutoff at the maximum energy cut \(\epsilon={10}^{3}{k}_{B}T\) (\({k}_{B}\) is Boltzmann’s constant and \(T\) is the effective temperature). The spring interaction \({U}_{{spr}}\) between neighboring monomers in a chain was described by the harmonic potential:
where \({r}_{i,i+1}\) is the distance between the \(i\)-th and \(\left(i+1\right)\)-th monomer centers, \({d}_{B}\) is the natural length of the springs, and \({\epsilon }_{{spr}}\) is the spring coefficient. We chose the parameters \({d}_{B}=\sigma\) and \({\epsilon }_{{spr}}={\epsilon }_{{cut}}\).
Nucleosome-nucleosome interactions via histone tails were modeled as attractive interactions between chromatin beads. The attractive interaction \({U}_{{attract}}\) between monomers was modeled by the harmonic potential:
for \({r}_{i,\, j}\, < \,{r}_{{cut}}\) and \(0\) elsewhere. In case there was nucleosome interaction, the interaction strength (\({k}_{{attract}}\)) and interaction cut-off distance (\({r}_{{cut}}\)) were set as \({k}_{{attract}}=11{k}_{B}T\) and \({r}_{{cut}}=1.4\sigma\), respectively. Weak \({k}_{{attract}}\) and/or shorter \({r}_{{cut}}\) resulted in spreading chromosomes. On the other hand, stronger \({k}_{{attract}}\) and/or longer \({r}_{{cut}}\) resulted in more compact chromosomes. In the absence of nucleosome interaction, \({k}_{{attract}}=0\).
Condensins I and II were modeled as a particle having the activity to form chromatin loops within the chromosome. We assumed that the condensins have no excluded volume because they are very elongated protein complexes. Each condensin I forms one loop with a loop length \(L=50\), which corresponds to several hundred kilobases of DNA and is consistent with available experiments25. Condensin I forms and stabilizes the loop structures by crosslinking two chromatin monomers separated on the chromosome with a sufficiently strong harmonic force. Since neighboring loops shared a base-point monomer, consecutive loops were realized. Thus, condensin I was described as the potential:
where \({r}_{{i}_{I},\, {j}_{I,\pm }}\) is the distance between the \({i}_{I}\)-th condensin I and the two chromatin monomers at the loop base, and \({M}_{I}\) is the number of condensin I (i.e., number of chromatin loops). Since we consider the consecutive loop structures in a chromosome by condensin I, \({M}_{I}=N/L\), where the \({i}_{I}\)-th condensin I binds to the \({j}_{I,-}=\left({i}_{I}-1\right)L\)-th and \({j}_{I,{+} }=\left({i}_{I}L-1\right)\)-th chromatin monomers to form a loop of length \(L\). Each condensin II binds four loops formed by condensin I25. Thus, condensin II was described as the potential:
where \({r}_{{i}_{{II}},\, {j}_{{II},\pm }}\) is the distance between the \({i}_{{II}}\)-th condensin II and the two chromatin monomers at the loop base, and \({M}_{{II}}\) is the number of condensin II.
Since we consider that each condensin II will bind the four consecutive chromatin loops formed by condensin I, then \({M}_{{II}}={M}_{I}/4\), where the \({i}_{{II}}\)-th condensin II binds to the \({j}_{{II},{-}}=4L\left({{i}_{{II}}-1}\right)\)-th and \({j}_{{II},{+} }=\left({4{i}_{II}L-1}\right)\)-th chromatin monomers to bind four chromatin loops. The crosslinking strength \({\epsilon }_{{cross}-I}\) and \({\epsilon }_{{cross}-{II}}\) of condensins I and II were set as \({\epsilon }_{{cross}-I}={\epsilon }_{{cross}-{II}}=100{k}_{B}T\).
MSDs were calculated under the following conditions. Since this study focused on chromosome structure after chromatin loops were formed, rod-shaped chromosomes with preformed loops (640 nm thick and 800 nm long) were prepared as the initial structures for the chromosome assembly simulations91 (Fig. 5a). The loop structure was stabilized by condensins at the base. From this initial state, time was evolved using a velocity-Verlet MD integrator with a time-step \({dt}={10}^{-5}\sec\), which reproduced the same average MSD as observed experimentally in metaphase. The mass and the friction of each chromatin monomer were set to \(m=1{k}_{B}T\) and \(\gamma=1{k}_{B}T\) in energy units. In the case with both types of condensins, the MSDs of chromatin beads were calculated at equilibration 10 min after the formation of a rod-shaped chromosome. The MSDs of the peripheral region were calculated for the 50% portion of monomers near the base of the chromatin loop, while the MSDs of the core region (axis region) were calculated for the remaining portion of the monomers. In the case of condensin removal with no restraint, a rod chromosome was first formed and left for 1 min. MSDs were calculated at equilibrium 10 min after one or both types of condensins were depleted from the chromosome. For modeling with restraint, the formed chromosome was placed in a cylinder (640 nm diameter and 800 nm long) whose size was similar to the chromosome. MSDs were calculated at equilibrium 10 min after condensin removal. All MSDs were obtained as the average of ten independent calculations, starting from rod-shaped chromosomes with different initial configurations.
The asymmetry coefficient (AC), the length of the condensin I or II axis, and the width of the chromatin in the direction orthogonal to the axis were calculated at the same time as the MSD calculation.
Statistics and reproducibility
The statistical tests used for data analyses were indicated in figure legends. The detailed information on research design is available in the Reporting Summary.
Below, we show how many times each experiment was repeated independently.
Figure 1a: We repeated this experiment three times and obtained consistent results.
Figure 1b: We repeated this experiment three times and obtained consistent results.
Figure 1c: We repeated this experiment twice and obtained consistent results.
Figure 2a-c: We repeated this experiment twice and obtained consistent results.
Figure 3a: We repeated this experiment twice and obtained consistent results.
Figure 4b: We repeated this experiment three times and obtained consistent results.
Figure 4c: We repeated this experiment three and obtained consistent results.
Figure 6b: We repeated this experiment twice and obtained consistent results.
Figure 6d: We repeated this experiment three times and obtained consistent results.
Supplementary Fig. 1e: We repeated this experiment three times and obtained consistent results.
Supplementary Fig. 3b: We repeated this experiment twice and obtained consistent results.
Supplementary Fig. 4a: We repeated this experiment three times and obtained consistent results.
Supplementary Fig. 5a: We repeated this experiment twice and obtained consistent results.
Supplementary Fig. 5b, e: We repeated this experiment three times and obtained consistent results.
Supplementary Fig. 5c, f: We repeated this experiment three times and obtained consistent results.
Supplementary Fig. 7a: We repeated this experiment twice and obtained consistent results.
Supplementary Fig. 7b: We repeated this experiment twice and obtained consistent results.
Supplementary Fig. 7c: We repeated this experiment three times and obtained consistent results.
Supplementary Fig. 8a: We repeated this experiment three times and obtained consistent results.
Supplementary Fig. 9c, d: We repeated this experiment twice and obtained consistent results.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this work are available in the main text, Methods, Supplementary Information, or Supplementary Data. Source data are provided with this paper.
Code availability
The numerical code in the chromatin simulation is publicly available on GitHub at https://doi.org/10.5281/zenodo.12659908106. The scripts for track-sorting and angle-distribution analysis are available at https://zenodo.org/records/12672197.
References
Eltsov, M., Maclellan, K. M., Maeshima, K., Frangakis, A. S. & Dubochet, J. Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc. Natl Acad. Sci. USA 105, 19732–19737 (2008).
Nishino, Y. et al. Human mitotic chromosomes consist predominantly of irregularly folded nucleosome fibres without a 30-nm chromatin structure. EMBO J. 31, 1644–1653 (2012).
Ou, H. D. et al. ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357, eaag0025 (2017).
Maeshima, K., Ide, S. & Babokhov, M. Dynamic chromatin organization without the 30-nm fiber. Curr. Opin. Cell Biol. 58, 95–104 (2019).
Chen J. K., et al. Nanoscale analysis of human G1 and metaphase chromatin in situ. bioRxiv, (2024).
McDowall, A. W., Smith, J. M. & Dubochet, J. Cryo-electron microscopy of vitrified chromosomes in situ. EMBO J. 5, 1395–1402 (1986).
Green, L. C. et al. Contrasting roles of condensin I and condensin II in mitotic chromosome formation. J. Cell Sci. 125, 1591–1604 (2012).
Hirano, T. Condensin-based chromosome organization from bacteria to vertebrates. Cell 164, 847–857 (2016).
Kireeva, N., Lakonishok, M., Kireev, I., Hirano, T. & Belmont, A. S. Visualization of early chromosome condensation: a hierarchical folding, axial glue model of chromosome structure. J. Cell Biol. 166, 775–785 (2004).
Liang, Z. et al. Chromosomes progress to metaphase in multiple discrete steps via global compaction/expansion (stress) cycles. Cell 161, 1124–1137 (2015).
Nagasaka, K., Hossain, M. J., Roberti, M. J., Ellenberg, J. & Hirota, T. Sister chromatid resolution is an intrinsic part of chromosome organization in prophase. Nat. Cell Biol. 18, 692–699 (2016).
Samejima, K. et al. Mitotic chromosomes are compacted laterally by KIF4 and condensin and axially by topoisomerase IIalpha. J. Cell Biol. 199, 755–770 (2012).
Shintomi, K. et al. Mitotic chromosome assembly despite nucleosome depletion in Xenopus egg extracts. Science 356, 1284–1287 (2017).
Uhlmann, F. SMC complexes: from DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 17, 399–412 (2016).
Nielsen, C. F., Zhang, T., Barisic, M., Kalitsis, P. & Hudson, D. F. Topoisomerase IIalpha is essential for maintenance of mitotic chromosome structure. Proc. Natl Acad. Sci. USA 117, 12131–12142 (2020).
Cuylen, S. et al. Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature 535, 308–312 (2016).
Takagi M. et al. Ki-67 and condensins support the integrity of mitotic chromosomes through distinct mechanisms. J. Cell Sci. 131, jcs212092 (2018).
Davidson, I. F. & Peters, J. M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 22, 445–464 (2021).
Meijering, A. E. C. et al. Nonlinear mechanics of human mitotic chromosomes. Nature 605, 545–550 (2022).
Hirano, T., Kobayashi, R. & Hirano, M. Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell 89, 511–521 (1997).
Ono, T. et al. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell 115, 109–121 (2003).
Paulson, J. R. & Laemmli, U. K. The structure of histone-depleted metaphase chromosomes. Cell 12, 817–828 (1977).
Maeshima, K. & Laemmli, U. K. A two-step scaffolding model for mitotic chromosome assembly. Dev. Cell 4, 467–480 (2003).
Ohta, S. et al. The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell 142, 810–821 (2010).
Walther, N. et al. A quantitative map of human Condensins provides new insights into mitotic chromosome architecture. J. Cell Biol. 217, 2309–2328 (2018).
Gibcus J. H. et al. A pathway for mitotic chromosome formation. Science 359, 6376 (2018).
Ono, T., Fang, Y., Spector, D. L. & Hirano, T. Spatial and temporal regulation of Condensins I and II in mitotic chromosome assembly in human cells. Mol. Biol. Cell 15, 3296–3308 (2004).
Gerlich, D., Hirota, T., Koch, B., Peters, J. M. & Ellenberg, J. Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr. Biol. 16, 333–344 (2006).
Goloborodko, A., Marko, J. F. & Mirny, L. A. Chromosome compaction by active loop extrusion. Biophys. J. 110, 2162–2168 (2016).
Ganji, M. et al. Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105 (2018).
Kim E., Barth R., Dekker C. Looping the genome with SMC complexes. Annu. Rev. Biochem. 92: 15–41 (2023).
Cheng, T. M. et al. A simple biophysical model emulates budding yeast chromosome condensation. eLife 4, e05565 (2015).
Gerguri, T. et al. Comparison of loop extrusion and diffusion capture as mitotic chromosome formation pathways in fission yeast. Nucleic Acids Res. 49, 1294–1312 (2021).
Tang, M. et al. Establishment of dsDNA-dsDNA interactions by the condensin complex. Mol. Cell 83, 3787–3800.e3789 (2023).
Mora-Bermudez, F., Gerlich, D. & Ellenberg, J. Maximal chromosome compaction occurs by axial shortening in anaphase and depends on Aurora kinase. Nat. Cell Biol. 9, 822–831 (2007).
Samejima K. et al. Functional analysis after rapid degradation of condensins and 3D-EM reveals chromatin volume is uncoupled from chromosome architecture in mitosis. J. Cell Sci. 131, jcs210187 (2018).
Schneider M. W. G. et al. A mitotic chromatin phase transition prevents perforation by microtubules. Nature 609, 183–190(2022).
Zhiteneva A. et al. Mitotic post-translational modifications of histones promote chromatin compaction in vitro. Open Biol. 7, 170076 (2017).
Cimini, D., Mattiuzzo, M., Torosantucci, L. & Degrassi, F. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol. Biol. Cell 14, 3821–3833 (2003).
Kruhlak, M. J. et al. Regulation of global acetylation in mitosis through loss of histone acetyltransferases and deacetylases from chromatin. J. Biol. Chem. 276, 38307–38319 (2001).
Wilkins, B. J. et al. A cascade of histone modifications induces chromatin condensation in mitosis. Science 343, 77–80 (2014).
Maeshima, K. A phase transition for chromosome transmission when cells divide. Nature 609, 35–36 (2022).
Gorisch, S. M., Wachsmuth, M., Toth, K. F., Lichter, P. & Rippe, K. Histone acetylation increases chromatin accessibility. J. Cell Sci. 118, 5825–5834 (2005).
Ricci, M. A., Manzo, C., Garcia-Parajo, M. F., Lakadamyali, M. & Cosma, M. P. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 160, 1145–1158 (2015).
Nozaki, T. et al. Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol. Cell 67, 282–293.e287 (2017).
Strickfaden, H. et al. Condensed chromatin behaves like a solid on the mesoscale in vitro and in living cells. Cell 183, 1772–1784.e1713 (2020).
Maeshima, K. et al. A transient rise in free Mg(2+) ions released from ATP-Mg hydrolysis contributes to mitotic chromosome condensation. Curr. Biol. 28, 444–451.e446 (2018).
Marsden, M. P. & Laemmli, U. K. Metaphase chromosome structure: evidence for a radial loop model. Cell 17, 849–858 (1979).
Earnshaw, W. C. & Laemmli, U. K. Architecture of metaphase chromosomes and chromosome scaffolds. J. Cell Biol. 96, 84–93 (1983).
Maeshima, K., Tamura, S., Hansen, J. C. & Itoh, Y. Fluid-like chromatin: toward understanding the real chromatin organization present in the cell. Curr. Opin. Cell Biol. 64, 77–89 (2020).
Iida, S. et al. Orientation-Independent-DIC imaging reveals that a transient rise in depletion attraction contributes to mitotic chromosome condensation. Proc. Natl Acad. Sci. USA. https://doi.org/10.1073/pnas.2403153121 (2024).
Dubochet, J. & Sartori Blanc, N. The cell in absence of aggregation artifacts. Micron 32, 91–99 (2001).
Mazza, D., Abernathy, A., Golob, N., Morisaki, T. & McNally, J. G. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. 40, e119 (2012).
Hihara, S. et al. Local nucleosome dynamics facilitate chromatin accessibility in living mammalian cells. Cell Rep. 2, 1645–1656 (2012).
Lerner, J. et al. Two-parameter mobility assessments discriminate diverse regulatory factor behaviors in chromatin. Mol. Cell 79, 677–688.e676 (2020).
Lakadamyali, M. Single nucleosome tracking to study chromatin plasticity. Curr. Opin. Cell Biol. 74, 23–28 (2022).
Ide, S., Tamura, S. & Maeshima, K. Chromatin behavior in living cells: lessons from single-nucleosome imaging and tracking. BioEssays 44, e2200043 (2022).
Ashwin, S. S., Nozaki, T., Maeshima, K. & Sasai, M. Organization of fast and slow chromatin revealed by single-nucleosome dynamics. Proc. Natl Acad. Sci. USA 116, 19939–19944 (2019).
Nozaki, T. et al. Condensed but liquid-like domain organization of active chromatin regions in living human cells. Sci. Adv. 9, eadf1488 (2023).
Iida, S. et al. Single-nucleosome imaging reveals steady-state motion of interphase chromatin in living human cells. Sci. Adv. 8, eabn5626 (2022).
Seeber, A., Hauer, M. H. & Gasser, S. M. Chromosome dynamics in response to DNA damage. Annu. Rev. Genet. 52, 295–319 (2018).
Parsons, G. G. & Spencer, C. A. Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. Mol. Cell Biol. 17, 5791–5802 (1997).
Chen, D. et al. Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins. J. Cell Biol. 168, 41–54 (2005).
Grimm, J. B. et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat. Methods 12, 244–250 (2015).
Kimura, H. & Cook, P. R. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153, 1341–1353 (2001).
Tokunaga, M., Imamoto, N. & Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 5, 159–161 (2008).
Betzig, E. et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006).
Rust, M. J., Bates, M. & Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795 (2006).
Selvin P. R. et al. Fluorescence imaging with one-nanometer accuracy (FIONA). CSH Protoc. 2007, pdb top27 (2007).
Jaqaman, K. et al. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5, 695–702 (2008).
Dion, V. & Gasser, S. M. Chromatin movement in the maintenance of genome stability. Cell 152, 1355–1364 (2013).
Kakui, Y., Rabinowitz, A., Barry, D. J. & Uhlmann, F. Condensin-mediated remodeling of the mitotic chromatin landscape in fission yeast. Nat. Genet. 49, 1553–1557 (2017).
Kakui, Y. et al. Fission yeast condensin contributes to interphase chromatin organization and prevents transcription-coupled DNA damage. Genome Biol. 21, 272 (2020).
Izeddin I. et al. Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. eLife 3, e02230 (2014).
Nagashima, R. et al. Single nucleosome imaging reveals loose genome chromatin networks via active RNA polymerase II. J. Cell Biol. 218, 1511–1530 (2019).
Manders, E. M., Kimura, H. & Cook, P. R. Direct imaging of DNA in living cells reveals the dynamics of chromosome formation. J. Cell Biol. 144, 813–821 (1999).
Hoencamp, C. et al. 3D genomics across the tree of life reveals condensin II as a determinant of architecture type. Science 372, 984–989 (2021).
Eykelenboom, J. K. et al. Live imaging of marked chromosome regions reveals their dynamic resolution and compaction in mitosis. J. Cell Biol. 218, 1531–1552 (2019).
Vassilev, L. T. et al. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl Acad. Sci. USA 103, 10660–10665 (2006).
Iemura K., Natsume T., Maehara K., Kanemaki M. T., Tanaka K. Chromosome oscillation promotes Aurora A-dependent Hec1 phosphorylation and mitotic fidelity. J. Cell Biol. 22, e202006116 (2021).
Stumpff, J., von Dassow, G., Wagenbach, M., Asbury, C. & Wordeman, L. The kinesin-8 motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment. Dev. Cell 14, 252–262 (2008).
Vladimirou, E. et al. Nonautonomous movement of chromosomes in mitosis. Dev. Cell 27, 60–71 (2013).
Natsume, T., Kiyomitsu, T., Saga, Y. & Kanemaki, M. T. Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 15, 210–218 (2016).
Yesbolatova, A. et al. The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun. 11, 5701 (2020).
Waizenegger, I. C., Hauf, S., Meinke, A. & Peters, J. M. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 103, 399–410 (2000).
Nozaki T., Hudson D. F., Tamura S., Maeshima K. Dynamic chromatin folding in the cell. In: Nuclear Architecture and Dynamics (eds. Lavelle C., Victor J.M.) (Academic Press, 2018).
Itoh, Y. et al. 1,6-hexanediol rapidly immobilizes and condenses chromatin in living human cells. Life Sci. Alliance 4, e202001005 (2021).
Gabriele, M. et al. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 376, 496–501 (2022).
Mach, P. et al. Cohesin and CTCF control the dynamics of chromosome folding. Nat. Genet. 54, 1907–1918 (2022).
Bailey M. L. P. et al. Loops and the activity of loop extrusion factors constrain chromatin dynamics. Mol. Biol. Cell mbcE23040119 (2023).
Sakai, Y., Mochizuki, A., Kinoshita, K., Hirano, T. & Tachikawa, M. Modeling the functions of condensin in chromosome shaping and segregation. PLoS Comput. Biol. 14, e1006152 (2018).
Shintomi, K. & Hirano, T. The relative ratio of condensin I to II determines chromosome shapes. Genes Dev. 25, 1464–1469 (2011).
Takagi, M., Natsume, T., Kanemaki, M. T. & Imamoto, N. Perichromosomal protein Ki67 supports mitotic chromosome architecture. Genes Cells 21, 1113–1124 (2016).
Yoshida, M., Kijima, M., Akita, M. & Beppu, T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 265, 17174–17179 (1990).
Hirano, T. Condensins and the evolution of torsion-mediated genome organization. Trends Cell Biol. 24, 727–733 (2014).
Hajjoul, H. et al. High-throughput chromatin motion tracking in living yeast reveals the flexibility of the fiber throughout the genome. Genome Res. 23, 1829–1838 (2013).
Samejima K. et al. Rules of engagement for condensins and cohesins guide mitotic chromosome formation. bioRxiv https://doi.org/10.1101/2024.04.18.590027 (2024).
Hancock, R. Structure of metaphase chromosomes: a role for effects of macromolecular crowding. PLoS One 7, e36045 (2012).
Miron E. et al. Chromatin arranges in chains of mesoscale domains with nanoscale functional topography independent of cohesin. Sci. Adv. 6 (2020).
Maeshima K., Iida S., Shimazoe M. A., Tamura S., Ide S. Is euchromatin really open in the cell? Trends Cell Biol. 34, 7–17 (2023).
Farr, S. E., Woods, E. J., Joseph, J. A., Garaizar, A. & Collepardo-Guevara, R. Nucleosome plasticity is a critical element of chromatin liquid-liquid phase separation and multivalent nucleosome interactions. Nat. Commun. 12, 2883 (2021).
Huertas, J., Woods, E. J. & Collepardo-Guevara, R. Multiscale modelling of chromatin organisation: Resolving nucleosomes at near-atomistic resolution inside genes. Curr. Opin. Cell Biol. 75, 102067 (2022).
Maeshima, K. et al. Cell-cycle-dependent dynamics of nuclear pores: pore-free islands and lamins. J. Cell Sci. 119, 4442–4451 (2006).
Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 (1970).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Sakai, Y. YSakaiGH/mitotic_chromatin_motions: v1.0.0 (v1.0.0). Zenodo, https://zenodo.org/records/12659908 (2024).
Vassilev, L. T. Cell cycle synchronization at the G2/M phase border by reversible inhibition of CDK1. Cell Cycle 5, 2555–2556 (2006).
Acknowledgements
We are grateful to Dr. M. Sasai, Dr. F. Uhlmann, Dr. S. Shinkai, Dr. Y. Itoh, Mr. A. Semeigazin, and Dr. K.M. Marshall for critical reading or editing of this manuscript, Dr. R. Nagashima for construction of some of the plasmids, Dr. T. Tanaka for providing the HT-1080 cells, and Ms. S. Iida for providing some published data. We thank Dr. K. Kinoshita, Dr. Y. Shimamoto, and Maeshima laboratory members for helpful discussions and support. The numerical computations have been performed with the RIKEN supercomputer system (HOKUSAI GreatWave). This work was supported by the Japan Society for the Promotion of Science (JSPS) and MEXT KAKENHI Grants (JP20H05936, JP21H02453, JP23K17398, and JP24H00061) to K. Maeshima, JSPS KAKENHI grant JP20K06594 and JST PRESTO Grant (JPMJPR21ED) to K.H., JSPS KAKENHI grant JP20K06482 to Y.S., and the JSPS KAKENHI grant (JP21H04719 and JP23H04925), the JST CREST grant (JPMJCR21E6) to M.T.K. K. Minami was a SOKENDAI Special Researcher (JST SPRING JPMJSP2104) and is a JSPS Fellow (JP23KJ0998). M.A.S. is a JSPS Fellow (JP24KJ1161).
Author information
Authors and Affiliations
Contributions
K. Maeshima, Y.S., and K.H. designed the project. K.H. generated H2B-Halo expressing cells, and performed single-nucleosome imaging, and analyses. Y.S. performed computational modeling. S.T. performed biochemical experiments, fluorescence imaging, and scientific illustrations. M.T. and N.I. contributed to the generation of condensin I- and II-AID and condensins I and II/ Ki-67-AID cell lines, and their fluorescence imaging and western blotting. M.A.S. and K. Minami contributed to nucleosome motion analysis. M.T.K. and T.N. contributed to SMC2-AID cell generation, western blot, and cell cycle analysis. K. Maeshima., K. Minami, K.H., S.T., Y.S., and M.A.S. wrote the manuscript with input from all other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Andrea Chiariello and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hibino, K., Sakai, Y., Tamura, S. et al. Single-nucleosome imaging unveils that condensins and nucleosome–nucleosome interactions differentially constrain chromatin to organize mitotic chromosomes. Nat Commun 15, 7152 (2024). https://doi.org/10.1038/s41467-024-51454-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51454-y
- Springer Nature Limited