

Abstract
The development of highly efficient and durable alkaline hydrogen evolution reaction (HER) catalysts is crucial for achieving high-performance practical anion exchange membrane water electrolyzer (AEMWE) at ampere-level current density. Herein, we report a design concept by employing Ga single atoms as an electronic bridge to stabilize the Ru clusters for boosting alkaline HER performance in practical AEMWE. Experimental and theoretical results collectively reveal that the bridged Ga sites trigger strong metal-support interaction for the homogeneous distribution of Ru clusters with high density, as well as optimize the Ru–H bond strength due to the electron transfer between Ru and Ga for enhanced intrinsic HER activity. Moreover, the oxophilic Ga sites near the Ru clusters tend to adsorb the hydroxyl species and accelerate the water dissociation for sufficient proton supplement in an alkaline medium. The Ru–GaSA/N–C catalyst exhibits a low overpotential of 4 ± 1 mV (10 mA cm−2) and high mass activity of 9.3 ± 0.5 A mg−1Ru at −0.05 V vs RHE. In particular, the Ru–GaSA/N–C-based AEMWE in 1 M KOH delivers a voltage of only 1.74 V to reach an industrial current density of 1 A cm−2, and can steadily operate at 1 A cm−2 for more than 170 h.
Similar content being viewed by others
Introduction
Electrocatalytic water splitting has been widely regarded as a promising route to generate green hydrogen from renewable electricity1,2,3,4,5. Liquid alkaline water electrolysis (LAWE) and proton exchange membrane water electrolysis (PEMWE) are the two mainstream water electrolysis technologies6,7. Considering that LAWE presents a poor match with renewable energy and PEMWE suffers from high system costs owing to the harsh acidic and strong oxidative operating conditions, the anion exchange membranes water electrolysis (AEMWE) is attracting growing attention in industrial hydrogen production8,9. However, the current AEMWE still presents much higher cell voltage than that of PEMWE, the main reason can be ascribed to the following aspects: (1) the membrane/ionomer conductivity of AEMWE is lower than that of PEMWE and (2) hydrogen evolution reaction (HER) kinetics in alkaline medium is more sluggish than that in acidic medium10. Therefore, designing highly efficient alkaline HER electrocatalysts with superior activity and durability is critical for achieving high-performance AEMWE. Pt-based electrocatalysts are widely used as alkaline HER electrocatalysts due to the proper hydrogen adsorption energy11,12,13. However, the weak water dissociation ability of Pt results in an insufficient supply of proton in the Volmer step, especially operating at industrial current density in AEM water electrolysis14. Ru-based catalysts, especially the Ru clusters (Ru/OMSNNC15, Ru@MWCNT16, Ru/HfO217, Ru/P–TiO218, Ru@C2N19) attract extensive attention owing to the stronger water dissociation ability, similar bond strength of Ru–H and much lower cost compared to Pt20,21,22,23. However, the current Ru clusters still suffer from insufficient activity and durability under the harsh conditions in practical AEMWE (> 500 mA cm−2) due to the following reasons: (1) the excessive Ru–H bond strength, insufficient water dissociation ability and low-density Ru clusters on the substrates lead to unsatisfactory performance especially at industrial current densities (> 500 mA cm−2)24, and (2) the weak cluster-support interaction results in the agglomeration and detachment of Ru clusters during the large current density operation in AEMWE25. Therefore, it is highly urgent to design new-type Ru cluster catalysts with optimized Ru–H bond strength, strong water dissociation ability, high density of cluster, and enhanced metal-support interaction for boosting the catalytic activity and stability of AEMWE, however, achieving such a target remains a grand challenge.
Herein, we selected electron-deficient and oxophilic Ga single atoms as electronic bridges to cooperate with the electron-rich Ru clusters, which induce strong metal-support interaction with Ru clusters and improve the water dissociation ability for highly efficient HER in AEMWE. We find that the Ga single atoms on the N-doped carbon can act as anchoring sites for stabilizing the Ru clusters, which leads to the homogeneous distribution of Ru clusters on carbon with high density, and also prevents the agglomeration and detachment of Ru clusters during the long-term electrolysis in AEMWE. The optimized Ru–GaSA/N–C catalyst exhibits outstanding performance with a quite small overpotential of only 4 ± 1 mV at 10 mA cm−2 and high mass activity of 9.3 ± 0.5 A mg−1Ru at −0.05 V vs RHE, surpassing that of PtRu/C and Pt/C catalysts. Remarkably, the Ru–GaSA/N–C-based AEMWE operated with 1 M KOH feed presents a low voltage of only 1.74 V at 1 A cm−2 with a low Ru loading of 0.08 mg cm−2 and can operate at 1 A cm−2 for more than 170 h with negligible voltage increase. Experimental and DFT calculation results collectively reveal that strong Ga–Ru bonds formed at the interface of Ru clusters and Ga–N4 substrate can optimize the Ru–H bond strength, and also oxophilic Ga sites near the Ru clusters can accelerate the water dissociation in an alkaline medium, both of which lead to the enhanced activity and stability of Ru–GaSA/N–C during the large current density operation in AEMWE.
Results
Materials synthesis and characterization
The Ru–GaSA/N–C catalyst was synthesized via a two-step route, as displayed in Fig. 1a. Briefly, Ga(acac)3@ZIF-8 was firstly prepared by encapsulating Ga(acac)3 molecule into the micropore of ZIF-8 due to the similar size of metal precursor and host26, followed by the pyrolysis at 900 °C to form the isolated Ga single atoms anchored on nitrogen-doped carbon support (GaSA/N–C). Afterward, the Ru–GaSA/N–C catalyst was synthesized by anchoring the Ru clusters on the GaSA/N–C support through an immersion combined thermal treatment process. Ru/N–C was also prepared by a similar process without the addition of Ga(acac)3.

a Schematic illustration of the synthesis of Ru–GaSA/N–C catalyst. b–d HAADF-STEM images of the Ru–GaSA/N–C catalyst, scale bar: 50 nm, 20 nm, and 2 nm, respectively. The inset in (c) is the size distribution of Ru clusters. e Amplified images from (d), scale bar: 1 nm. f Line-scanning intensity profiles along with the red and blue directions in (e). g EDS elemental mapping images of Ru–GaSA/N–C, scale bar: 5 nm (the corresponding HAADF-STEM image is shown in Supplementary Fig. 5).
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images show the dodecahedral morphology of GaSA/N–C catalyst with porous structure, and no metal nanoparticles are formed during the pyrolysis process (Supplementary Figs. 1, 2). The aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of GaSA/N–C present the existence of Ga single atoms on nitrogen-doped carbon support (Supplementary Fig. 3). After the incorporation of Ru, the catalyst remains the dodecahedral morphology with apparent bright particles (~1.8 nm) anchoring on carbon support, indicating the successful incorporation of Ru clusters on the GaSA/N–C substrate (Fig. 1b, c and Supplementary Fig. 4). Further HAADF-STEM image exhibits the coexistence of Ru clusters and Ga single atoms on the carbon support (Fig. 1d). Atomic line profiles show that Ga atoms are partially distributed around the Ru clusters (Fig. 1e, f). EDS mapping images display that Ru mainly distributes on the position of the clusters, while Ga appears randomly on the whole carbon support (Fig. 1g and Supplementary Fig. 5). Brunauer–Emmett–Teller (BET) test of Ru–GaSA/N–C indicates that it possesses a large specific surface area of 1452.75 m2 g−1 (Supplementary Fig. 6). Inductively coupled plasma-optical emission spectrometer (ICP-OES) shows that the Ru and Ga contents of Ru–GaSA/N–C are 7.98 wt% and 0.94 wt%, respectively (Supplementary Table 1). All these results indicate the successful construction of Ru clusters and Ga single atoms on N-doped carbon. By contrast, the Ru/N–C catalyst absence of Ga single atoms exhibits a larger particle size of around 5.5 nm, demonstrating the critical role of Ga single atoms in stabilizing Ru clusters (Supplementary Fig. 7).
The crystal structure of catalysts was analyzed by X-ray diffraction (XRD). GaSA/N–C and pure N–C catalysts display two broad peaks at around 25° and 43°, assigned to the (002) and (101) planes of C (Fig. 2a and Supplementary Fig. 8), which reveals the isolated single atoms of Ga on carbon support, consistent with the HAADF-STEM results. After the incorporation of Ru, only a broad peak at 44° can be observed for the Ru–GaSA/N–C catalyst, while distinct peaks at 38°, 42°, and 44° appear for the Ru/N–C sample, illustrating the reduced particle size of Ru by the assistance of Ga single atoms15. This result further indicates the existence of a strong interaction between Ga and Ru for stabilizing the Ru clusters. X-ray photoelectron spectroscopy (XPS) analysis was conducted to investigate the electronic structures and valence states of the catalysts. As displayed in the high-resolution Ru 3p spectra (Fig. 2b), the Ru0 peak of Ru–GaSA/N–C exhibits a positive shift relative to that of Ru/N–C, indicating the lowered electron density state27. In addition, the peak of Ru–GaSA/N–C in the high-resolution Ga 2p spectra presents a negative shift compared to GaSA/N–C, suggesting the electron transfer between Ru and Ga (Fig. 2c). The above XPS analysis confirms that Ga single atoms can act as an electronic bridge to induce strong cluster-substrate interaction and regulate the electronic structure of Ru clusters for enhanced HER performance28.

a XRD patterns of Ru–GaSA/N–C, Ru/N–C, and GaSA/N–C. High-resolution XPS spectra of b Ru 3p and c Ga 2p. d Ru K-edge XANES spectra of Ru–GaSA/N–C, Ru/N–C, Ru foil, and RuO2. e The corresponding Fourier-transformed Ru K-edge EXAFS spectra. f Ga K-edge XANES spectra of Ru–GaSA/N–C, GaSA/N–C, Ga foil, and Ga2O3. g The corresponding Fourier-transformed Ga K-edge EXAFS spectra. h Wavelet transforms for the k3-weighted Ru K-edge EXAFS signals of Ru–GaSA/N–C, Ru foil, and RuO2.
X-ray absorption fine structure spectroscopy (XAFS) was employed to further investigate the coordination structure and electronic state of Ru and Ga for the Ru–GaSA/N–C catalyst. The Ru K-edge X-ray absorption near edge structure (XANES) spectra reveal that the absorption edges of Ru–GaSA/N–C and Ru/N–C approach that of Ru foil, indicating the metallic feature of Ru (Fig. 2d). The positive edge shift of Ru–GaSA/N–C relative to Ru/N–C suggest the increased Ru oxidation state of Ru–GaSA/N–C, further indicating the electron transfer from Ru to Ga. In addition, the extended X-ray absorption fine structure (EXAFS) spectra of Ru–GaSA/N–C show a dominant peak at 2.4 Å, ascribed to the Ru–Ru bonding (Fig. 2e, Supplementary Fig. 9, and Supplementary Table 2), and the weak peak at 1.5 Å, attributed to the Ru–N/C coordination due to the interaction between Ru clusters and N–C substrate. The coordination structures can also be confirmed by the wavelet transform (WT) analysis (Fig. 2h). The XANES spectra of Ga displays that the absorption edges of Ru–GaSA/N–C and GaSA/N–C are close to that of Ga2O3, representing that the oxidation state of Ga is around +3 (Fig. 2f). The EXAFS spectra of Ru–GaSA/N–C and GaSA/N–C presents a distinct peak at around 1.4 Å, corresponding to the Ga-N/C coordination (Fig. 2g, Supplementary Figs. 10 and 11, and Supplementary Tables 2 and 3). In addition, a peak at around 2.5 Å exists for Ru–GaSA/N–C rather than GaSA/N–C, indicating the interaction between Ga atoms and Ru clusters to form the Ga–Ru bond23.
Electrocatalytic HER performance
The HER performance of the Ru–GaSA/N–C catalysts was examined in the N2-saturated 1 M KOH solution (Fig. 3a and Supplementary Fig. 12). The optimized Ru–GaSA/N–C delivers quite low overpotentials of 4 ± 1 mV and 46 ± 2 mV to reach 10 mA cm−2 and 100 mA cm−2, superior to that of Ru/N–C (18 ± 2 mV and 102 ± 5 mV at 10 mA cm−2 and 100 mA cm−2), PtRu/C (18 ± 1 mV and 82 ± 5 mV at 10 mA cm−2 and 100 mA cm−2), Pt/C (29 ± 2 mV and 149 ± 6 mV at 10 mA cm−2 and 100 mA cm−2) and GaSA/N–C (551 ± 3 mV at 10 mA cm−2) (Supplementary Fig. 13). The performance of Ru–GaSA/N–C even surpasses the PtRu/C catalyst with a higher loading of 36 μgRu+Pt cm−2 (Supplementary Fig. 14). The Ru–GaSA/N–C displays the lowest Tafel slope among all the investigated catalysts, indicating the enhanced HER kinetics of Ru–GaSA/N–C (Supplementary Fig. 15). The Ru–GaSA/N–C also exhibits extremely high mass activity of 9.3 ± 0.5 A mg−1Ru at an overpotential of 50 mV, higher than those of Ru/N–C (2.9 ± 0.3 A mg−1Ru) and PtRu/C (8.4 ± 0.2 A mg−1Ru) (Fig. 3b). Moreover, the Ru–GaSA/N–C shows the largest electrochemical active surface area (ECSA) among different catalysts tested by CO stripping (Supplementary Fig. 16). To further examine the intrinsic HER activity, turnover frequency (TOF) values of catalysts were calculated29,30. As shown in Supplementary Fig. 17, Ru–GaSA/N–C presents a high TOF of 18.4 ± 1.1 s−1 at an overpotential of 50 mV, better than those of Ru/N–C (12.1 ± 1.1 s−1), PtRu/C (15.8 ± 1.1 s−1) and Pt/C (6.0 ± 0.4 s−1). The electrochemical impedance spectroscopy (EIS) measurements show the lowest charge-transfer resistance of Ru–GaSA/N–C among the contrast catalysts, further supporting the promoted HER activity (Supplementary Fig. 18). The performances of Ru–GaSA/N–C is well-placed among select recent state-of-art catalysts from the aspects of overpotential and mass activity (Supplementary Fig. 19 and Supplementary Table 4).

a iR-corrected polarization curves (R was 5.7 ± 0.1 Ω) tested by rotating disk electrode (RDE) at a scan rate of 5 mV s−1 under a rotating rate of 1600 rpm (noble metal loading was about 12 μg cm−2). b Mass activities of Ru–GaSA/N–C, Ru/N–C, and PtRu/C catalysts normalized to Ru mass at an overpotential of 50 mV. c iR-corrected polarization curves of Ru–GaSA/N–C, Ru/N–C, PtRu/C, and Pt/C before and after 10,000 cycling tests by RDE. d Chronopotentiometry tests of Ru–GaSA/N–C, Ru/N–C, PtRu/C, and Pt/C coated on carbon paper at 10 mA cm−2. e iR-corrected polarization curves (R was 1.7 ± 0.1 Ω) of Ru–GaSA/N–C, Ru/N–C, PtRu/C, and Pt/C catalysts coated on carbon paper (noble metal loading was about 40 μg cm−2). f Stability test of Ru–GaSA/N–C coated on carbon paper at 1 A cm−2 for 100 h.
Besides the HER catalytic activity, stability is another essential factor in practical application. As presented in Fig. 3c, the Ru–GaSA/N–C exhibits excellent stability in alkaline electrolytes with a negligible increase of overpotential after 10,000 cycling tests compared to Ru/N–C, PtRu/C, and Pt/C catalysts. Ru–GaSA/N–C also presents stable potential at 10 mA cm−2 for 100 h, while Ru/N–C, PtRu/C and Pt/C catalysts show obvious potential increase in the chronopotentiometry (CP) tests (Fig. 3d). TEM and XPS characterization after electrolysis confirm the maintained structure and valence state of the Ru–GaSA/N–C catalyst, further evidencing the excellent catalytic durability (Supplementary Figs. 20 and 21). However, Ru/N–C and Pt/C catalysts show obvious agglomeration of metal nanoparticles (Supplementary Figs. 22 and 23) due to the weak metal-support interaction11,31.
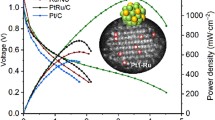
We also evaluated the HER performance of Ru–GaSA/N–C at ampere-level current density by directly coating the catalysts on carbon paper. As shown in Fig. 3e and Supplementary Fig. 24, the Ru–GaSA/N–C only requires an overpotential of 178 ± 4 mV to reach an industrial current density of 1 A cm−2, much lower than those of Ru/N–C (354 ± 5 mV), PtRu/C (260 ± 8 mV) and Pt/C (583 ± 7 mV), and can steadily operate at 1 A cm−2 for 100 h with negligible performance decay (Fig. 3f).
Performance of AEMWE devices
Encouraged by its superior HER activity and durability, we further assembled a membrane-electrode-assembly (MEA)-based AEMWE device using Ru–GaSA/N–C as cathode catalyst and RuO2 as anode catalyst to evaluate its practical application potential in 1 M KOH (Fig. 4a and Supplementary Fig. 25). The polarization curves show that Ru–GaSA/N–C-based AEMWE device only needs 1.61 V and 1.74 V to reach an industrial current density of 0.5 A cm−2 and 1 A cm−2 with a low Ru loading of about 0.08 mg cm−2, much lower than that of Pt/C-based AEMWE with a Pt loading of about 0.2 mg cm−2 and the reported state-of-the-art Ru, Pt-based AEMWE catalysts (Fig. 4b, c and Supplementary Table 5). It should be mentioned here that, the cell voltages of most reported AEMWE devices in literature are much higher than that of PEMWE devices using Pt/C and IrO2 as cathode and anode catalysts. By using the Ru–GaSA/N–C as a cathode catalyst, the cell voltage of our AEMWE can even reach the standard of the PEMWE device (Supplementary Fig. 26). The stability curve of Ru–GaSA/N–C-based electrolyzer exhibits the well-maintained cell voltage over 170 h at a large current density of 1 A cm−2 with a degradation rate of only 49.7 μV h−1 (Fig. 4d). Compared with previously reported state-of-the-art Ru, Pt-based catalysts (Fig. 4e and Supplementary Table 5), the Ru–GaSA/N–C catalyst exhibits superior comprehensive advantages for AEMWE performance from the aspects of overpotential, Tafel slope, mass activity, cell voltage, operation current density and degradation rate, indicating its potential in practical application.

a Scheme diagram of the AEMWE device. b Polarization curves of the AEMWE using Ru–GaSA/N–C and Pt/C as cathode catalysts without iR-correction (The Ru and Pt loading was about 80 μg cm−2 and 200 μg cm−2 for the cathode). c Comparison of the cell voltage of Ru–GaSA/N–C-based AEMWE with other reported state-of-the-art Ru, Pt-based catalysts at various current densities. d CP curve of the Ru–GaSA/N–C-based AEMWE tested at 1 A cm−2. e Comprehensive performance comparison of the Ru–GaSA/N–C with other reported state-of-the-art Ru, Pt-based catalysts.
Origin of the excellent performance of Ru–GaSA/N–C
Density functional theory (DFT) calculations were performed to investigate the origin of the enhanced HER performance of the Ru–GaSA/N–C catalyst. Based on the experimental data and theoretical optimization, the Ru–GaSA/N–C basic model consists of two Ga–N4 sites and one Ru13 cluster, where one Ga–N4 site combined with the Ru13 cluster and another Ga-N4 site is close to the Ru13 cluster, while the Ru/N–C basic model was built by anchoring Ru13 cluster on N–C slab (Fig. 5a and Supplementary Data 1 and 2). The optimized interatomic distance of Ga–Ru is 2.83 Å, consistent with the EXAFS fitting results. Electron localization function (ELF) analysis reveals that the Ru13 cluster bonded with Ga and C/N atoms for Ru–GaSA/N–C, and the Ru13 cluster only bonded with C/N atoms for Ru/N–C (Fig. 5b, c). The subsequent binding energy analysis displays the superior stability of the Ru–GaSA/N–C system relative to the Ru/N–C due to the contribution of the Ga–Ru bond (Supplementary Table 6). Barde charge analysis indicates that the Ru in Ru–GaSA/N–C loses more electrons than that in Ru/N–C, consistent with the XANES and XPS results. The main reason can be ascribed to the electron deficiency of Ga single atoms due to the abundant empty p orbits of Ga32,33, which leads to the electron transfer from electron-rich Ru clusters to electron-deficient Ga single atoms for the optimized electronic structure of Ru.

a The side views (top) and top views (bottom) of Ru–GaSA/N–C and Ru/N–C models. The ELF of b Ru–GaSA/N–C and c Ru/N–C. The energy barrier for breaking the OH-H bond in the Volmer step (water dissociation) of d Ru–GaSA/N–C and e Ru/N–C. The optimized atomic geometries of the intermediate state are shown in the insets. f Kinetic isotope effect (KIE) values of Ru–GaSA/N–C and Ru/N–C at various potentials obtained in 1 M KOH/H2O and KOD/D2O. g In situ ATR-SEIRAS analysis of Ru–GaSA/N–C and Ru/N–C at various potentials. h Density of states (DOS) of Ru–GaSA/N–C and Ru/N–C. i Gibbs free energy diagram of H+ to H2 for Ru–GaSA/N–C and Ru/N–C. The inset is the charge density distribution of H adsorption on the Ru–GaSA/N–C and Ru/N–C.
Water dissociation on the catalyst surface is a critical step during the alkaline HER process. The energy-limiting step is dramatically decreased with the assistance of interfacial Ga atoms (Fig. 5d, e), indicating the stronger water dissociation ability of Ru–GaSA/N–C. Detailed reaction pathway for H2O dissociation on the Ru–Ga site is shown in the insets of Fig. 5d and Supplementary Data 3 to 6. At the initial state, H2O is adsorbed on the Ga-N4 site near the Ru13 cluster with the OH groups bonded with Ga single atoms and the H atoms orientated toward the Ru cluster. Upon the OH–H bond breakage, the final states show OH groups bonded with Ga single atoms and H atoms bonded with Ru clusters. Kinetic isotope effect (KIE) measurement was also conducted to investigate the water dissociation kinetics of HER. In alkaline medium, the dissociation of water (Volmer step) is the RDS of HER and the lower value of KIE indicates easier dissociation of H–OH34. The KIE value of Ru–GaSA/N–C is much lower than that of Ru/N–C, demonstrating the improved water dissociation kinetics of the Ru–GaSA/N–C catalyst (Fig. 5f and Supplementary Fig. 27). In situ surface-enhanced infrared absorption spectroscopy with attenuated total reflection (ATR-SEIRAS) spectra reveal that the frequency of O–H stretching mode (νO–H) of Ru–GaSA/N–C catalyst shows a positive shift compared with that of Ru/N–C catalyst (Fig. 5g), which leads to the increased mobility of interfacial water for facilitating the water dissociation process35,36.
Moreover, the adsorption energy of proton H* (ΔGH*) is also a vital descriptor for evaluating HER activity. According to previous reports, Ru possesses lower intrinsic HER activity compared to Pt due to the excessive H adsorption, which hinders the H2 formation for reduced HER activity. The density of states (DOS) plots reveal that the d band center of the Ru–GaSA/N–C (−1.901 eV) shows an obvious downshift relative to that of Ru/N–C (−1.845 eV), which weakens the H binding strength to enable more feasible H transfer (Fig. 5h). The subsequent analysis of free energy of H adsorption displays the lower free energy of H adsorption of Ru–GaSA/N–C relative to Ru/N–C, suggesting the significant role of Ga on tuning H adsorption (Fig. 5i). The reason for the optimization of H adsorption was further investigated from a microscopic electronic perspective by ELF, revealing that the Ru–GaSA/N–C possesses weaker electron localization degree in the Ru–H bonding region than that of Ru/N–C for reduced Ru–H bonding strength and finally improve the intrinsic HER performance (inset of Fig. 5i).
Discussion
In conclusion, we report a class of Ru–GaSA/N–C catalyst by employing Ga single atoms on the N-doped carbon as an electronic bridge to anchor the Ru clusters for practical AEMWE. The Ru–GaSA/N–C catalyst exhibits a small overpotential of only 4 ± 1 mV at 10 mA cm−2, high mass activity of 9.3 ± 0.5 A mg−1Ru at an overpotential of 50 mV, and excellent operation durability of over 100 h at 1 A cm−2, superior to the PtRu/C catalyst and is well-placed among select recently reported state-of-the-art Ru, Pt-based catalysts. The Ru–GaSA/N–C-based AEMWE in 1 M KOH requires only 1.74 V to reach 1 A cm−2 with a low Ru loading of 0.08 mg cm−2, and can operate at 1 A cm−2 for more than 170 h with negligible voltage increase. Experimental and DFT calculation results indicate that the formation of strong Ga–Ru bonds at the interface of Ru clusters and Ga–N4 substrate can tune the H adsorption energy for improved intrinsic HER activity and meanwhile enhance the stability of Ru–GaSA/N–C in AEMWE, and the oxophilic Ga sites adjacent to the Ru clusters of Ru–GaSA/N–C catalyst can enhance the water dissociation ability due to the favorable binding with OH species for improved activity and stability during the industrial current density operation in AEMWE.
Methods
Materials
Ruthenium acetylacetonate (Ru(acac)3, 98%) was obtained from Adamas-beta. Gallium acetylacetonate (Ga(acac)3, 99.9%) and 2-methylimidazole (C4H6N2, 98%) were purchased from Shanghai Aladdin Biochemical Co., Ltd. Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99%) was obtained from Sinopharm Chemical Reagent Co., Ltd. Methanol (CH3OH, 99.8%) and ethanol (C2H5OH, 99.8%) were purchased from Beijing Tong Guang Fine Chemicals Company. AEM membrane (X37-50, 0.05 mm) was acquired from Dioxide Materials Sustainion. PEM membrane (Nafion 117, 0.18 mm) was obtained from DuPont Co. All reagents were used without further purification.
Synthesis of GaSA/N–C and N–C
The GaSA/N–C was synthesized by a chemical and pyrolysis strategy. Typically, Zn(NO3)2·6H2O (3 g) and Ga(acac)3 (200 mg) were dissolved in 40 mL methanol as solution A. 2-methylimidazole (6.5 g) was dissolved in 80 mL methanol as solution B. Then, two solutions were rapidly mixed under magnetic stirring for 24 h at room temperature. The precipitate was centrifuged, washed with methanol for three times and dried in an oven at 60 °C, followed by annealing at 900 °C for 2 h in a 5% H2/Ar atmosphere. The N–C sample was prepared without the addition of Ga(acac)3.
Synthesis of Ru–GaSA/N–C and Ru/N–C
In a typical procedure, GaSA/N–C (50 mg) was mixed with Ru(acac)3 (18 mg) in a 100 mL beaker containing 40 mL ethanol. After evaporation, the dried powder was then subjected to high-temperature H2-reduction treatment in 5% H2/Ar at 400 °C for 2 h. The synthetic process of Ru/N–C was similar to that of Ru–GaSA/N–C except using N–C instead of GaSA/N–C as support.
Characterization
Transmission electron microscope (TEM) was conducted on JEOL JEM-2010F. HAADF-STEM and energy dispersive X-ray spectroscopy (EDS) element mappings were taken on the JEOL 2200FS STEM/TEM microscope at 300 kV. XRD was performed on Bruker D8 Advance diffractometer at 40.0 kV and 120 mA with Cu–Kα radiation. XPS was carried out on a Thermo Scientific K-Alpha spectrometer. ICP-OES was applied to the Agilent 7800 instrument. XAFS spectra were applied at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF) and BL1W1B in the Beijing Synchrotron Radiation Facility (BSRF). The Athena module of the IFEFFIT software package was conducted to analyze the XAFS raw data according to standard procedures, and the EXAFS data fitting was performed on the Artemis program.
Electrochemical measurements
Electrochemical measurements were conducted in a three-electrode cell on an electrochemical workstation (CHI 760E) setup with a rotating disk electrode (RDE). Hg/HgO (1 M KOH) and graphite rod (Φ = 6 mm) were used as reference and counter electrodes, respectively. The catalyst-coated glassy carbon (GC) electrode (diameter: 5 mm) was conducted as a working electrode. 1 M KOH solution was used as an electrolyte and the electrolytic cell volume was about 100 mL. Before testing, the Hg/HgO electrode was calibrated by cyclic voltammetry using a purified Pt wire as the working and counter electrode in H2-saturated 1 M KOH electrolyte. The average potential at which the current crosses zero was regarded as the thermodynamic potential relative to Hg/HgO37. All recorded potentials (E) were converted to the reversible hydrogen electrode (RHE) based on the following equation: ERHE = EHg/HgO + 0.913. To prepare the working electrode, 5 mg as-prepared catalyst was firstly dispersed into 1 mL solution containing 975 μL of isopropanol and 25 μL of Nafion solution (D521), followed by ultrasonication for 30 min to form a homogenous ink. Afterward, 6 μL of catalyst ink was dropped onto the freshly polished GC electrode with a Ru loading of about 12 μg cm−2 and naturally dried in air. The noble metal loading of commercial PtRu/C and Pt/C catalysts was also controlled to be about 12 μg cm−2, respectively. To assess the performance of the PtRu/C catalyst with the same Ru loading as Ru–GaSA/N–C and Ru/N–C catalysts, we also prepared PtRu/C catalyst with higher loading on the GC electrode (Ru + Pt of 36 μg cm−2 and Ru of 12 μg cm−2). Before the HER test, the electrolyte was purged with N2 with a flow rate of 30 mL min−1 to form the N2-saturated 1 M KOH solution (pH is 13.8 ± 0.1). Linear sweep voltammetry polarization curves were tested three times independently at a scan rate of 5 mV s−1 with 95% iR-compensation under a rotating rate of 1600 rpm. Accelerated durability tests (ADT) were performed on RDE by cycling between 0 to −0.2 V vs RHE for 10,000 cycles at room temperature. The durability of the catalysts was also assessed by CP at a current density of 10 mA cm−2 for 100 h via coating the catalysts on carbon paper with a noble metal loading of about 24 μg cm−2. EIS measurements were recorded at an overpotential of 27 mV with a 5 mV amplitude and a frequency range from 100,000 to 0.1 Hz on RDE. To evaluate the HER performance of Ru–GaSA/N–C at ampere-level current density, we directly coated the catalysts on the carbon paper with a noble metal loading of about 40 μg cm−2.
CO stripping test
The ECSA of the catalysts was evaluated via a CO stripping test. Briefly, CO adsorption was firstly conducted at 0.1 V vs RHE for 10 min in a CO-saturated 0.1 M HClO4 solution. Then, the electrode was transferred into an N2-saturated 0.1 M HClO4 solution. The CO stripping current was obtained by CV in the potential range of 0–1.0 V vs RHE at a scan rate of 50 mV s−1.
The ECSA was calculated according to the following equation:
where Q is the charge constant of CO stripping (420 μC cm−2), and SCO, V, and M represent the integration area of CO stripping, sweep speed, and noble metal loading, respectively.
Calculation method of TOF
The TOF, s−1 was calculated according to the following equation:
where I, F, and n represent the measured current, Faraday constant (96,485 C mol−1), and molar number of active sites.
n was calculated according to the following equation:
where SCO and V represent the integration area of CO desorption and sweep speed, respectively.
Electrochemical measurements in AEMWE device
The AEMWE device was composed of the cathode (Ru–GaSA/N–C or Pt/C coated on carbon paper), anode (RuO2 coated on Ni foam), and commercial AEM membrane (X37-50, Dioxide Materials Sustainion). The membrane was immersed in 1 M KOH solution for at least 24 h prior to being employed as an electrolyte. The cathode and anode were prepared by air-spraying catalyst ink onto the carbon paper and Ni foam. The Ru and Pt loading was controlled to be about 0.08 mg cm−2 and 0.2 mg cm−2 for the cathode. The RuO2 loading was controlled to be about 1 mg cm−2 for the anode. The MEA was prepared by integrating the cathode, membrane, and anode between two Ti bipolar plates with a torque of 10 N m to complete an AEMWE device. The cell size is 5 × 5 × 4 cm3 with an active area of 1 cm2. 1 M KOH was circulated through the anodic side with a flow rate of 20 mL min−1 by a peristaltic pump (YZ1525, RONGBAI PUMP). The cell was activated at 100 mA cm−2 for 1 h prior to the test. The performance of AEMWE was evaluated by measuring polarization curves from 0.0 A cm−2 to 1.0 A cm−2 at 60 °C on a battery test system (CT-4008T-5V12A-S1-F, NEWARE). The stability of the AEMWE was evaluated by CP test at 1.0 A cm−2 for 170 h. All measurements in AEMWE were recorded without iR-correction.
Electrochemical measurements of the PEMWE device using commercial Pt/C and IrO2 catalysts
To prepare the membrane electrode assembly (MEA), commercial Pt/C and IrO2 were used as cathode and anode catalysts, Nafion 117 membrane was employed as electrolyte, and Pt-coated Ti fiber was used as gas diffusion layers (GDL). Nafion 117 was firstly treated with H2O2 and 0.5 M H2SO4 at 80 °C for 1 h, respectively. The cathode and anode catalysts were directed air sprayed onto the Nafion 117 membrane with a Pt loading of about 0.4 mg cm−2 and IrO2 loading of about 1.5 mg cm−2. The MEA with an active area of 1 cm2 was prepared by hot-pressing the catalyst-loaded membrane and GDL at 130 °C for 2 min under a pressure of 3 MPa, which was sandwiched by two Ti bipolar plates with a torque of 10 N m to complete a PEMWE device. DI water was circulated through the anodic side with a flow rate of 20 mL min-1 by a peristaltic pump (YZ1525, RONGBAI PUMP). The cell was activated at 100 mA cm−2 for 1 h prior to the test. The performance of PEMWE was evaluated by measuring polarization curves from 0.0 to 1.0 A cm−2 at 60 °C on a battery test system (CT-4008T-5V12A-S1-F, NEWARE). All measurements in PEMWE were recorded without iR-correction.
KIEs experiments
The KIE experiments were conducted in 1.0 M KOH/H2O and 1.0 M KOD/D2O solution, respectively. The corresponding current densities at the same overpotential were donated as jH and jD, respectively. The KIE was calculated using the following equation:
Calculation details
We perform total energy and electronic properties calculations using the Vienna Ab initio Simulation Package based on DFT38,39,40. To describe electron-ion interaction, we employ the projector augmented wave method, while the exchange-correlation kernel is treated with the Perdew Burke Ernzerhof form of the generalized gradient approximation41. We also include the DFT + D3 method to effectively characterize weak interactions, which integrates empirical corrections following Grimme’s scheme. A cutoff energy of 500 eV is expanded for the plane wave basis set, ensuring a total energy convergence of less than 1 meV/atom.
Our slab models are constructed with reference to experimental observations. The geometric structures are constructed using a supercell of graphene with dimensions of \(\left(6\times 6\times 1\right)\). Experimental observations suggest an even number of nitrogen atoms coordinated in the systems. In our models, we substituted carbon atoms with nitrogen, resulting in substrates containing four nitrogen atoms. To explore the targeting effect of Ga atoms, we also construct slabs by introducing Ga atoms into the nitrogen-based graphene slab. For simplicity, we label the slab configurations N4, while those containing Ga are denoted as GaN4. Afterward, the Ru13 nanoparticle is absorbed into these slabs. A vacuum slab of 16 Å is employed to separate periodic images along the c direction. We utilize a Monkhorst-Pack k-mesh42 initial set with dimensions (1 × 1 × 1), which is subsequently increased to (3 × 3 × 1) for DOS calculations. Geometry optimization is performed with full relaxation, maintaining the shape and volume constraints until the residual force on each atom is smaller than 0.01 eV/Å. In this study, the catalytic reaction is facilitated by the presence of the Ru13 nanoparticle. Therefore, the corresponding structure-properties relationship, electronic properties, and catalytic performance are calculated upon these configurations containing Ru13 nanoparticles. The properties considered include ELF, HER activity, d band center, and DOS.
Following the optimization of structures, we evaluate catalytic properties by computing the hydrogen adsorption free energy, as determined by Eq. (5) for the Gibbs free energy of H* adsorption43:
Here, \({\Delta E}_{{{ZPE}}}\) is the correction for zero-point energy, and \({\Delta S}_{H}\) denotes the entropy difference between H* adsorption and the H2 molecule. T represents the temperature at 298.15 K. \({\Delta E}_{H}\) refers to the total energy of H adsorption on the system, defined as:
where \({E}_{{H}^{*}}\), \({E}_{{H}_{2}}\), and \({E}_{system}\) represent the total energy of the system with H* adsorption, the H2 molecule, and systems without H*, respectively.
The expression for \({\Delta E}_{{{\rm{ZPE}}}}\) is as follows:
where \({E}_{{{ZPE}}}^{H}\) is calculated based on the vibration frequency of the systems with H adsorption, and \({E}_{{{ZPE}}}^{{H}_{2}}\) denotes the zero-point energy of the final states.
Next, we can express \({\Delta S}_{H}\) conveniently as half of the entropy of the final states under ambient conditions:
\({\Delta E}_{{{ZPE}}}-{T\Delta S}_{H}\) equals −0.24 eV at a temperature of 298.15 K. Consequently, Eq. (5) can be simplified to:
Considering the predominant electron donation originating from the d states of Ga atoms among these configurations, DOS projected onto these d states can be accurately characterized by the d band center. The expression is specified by Eq. (10):
where \(D\left(E\right)\) refers to the DOS corresponding to energy \(E\).
Data availability
The data that support the conclusions of this study are available within the paper and Supplementary information. Source data are provided in this paper.
References
Chong, L. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609–616 (2023).
Shi, Z. et al. Phase-dependent growth of Pt on MoS2 for highly efficient H2 evolution. Nature 621, 300–305 (2023).
Xie, H. et al. A membrane-based seawater electrolyser for hydrogen generation. Nature 612, 673–678 (2022).
Wu, Z.-Y. et al. Non-iridium-based electrocatalyst for durable acidic oxygen evolution reaction in proton exchange membrane water electrolysis. Nat. Mater. 22, 100–108 (2023).
Guo, J. et al. Direct seawater electrolysis by adjusting the local reaction environment of a catalyst. Nat. Energy 8, 264–272 (2023).
Liu, R.-T. et al. Recent advances in proton exchange membrane water electrolysis. Chem. Soc. Rev. 52, 5652–5683 (2023).
Du, N. et al. Anion-exchange membrane water electrolyzers. Chem. Rev. 122, 11830–11895 (2022).
Zeng, L. et al. Anti-dissolution Pt single site with Pt(OH)(O3)/Co(P) coordination for efficient alkaline water splitting electrolyzer. Nat. Commun. 13, 3822 (2022).
Yang, C. et al. Mn-oxygen compounds coordinated ruthenium sites with deprotonated and low oxophilic microenvironments for membrane electrolyzer-based H2-production. Adv. Mater. 35, 2303331 (2023).
Li, Z. et al. Stable rhodium (IV) oxide for alkaline hydrogen evolution reaction. Adv. Mater. 32, 1908521 (2020).
Zeng, L. et al. Single-atom Cr–N4 sites with high oxophilicity interfaced with Pt atomic clusters for practical alkaline hydrogen evolution catalysis. J. Am. Chem. Soc. 145, 21432–21441 (2023).
Zhang, R. et al. Single-atomic platinum on fullerene C60 surfaces for accelerated alkaline hydrogen evolution. Nat. Commun. 14, 2460 (2023).
Zhang, H. et al. Dynamic traction of lattice-confined platinum atoms into mesoporous carbon matrix for hydrogen evolution reaction. Sci. Adv. 4, eaao6657 (2018).
Wan, C. et al. Amorphous nickel hydroxide shell tailors local chemical environment on platinum surface for alkaline hydrogen evolution reaction. Nat. Mater. 22, 1022–1029 (2023).
Wu, Y.-L. et al. Ordered macroporous superstructure of nitrogen-doped nanoporous carbon implanted with ultrafine Ru nanoclusters for efficient pH-universal hydrogen evolution reaction. Adv. Mater. 33, 2006965 (2021).
Kweon, D. H. et al. Ruthenium anchored on carbon nanotube electrocatalyst for hydrogen production with enhanced Faradaic efficiency. Nat. Commun. 11, 1278 (2020).
Li, G. et al. The synergistic effect of Hf-O-Ru bonds and oxygen vacancies in Ru/HfO2 for enhanced hydrogen evolution. Nat. Commun. 13, 1270 (2022).
Zhou, S. et al. Boosting hydrogen evolution reaction by phase engineering and phosphorus doping on Ru/P–TiO2. Angew. Chem. Int. Ed. 61, e202212196 (2022).
Mahmood, J. et al. An efficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction. Nat. Nanotechnol. 12, 441–446 (2017).
Zhang, J. et al. Competitive adsorption: reducing the poisoning effect of adsorbed hydroxyl on Ru single-atom site with SnO2 for efficient hydrogen evolution. Angew. Chem. Int. Ed. 61, e202209486 (2022).
Zhou, C. et al. RuCoOx nanofoam as a high-performance trifunctional electrocatalyst for rechargeable zinc-air batteries and water splitting. Nano Lett. 21, 9633–9641 (2021).
Rong, H., Ji, S., Zhang, J., Wang, D. & Li, Y. Synthetic strategies of supported atomic clusters for heterogeneous catalysis. Nat. Commun. 11, 5884 (2020).
Zhao, X. et al. A double atomic-tuned RuBi SAA/Bi@OG nanostructure with optimum charge redistribution for efficient hydrogen evolution. Angew. Chem. Int. Ed. 62, e202300879 (2023).
Wang, L. et al. Theory-guided design of electron-deficient ruthenium cluster for ampere-level current density electrochemical hydrogen evolution. Nano Energy 115, 108694 (2023).
Gao, T. et al. Understanding the atomic and defective interface effect on ruthenium clusters for the hydrogen evolution reaction. ACS Catal. 13, 49–59 (2023).
Li, Z. et al. Iridium single-atom catalyst on nitrogen-doped carbon for formic acid oxidation synthesized using a general host-guest strategy. Nat. Chem. 12, 764–772 (2020).
Su, P. et al. Exceptional electrochemical HER performance with enhanced electron transfer between Ru nanoparticles and single atoms dispersed on a carbon substrate. Angew. Chem. Int. Ed. 60, 16044–16050 (2021).
Zhao, Y. et al. Modulating Pt–O–Pt atomic clusters with isolated cobalt atoms for enhanced hydrogen evolution catalysis. Nat. Commun. 13, 2430 (2022).
Kuang, Y., Qiao, W., Yang, F. & Feng, L. Electrochemical hydrogen evolution efficiently boosted by interfacial charge redistribution in Ru/MoSe2 embedded mesoporous hollow carbon spheres. J. Energy Chem. 85, 447–454 (2023).
Hu, J. et al. Electrocatalysis induced reconstruction of RuNiPO for highly efficient hydrogen evolution reaction. J. Energy Chem. 87, 24–31 (2023).
Liang, L. et al. Ultra-small platinum nanoparticles segregated by nickle sites for efficient ORR and HER processes. J. Energy Chem. 65, 48–54 (2022).
Jiang, W. et al. Photocatalyst for high-performance H2 production: Ga-doped polymeric carbon nitride. Angew. Chem. Int. Ed. 60, 6124–6129 (2021).
Xie, Y. et al. The nature of the gallium–gallium triple bond. J. Am. Chem. Soc. 120, 3773–3780 (1998).
Zhu, T. et al. Amorphous ruthenium–selenium nanoparticles as a pH-universal catalyst for enhanced hydrogen evolution reaction. ACS Catal. 14, 1914–1921 (2024).
Sun, K. et al. Interfacial water engineering boosts neutral water reduction. Nat. Commun. 13, 6260 (2022).
Mu, X. et al. Constructing symmetry-mismatched RuxFe3−xO4 heterointerface-supported Ru clusters for efficient hydrogen evolution and oxidation reactions. Nano Lett. 24, 1015–1023 (2024).
Burke, M. S., Kast, M. G., Trotochaud, L., Smith, A. M. & Boettcher, S. W. Cobalt-iron (Oxy)hydroxide oxygen evolution electrocatalysts: the role of structure and composition on activity, stability, and mechanism. J. Am. Chem. Soc. 137, 3638–3648 (2015).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Nørskov, J. K. et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 152, J23–J26 (2005).
Acknowledgements
This study was financially supported by National Science Fund for Distinguished Young Scholars (no. 52025133 (S.G.)), National Key R&D Program of China (no. 2022YFE0128500 (S.G.)), the National Natural Science Foundation of China (nos. 22305010 (C.Z.), 52261135633 (S.G.), 52302207 (L.L.), and 22309004 (F.L.)), the China National Petroleum Corporation-Peking University Strategic Cooperation Project of Fundamental Research, the Beijing Natural Science Foundation (no. Z220020 (S.G.)), Tencent Foundation through the XPLORER PRIZE, CNPC Innovation Found (no. 2021DQ02-1002 (S.G.)) and the China Postdoctoral Science Foundation (nos. 2023M730051 (C.Z.) and 2023M730029 (L.L.)). The authors thank the photoemission photo endstations BL14W1 in the Shanghai Synchrotron Radiation Facility (SSRF) and the 1W1B beamline at BSRF for the help with XAFS characterizations.
Author information
Authors and Affiliations
Contributions
S.G. conceived the project. C.Z. designed the research, and performed the material synthesis, characterization, and electrochemical tests. C.Z., Z.D., Y.C., and L.Z. participated in the assembly and tests of AEMWE. Q.Z. and L.G. performed the HAADF-STEM characterization. J.S. conducted the DFT calculations. Y.H., L.L., W.Z., and F.L. participated in part of the basic experiments. C.Z. wrote the paper. S.G. and M.L. guided the paper writing. All authors participated in the project discussions and gave comments on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ligang Feng, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, C., Shi, J., Dong, Z. et al. Oxophilic gallium single atoms bridged ruthenium clusters for practical anion-exchange membrane electrolyzer. Nat Commun 15, 6741 (2024). https://doi.org/10.1038/s41467-024-51200-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51200-4
- Springer Nature Limited