

Abstract
The skeleton has been suggested to function as an endocrine organ controlling whole organism energy balance, however the mediators of this effect and their molecular links remain unclear. Here, utilizing Schnurri-3−/− (Shn3−/−) mice with augmented osteoblast activity, we show Shn3−/−mice display resistance against diet-induced obesity and enhanced white adipose tissue (WAT) browning. Conditional deletion of Shn3 in osteoblasts but not adipocytes recapitulates lean phenotype of Shn3−/−mice, indicating this phenotype is driven by skeleton. We further demonstrate osteoblasts lacking Shn3 can secrete cytokines to promote WAT browning. Among them, we identify a C-terminal fragment of SLIT2 (SLIT2-C), primarily secreted by osteoblasts, as a Shn3-regulated osteokine that mediates WAT browning. Lastly, AAV-mediated Shn3 silencing phenocopies the lean phenotype and augmented glucose metabolism. Altogether, our findings establish a novel bone-fat signaling axis via SHN3 regulated SLIT2-C production in osteoblasts, offering a potential therapeutic target to address both osteoporosis and metabolic syndrome.
Similar content being viewed by others

Introduction
Thermogenic beige and brown adipocytes dissipate heat through uncoupled respiration1,2,3. It had been widely appreciated that beiging or browning of white adipose tissue (WAT), induced by external stimuli such as cold exposure, has beneficial impact on whole body energy homeostasis and protects from the development of metabolic syndrome. A growing body of evidence suggests that inter-organ crosstalk also plays a vital role in regulation of WAT browning under physiological and pathological conditions. For instance, FGF21, a hepatic cytokine, acts on the central nervous system (CNS) to indirectly activate WAT browning against metabolic syndrome through sympathetic innervation4. Similarly, another immune-adipose crosstalk mediator termed meteorin-like (Metrnl) increased eosinophil-dependent interleukin 4 and 13 synthesis which induces adipocytes to assume a thermogenic phenotype, consequently improving glucose tolerance and reducing weight gain5.
In addition to these mediators, bone forming osteoblasts in the skeleton have been suggested to affect adipose tissue and systemic energy metabolism through the secretion of bone derived hormones, or osteokines6, such as osteocalcin7,8. Also, loss of mTORC1 signaling in osteoblasts results in significantly decreased adiposity and protects mice from diet-induced-obesity (DIO)9,10. Interestingly, this effect occurred even in osteocalcin deficient mice7, leading us to hypothesize that additional undiscovered skeletal regulators of systemic energy metabolism exist6,11. Upregulated hedgehog signaling in osteoblasts led to a reduction in adipose tissue through bone-derived PTHrP, influencing systemic energy expenditure12. In another example, osteocyte-derived sclerostin (SOST) was observed to regulate adipocyte hypertrophy and beige adipogenesis as seen in loss-of-function mice or after treatment with recombinant SOST protein13.
Schnurri-3 (SHN3, also Hivep3) is an adapter protein that acts in a cell intrinsic manner in osteoblasts to suppress their bone forming activity14,15. Previously, we reported that not only does loss of Shn3 augment osteoblast activity, but it also promotes the ability of osteoblasts to reshape their local vascular microenvironment by increasing the production of SLIT3, which in this context functioned as a novel osteoblast-derived angiogenic factor16. In studying this, we noted that in addition to regulating Slit3, deletion of Shn3 was also able to regulate the expression of multiple secreted factors in osteoblasts. Given that the only phenotype of Shn3-null mice (Shn3−/− mice) observed to date is a markedly high bone mass due primarily to elevated osteoblast activity, with no other apparent developmental abnormalities outside of the skeleton14,16, we hypothesized that SHN3-deficient mice represented an ideal tool to identify osteolineage-derived osteokines regulating systemic health, especially including putative yet-to-be identified osteokines regulating energy metabolism.
In this study, we utilized Schnurri-3−/− (Shn3−/−) mice to probe the general relationship between bone formation and metabolic syndrome. Shn3−/−mice display resistance against high-fat diet-induced obesity (DIO) with improved glucose homeostasis and insulin sensitivity associated with enhanced browning of white adipose tissue (WAT). By generating osteoblastic (Shn3Osx mice) and adipocytic (Shn3Adipoq mice) conditional Shn3 knockout murine model, we found Shn3Osx mice but not Shn3Adipoq mice recapitulates the obesity resistance phenotype seen in Shn3−/− mice, suggesting that this phenotype is driven by the skeleton. Through in vitro co-culture and in vivo fat-pad transplantation assays, we interpret that cytokines secreted by activated osteoblasts lacking Shn3 expression are ablet to promote WAT browning. Among them, we identify a C-terminal fragment of SLIT2 (SLIT2-C) as a Shn3-regulated factor secreted by osteoblasts that regulates WAT browning, with osteoblasts serving as the major source of systemic circulating SLIT2-C during HFD exposure. Lastly, we take advantage of AAV-mediated in vivo silencing of Shn3 in mice and found these mice phenocopied the lean phenotype and augmented glucose metabolism in Shn3-deficient mice. We here identify therapeutic value of SHN3 silencing in treating diseases relate to both bone loss and obesity-related metabolic disorders by skeleton-derived Slit2 production.
Results
Shn3 −/− mice are resistant to DIO and impaired glucose homeostasis
To first examine the impact of SHN3 on global energy metabolism, we challenged adult Shn3−/− mice on the C57BL6/J background and their littermate controls with high-fat-diet (HFD) feeding. As expected, Shn3−/− mice on HFD still displayed a strikingly high bone mass phenotype (Supplementary Fig. 1a). More interestingly, Shn3−/− mice displayed resistance to DIO relative to control-fat-diet (CFD) condition (Fig. 1a and Supplementary Figs. 1b–c). In line with this, inguinal, epididymal WAT and intrascapular brown adipose tissue (BAT) depots were smaller in Shn3−/− mice, with reduced adipocyte size noted on histology (Fig. 1b, c). There was also significant alleviation of hepatic steatosis in Shn3−/−mice (Fig. 1c). This protection from DIO was further supported by an assessment of body composition showing significant differences in fat mass between groups without significant alteration in lean mass (Fig. 1d, e). The same studies were conducted in Shn3−/− mice on both the female C57BL6/J and BALB/c background exposed to HFD with similar results (Supplementary Figs. 1c–g).

All samples were harvested from C57BL6/J background male WT (n = 7) and Shn3−/−mice (n = 6) after HFD feeding for 12 weeks. a Body weight curve obtained during 12 weeks HFD feeding starting from 6-weeks of age (P values directly labeled above the time points). b Representative general view of liver, BAT, iWAT and eWAT of WT and Shn3−/− mice. c Representative histological sections of liver, BAT, iWAT and eWAT with H&E staining from 3 individual repeated experiments. Scale bar=200um. d Tissue wet weight of iWAT, eWAT, BAT and liver. e Body composition quantified through Echo-MRI. f Results of intraperitoneal glucose tolerance test, insulin tolerance test and quantification of area under curve (AUC) (n = 6 for each group). g Representative histological sections of pancreas islets with H&E staining from 3 individual slides. Scale bar=100um. Results were presented as mean ± s.d. and analyzed by two-way ANOVA with Bonferroni’s multiple comparisons test (a, f) or unpaired, two-tailed Student’s t test (d–e). *P < 0.05, **P < 0.005, ***P < 0.0005. Source data are provided as a Source Data file.
Since obesity is an important independent risk factor for type-2-diabetes17, we next examined whether Shn3 ablation can also rescue DIO-induced insulin resistance. Indeed, Shn3−/−mice displayed an improved glucose tolerance and reduced insulin resistance relative to the littermate controls (Fig. 1e, f). Consistent with this, the size of the pancreatic islets was reduced in Shn3−/−mice (Fig. 1g)18,19. Altogether, these results revealed that in addition to mineralization, SHN3 functions as a critical regulator of adiposity and glucose homeostasis after HFD exposure, indicating a SHN3-mediated link between skeletal homeostasis and systemic energy metabolism.
Increased energy expenditure and WAT browning in Shn3 −/− mice
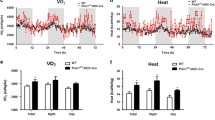
To understand the basis of the resistance to DIO seen in Shn3−/−mice, HFD-fed Shn3−/−mice and littermate controls were individually housed in metabolic cages for monitoring. Through indirect calorimetric analyzes, we discovered significantly increased energy expenditure (EE) in Shn3−/− mice relative to littermate controls during both the light and dark cycle (Fig. 2a–c). In addition, we did not detect differences in respiratory exchange ratio and locomotor activity between Shn3−/− mice and controls (Supplementary Figs. 2a, b). Intriguingly, despite their obesity resistance phenotype, Shn3−/− mice displayed significantly increased food intake (Supplementary Fig. 2c), indicating that regulation of appetite is not responsible for and is most likely secondary to the overall metabolic phenotype.

Individuals measured by indirect calorimetry were WT (n = 7) and Shn3−/− mice (n = 6) after 12-week-HFD exposure. a Calculated energy expenditure of WT and Shn3−/−mice under room temperature within a 120-hour-period. b Bar graph of overall, light cycle and dark cycle energy expenditure of WT and Shn3−/−mice under room temperature. c Regression-based analysis of energy expenditure against total body mass. d Gross view of iWAT from WT and Shn3−/−mice after 12-week-HFD exposure, black arrow pointing to inguinal lymph nodes. e Representative H&E staining of iWAT sections obtained from WT and Shn3−/− mice after 12-week-HFD exposure. Scale bar=200um. Results were selected from 3 repeated experiments. f Thermogenic gene expression level relative to Gapdh of iWAT quantified by qPCR in WT and Shn3−/− mice. g Average adipocyte surface area of WT and Shn3−/− mice. Results were presented as mean ± s.d. and analyzed by unpaired, two-tailed Student’s t test (b and f) or two-way ANOVA with Bonferroni’s multiple comparisons test (a and g). Source data are provided as a Source Data file.
Having excluded these other possible explanations for the resistance to DIO, we next considered whether increases in the browning of white adipose tissue could account for this phenotype. Thus, we next assessed whether increases in WAT beige adipocytes contributed to the DIO resistance phenotype of Shn3−/− mice. Inguinal WAT (iWAT) in Shn3−/− mice displayed a darker color upon gross examination suggestive of increased browning (Fig. 2d). Upon histologic examination, the per cell cross-sectional area of iWAT adipocytes was markedly decreased, and an increased number of multilocular adipocytes morphologically consistent with beige adipocytes were noted in Shn3−/− iWAT (Fig. 2e, g). Consistent with this, marker genes linked to adipocyte thermogenesis and beige adipocytes such as Ucp1, Pgc1a, Cidea and Cox8b were highly upregulated in iWAT from Shn3−/− mice compared to controls (Fig. 2f). In parallel with these alterations in gene expression, mitochondrial respiration was enhanced in primary iWAT cells of Shn3−/− mice (Supplementary Fig. 2d).
To determine the functional contribution of brown and beige adipocytes to DIO resistance in Shn3−/− mice, mice were challenged with HFD under thermoneutral housing conditions (30˚C), which will blunt the metabolic contribution of thermogenic adipocytes. The resistance of Shn3−/− mice to DIO seen under standard housing conditions (24˚C) was strikingly dampened under thermoneutral housing, as the body mass curve showed no significant differences between groups (Supplementary Fig. 2e). Similarly, in contrast to observations under standard housing conditions, a similar level of adiposity and expression level of thermogenic genes in WAT was seen in Shn3−/− mice in thermoneutral housing relative to littermate controls (Supplementary Figs. 2f–g). Histological examination of iWAT indicates that the increase in beige adipocytes seen under standard housing in Shn3−/−mice disappeared, demonstrating that beige adipocytes remain responsive to thermal suppression (Supplementary Fig. 2h). The protection of Shn3−/− mice from DIO-linked insulin resistance was also suppressed under thermoneutral housing conditions (Supplementary Figs. 2i–j). Taken together, these results suggested that enhanced WAT browning is a key effector of resistance to DIO in Shn3−/− mice.
Deletion of Shn3 in osteoblasts but not adipocytes recapitulates resistance to DIO
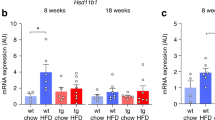
Shn3 has previously been shown to function in a cell intrinsic manner in osteoblasts to regulate bone formation15,16. Here, we sought to determine if the effect of SHN3 to regulate systemic energy metabolism was also intrinsic to osteoblasts, and therefore tied to regulation of bone anabolism, or reflected a novel function of SHN3 in adipocytes. To this end, a Shn3-floxed conditional allele was bred to both Osterix-Cre (Osx-Cre) targeting osteoblast-lineage cells and Adiponectin-Cre (Adipoq-Cre) targeting adipocytes20. The resulting Shn3Osx and Shn3Adipoq mice were then subjected to metabolic studies. Separately, a separate cohort including Osx-Cre controls was also utilized given suggested best practices when working with this cre line21. We also assessed the Shn3 deletion efficiency in Shn3Osx and Shn3Adipoq mice. As shown in Supplementary Figs. 3a–d, Shn3Osx mice displayed marked decrease of Shn3 expression in bone (Supplementary Fig. 3a) but not adipose tissue (Supplementary Fig. 3b). Shn3Adipoq mice showed a significant decrease of Shn3 expression in iWAT (Supplementary Fig. 3c), but not in osteolineage cells of the skeleton (Supplementary Fig. 3d).
Similar to findings in Shn3−/− mice, Shn3Osx mice exhibited significantly increased bone mass, decreased body weight gain, reduced peripheral WAT (inguinal and epididymal) accumulation and were protected from the development of hepatic steatosis after HFD exposure, whereas Shn3Adipoq mice did not recapitulate the obesity resistance phenotype seen in Shn3−/− mice after 12 weeks of exposure to HFD (Fig. 3a–c and Supplementary Figs. 3e–j). This anti-obesity effect of Shn3 in bone was further confirmed through histological examination of eWAT, iWAT and liver, showing adipocytes with a markedly reduced cross-sectional area and an attenuation of hepatic steatosis (Fig. 3d and Supplementary Fig. 3k). Consistent with the overall effects on weight gain after HFD, none of these metabolic phenotypes observed in Shn3Osx mice were seen in Shn3Adipoq mice (Fig. 3c, lower and Supplementary Fig. 3g). This overall indicated that SHN3 acts in osteoblasts to mediate bone to adipose inter-organ communication regulating systemic energy balance.

a Body weight curve of male Shn3OSX and Shn3f/f mice after HFD for 12 weeks (n = 10 for each group, P values directly labeled above the time points). b Body weight curve of Shn3Adipoq and Shn3f/f mice (n = 10) after HFD for 12 weeks. c Tissue wet weight of iWAT, eWAT, BAT and liver (Shn3OSX and Shn3f/f, upper, n = 10 for Shn3f/f group and n = 8 for Shn3OSX group. Shn3Adipoq and Shn3f/f, lower, n = 8 for Shn3f/f group and n = 7 for Shn3Adipoq group.). d Representative histological sections of iWAT, eWAT and liver from 3 slides of Shn3OSX and Shn3f/f mice, respectively. Scale bar=200 um. e–f Results of intraperitoneal glucose tolerance and insulin tolerance tests (P values directly labeled above the time points) and the area under the curve for Shn3Osx and Shn3f/f mice is shown (n = 6 for each group). g Serum C-peptide level measured by sandwich ELISA of Shn3Osx (n = 8) and Shn3f/f (n = 10) individuals. h Representative H&E and insulin IHC of pancreas islet from 3 individual slides. Scale bar=200 um. i–j Histomophometric quantification result of islet area/tissue area and islet number (n = 5 for each group). (k) Quantification of serum triacylglycerol (TG, n = 7 for Shn3Osx group and n = 8 for Shn3f/f group), free fatty acid (FFA, n = 8 for each group) level and adiponectin (n = 8 for each group) of Shn3OSX and Shn3f/f mice through ELISA assays. l Representative H&E sections (Scale bar=500 um) and Ucp-1 IHC results (Scale bar=200 um) of iWAT from 3 individual slides of Shn3Osx and Shn3f/f individuals, respectively. m Representative immunoblots of UCP1 of iWAT obtained from HFD-fed Shn3Osx and Shn3f/f mice. n Quantification of Ucp-1 expression level in iWAT from HFD-fed Shn3Osx (n = 7) and Shn3f/f (n = 8) mice. o Thermogenic gene expression level relative to Gapdh in iWAT of Shn3Osx (n = 10) and Shn3f/f mice (n = 7). p Lipolytic gene expression in iWAT harvested from HFD-fed Shn3Osx and Shn3f/f mice (n = 5 for each group). Results were presented as mean ± s.d., analyzed by unpaired, two-tailed Student’s t test (lower of c, g, i–k, n–p), one-way ANOVA with Tukey’s test (upper of c), two-way ANOVA with Bonferroni’s multiple comparisons test (a, b, e, f). *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001 (Shn3f/f vs. Shn3Osx). #P < 0.05, ##P < 0.005, ###P < 0.0005 (Osx Cre vs. Shn3Osx). Source data are provided as a Source Data file.
Shn3osx mice similarly displayed a rescue of HFD induced metabolic dysfunction. After HFD challenge, Shn3Osx mice exhibited enhanced glucose clearance compared to Shn3f/f controls in an intraperitoneal glucose tolerance test (IGTT) (Fig. 3e, Fig. 3f and Supplementary Fig. 3l, left). Similarly, Shn3Osx mice exhibited improved insulin sensitivity during an insulin tolerance test (ITT) (Fig. 3e, f and Supplementary Fig. 3l, right). Consistent with this, levels of endogenous insulin C-peptide were decreased in Shn3Osx mice (Fig. 3g), and hyperplasia of pancreatic islets was attenuated without alteration in islet numbers in Shn3Osx mice (Fig. 3h–j)17. Moreover, Shn3-deficiency in osteoblasts also improved circulating lipid profiles and increased levels of circulating adiponectin (Fig. 3k). These effects were not seen in Shn3Adipoq mice (Supplementary Fig. 3h), indicating that SHN3 acts in osteoblast and not adipogenic lineage cells to regulate energy metabolism.
Mirroring findings in Shn3−/− mice, enhanced WAT browning was detected in Shn3Osx mice, including increased expression of the browning-associated marker UCP1 by both iWAT immunohistochemistry (IHC) and immunoblotting (Fig. 3l–n). In addition to UCP1, the expression level of the other thermogenic or beige adipocyte markers was also elevated in iWAT of Shn3Osx mice (Fig. 3o). In parallel with this, elevated lipolytic gene expression in WAT of Shn3Osx mice was seen, consistent with the attenuation of hyperlipidemia in these mice (Fig. 3p). Collectively, these observations indicate that an osteoblast mediated bone to fat signaling axis regulates systemic energy metabolism, resistance to DIO and metabolic syndrome.
SHN3-regulated secreted factors produced by osteoblasts mediate WAT browning
The functional interaction observed between bone and white adipose tissue raised interest in identifying SHN3 regulated hormones produced by osteoblasts that mediate adipose tissue browning as bone and subcutaneous WAT are anatomically separate tissues. To test our hypothesis that bone is able to generate secreted mediators of adipocyte browning, we set up both in vitro (diagram in Fig. 4a) and in vivo assays (diagram in Fig. 4c) to evaluate whether SHN3 regulates an endocrine pathway in osteoblasts controlling WAT browning. In vitro, co-culture of differentiating adipocytes with Shn3Osx osteoblasts enhanced the expression of gene programs associated with beige adipocytes, including Ucp1, Pgc1a, Cidea, Dio2 and Cox8b, compared with co-culture with control osteoblasts (Fig. 4a, b). Thus, SHN3 deficiency results in the ability of osteoblasts to directly promote adipocyte browning in vitro.

a Schematic diagram of the co-culture assay. b Thermogenic gene expression in co-cultured differentiated beige adipocytes (n = 4 for each group). Genotypes of osteoblasts are indicated. Results were collected from 3 repeated experiments. c Schematic diagram of transplantation assay. d Graft weight of fat graft transplanted into Shn3Osx and Shn3f/f hosts. e Representative H&E sections and UCP-1 IHC results of fat allografts implanted into Shn3Osx and Shn3f/f hosts for 4 weeks. respectively. Results were collected from 3 individual slides. f Ucp1 gene expression level relative to Gapdh of fat graft transplanted into Shn3Osx and Shn3f/f hosts (n = 4 for each group). Results were presented as mean ± s.d., analyzed by unpaired, two-tailed Student’s t test (b, d, f). Diagram (a) and (c) were generated through utilizing BioRender Apps. Source data are provided as a Source Data file.
To then extend this model and ensure that the increased browning phenotypes observed in Shn3Osx mice reflect ongoing bone to adipose crosstalk, intact iWAT fat pads (allograft input weight around 400 ± 10 mg) were dissected from WT donors and transplanted into dorsal subcutaneous pockets in Shn3Osx and control hosts for 4 weeks before analysis (Fig. 4c). As expected, the graft weight of iWAT transplanted into Shn3Osx hosts was significantly less relative to grafts in control hosts (Fig. 4d). Consistent with this, adipose grafts implanted into Shn3Osx hosts exhibited smaller adipocyte cross sectional area and expressed higher levels of UCP1 as seen by both IHC and transcript analysis, as well as the other beige adipocyte markers including Pgc1a, Cidea, Dio2 and Cox8b (Fig. 4e, f). Thus, SHN3-regulated osteokines, osteoblast secreted hormones, remotely regulate WAT browning and systemic energy metabolism.
SLIT2 is an osteokine secreted by osteoblasts that protects mice from DIO
To identify osteoblast-derived factors associated with WAT browning, we analyzed RNA-sequencing (RNA-seq) transcriptional profiling of Shn3-deficient and control primary osteoblasts16, filtering differentially expressed genes to focus solely on secreted proteins. Among them, robust expression of Slit2, an axon cue which has been studied as a thermogenic factor acting on beige adipocytes22, was noted in control osteoblasts, and Slit2 expression was increased by SHN3 deficiency (Fig. 5a).

a Heatmap showing relative RNA levels of several cytokines in primary osteoblasts isolated from WT and Shn3−/− mice (n = 3). b Immunoblotting for SLIT2 in osteoblast lysates, serum, osteoblast supernatant. The red and bold blue boxes indicate regions that were cut for further LC-MS studies. Results were selected from 3 repeated experiments. c Matched peptides of SLIT2 and the related amino acid sequence detected in murine sera. d ELISA measurement of SLIT2 in conditioned media from WT and Shn3−/− osteoblasts (n = 4 for each group). (e) Immunoblotting demonstrating the levels of SLIT2-C in the serum of HFD-fed Shn3−/− and WT mice and (f) ELISA measurement of circulating SLIT2 levels in Shn3−/− and WT mice after 12-weeks of HFD exposure (n = 9 for WT group and n = 11 for Shn3−/−group). g Immunoblotting for SLIT2-C in the serum of HFD-exposed Slit2Osx and Slit2f/f mice. h ELISA results of circulating SLIT2 levels in Slit2f/f and Slit2Osx mice (n = 6 for each group). i Body weight curve of HFD-exposed Slit2Osx and Slit2f/f mice (n = 6 for each group). j Representative gross images of iWAT obtained from Slit2Osx and Slit2f/f mice. k tissue wet weight of iWAT, eWAT, liver and BAT obtained from Slit2Osx and Slit2f/f mice (n = 6 for each group). l Representative histological sections of iWAT, eWAT and liver from 3 individual slides of Slit2Osx and Slit2f/f mice after a 10-week-HFD challenge. Scale bar=100 um. m Thermogenic gene expression level relative to Gapdh in iWAT of HFD-fed Slit2Osx and Slit2f/f mice (n = 4 for each group). Results were presented as mean ± s.d., analyzed by unpaired, two-tailed Student’s t test (e, f, h, k and m) or two-way ANOVA with Bonferroni’s multiple comparisons test (i). *P < 0.05. Source data are provided as a Source Data file.
To demonstrate that osteoblasts secrete SLIT2, we first analyzed Slit2 expression patterns in mice23. Bone and differentiated osteoblasts were both noted to express Slit2 (Supplementary Figs. 4a–b). Furthermore, we performed immunoblotting using a previously validated anti-SLIT2 antibody22 combined with mass spectrometry analysis, demonstrating that in addition to a full length SLIT2, osteoblasts produce a cleaved C-terminal fragment (SLIT2-C) (Fig. 5b, c and Supplementary Figs. 4b–c). Likewise, the same C-terminal fragment was present in circulation. The specific C-terminal SLIT2 peptides identified mirrored those identified in a prior study22. In line with the SHN3-mediated suppression of Slit2 expression observed here, SHN3-deficient osteoblasts secreted greater amounts of SLIT2-C than WT controls, quantified by an ELISA assay validated with a spike and recovery test (Fig. 5d and Supplementary Figs. 4d–f). Considering SHN3 is a critical suppressor of Wnt signaling in osteogenesis, we further found that activation of Wnt signaling indeed drove increased Slit2 expression in osteoblasts (Supplementary Fig. 4g). More importantly, Shn3−/− animals displayed increased circulating serum levels of SLIT2 (Fig. 5e, f). Given that SHN3 largely functions in osteoblasts, this raises the possibility that osteoblasts are the major source of circulating SLIT2 and thereby the key cell type determining overall serum SLIT2 levels.
To address this, we generated mice with osteolineage-specific and adipolineage-specific ablation of Slit2 (Slit2Osx mice and Slit2Adipoq mice, respectively) showing that only Slit2Osx mice exhibited a significant decrease in circulating SLIT2-C (Fig. 5g, h and Supplementary Figs. 4h–i). Meanwhile, specific deletion of Slit2 in osteoblasts did not indirectly alter the expression level of Slit2 or its receptors (Robo1 and Robo2) in adipose tissue (Supplementary Figs. 4j–l). These results indicated that osteoblasts are the major physiological source of circulating SLIT2.
Functionally, osteoblast-derived SLIT2 mediated systemic obesity resistance as Slit2Osx mice gained more weight compared to littermate controls in response to a HFD challenge, despite not displaying a bone phenotype (Fig. 5i and Supplementary Figs. 4m–p). WAT from inguinal or epididymal depots was increased in weight, and increased levels of hepatic steatosis were observed in Slit2Osx mice (Fig. 5j–l and Supplementary Figs. 4q–r). Upon histologic analysis, iWAT and eWAT adipocytes displayed increased cross-sectional area in Slit2Osx mice (Fig. 5l and Supplementary Fig. 4s). Additionally, there was a notable decrease in the expression of thermogenic genes including Ucp1, Pgc1a and Dio2 (Fig. 5m and Supplementary Fig. 4t). Collectively, our results identified that SLIT2 is a novel osteokine produced by osteoblasts in a SHN3 regulated manner that mediates WAT browning to increase energy expenditure.
AAV-mediated skeletal SHN3 depletion reduced development of DIO and metabolic syndrome
To investigate whether in vivo gene therapy mediating Shn3 silencing could reduce adiposity and related metabolic disorders during the development of diet-induced obesity, we used a replication-defective AAV serotype-9 (rAAV-9) to deliver a Shn3-silencing construct in osteoblastic lineage cells, following previously reported approaches24. We created rAAV-9 vectors containing plasmids carrying either a Shn3-targeting artificial microRNA (rAAV9-amiR-Shn3) or a miRNA-control (vector control, rAAV9-amiR-Ctrl) as described before24,25 and administered these vectors to HFD-fed WT mice. In vitro IVIS imaging revealed that rAAV9 effectively targeted skeletal sites, with the highest level of eGFP expression observed in lower limbs, while no fluorescence signal was detected in adipose tissue (Fig. 6a, b). Furthermore, mice treated with rAAV9-amiR-Shn3 exhibited a significant increase in bone mass compared to mice treated with rAAV9-amiR-Ctrl, consistent with previous reports24 (Fig. 6c, d, Supplementary Fig. 5a). In line with the metabolic phenotype seen in Shn3-deficient mice, treatment with rAAV9-amiR-Shn3 also elevated circulating SLIT2-C levels, and significantly decreased body weight in HFD-fed WT mice (Fig. 6e and Supplementary Figs. 5b–c). Additionally, mice in the rAAV9-amiR-Shn3 group showed decreased adiposity in various depots, including iWAT, eWAT, and BAT, and liver (Fig. 6f, g). Importantly, similar to our observations in Shn3−/− mice and Shn3Osx mice, animals treated with rAAV9-amiR-Shn3 exhibited increased numbers of beige adipocytes, reduced adiposity in iWAT, and significantly improved steatosis (Fig. 6h). Moreover, the expression of thermogenic genes in WAT derived from the rAAV9-amiR-Shn3 group was markedly elevated (Fig. 6i). Finally, rAAV9-amiR-Shn3 injection improved glucose tolerance and insulin sensitivity in diet-induced obese mice (Fig. 6j). In conclusion, our results provide proof-of-concept demonstrating that postnatal ablation of Shn3 via viral gene therapy or alternative methods might be able to treat osteoporosis, obesity, and related metabolic disorders simultaneously.

Representative IVIS imaging of (a) whole body and (b) major organs from murine models after the injection of a single dose of 4 × 1011 GC rAAV9-amiR-Shn3 or rAAV9-amiR-Ctrl (Fluorescence signal intensity labeled as colored symbols on the right. Yellow represents strong signal and red represents dim signal). μCT reconstruction and quantification of (c) femoral trabecular and (d) cortical bone of rAAV9-amiR-Shn3 or rAAV9-amiR-Ctrl administered mice (n = 5 for each group). Scale bar=1 mm. e Body weight curve of rAAV9-amiR-Shn3 and rAAV9-amiR-Ctrl injected mice over a 12-week HFD exposure (n = 8 for each group). f General view of inguinal adipose fat pad and intrascapular brown adipose tissue. g Tissue wet weight of iWAT, eWAT, liver and BAT obtained from rAAV9-amiR-Shn3 and rAAV9-amiR-Ctrl injected mice (n = 8 for each group). h Representative histological H&E results of iWAT and liver from rAAV9-amiR-Shn3 and rAAV9-amiR-Ctrl group. Scale bar=100 um. Results were selected from 3 repeated experiments. i Thermogenic gene expression level relative to Gapdh in iWAT of mice treated with rAAV9-amiR-Shn3 or rAAV9-amiR-Ctrl (n = 6 for each group). j IGTT and ITT results at 10 weeks after rAAV injection (n = 8 for each group). Results were presented as mean ± s.d., analyzed by unpaired, two-tailed Student’s t test (c–d, g, i) or two-way ANOVA with Bonferroni’s multiple comparisons test (e, j). *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001. Source data are provided as a Source Data file.
Discussion
Disrupted energy balance is central to the pathogenesis of obesity26,27. Multiple organs and tissues work together to sense and respond to nutritional challenges to maintain normal systemic metabolic homeostasis. Previous studies have identified genetic alterations that concurrently regulate both bone volume and systemic energy homeostasis, such as SOST and mTORC19,10,13. Clinical data on the association between osteoporotic/osteopetrotic diseases and adiposity/BMI further suggests a relationship between skeletal metabolism and systemic energy metabolism28,29,30,31. Despite evidence for this bone to adipose link regulating energy metabolism, the skeletal derived hormones acting on adipose tissue and the mechanism by which bone impacts adipose function remain poorly understood. Herein, we identified that, in addition to the known role of Shn3 in the skeleton to suppress bone formation, Shn3 further acts in bone to suppress a bone to fat signaling axis regulating WAT browning. In addition to increasing bone mass, ablation or inhibition of Shn3 in osteoblasts protected mice from DIO and associated impairments in glucose homeostasis, partially through the secretion of a bone-derived thermogenic factor, SLIT2. Thus, this study identifies a SHN3-SLIT2 axis in osteoblasts mediating functional interactions between bone and fat.
Bones produce secreted factors that both act locally to regulate skeletal function itself and, more intriguingly, remotely act as hormones to control the physiology of other systems in an endocrine manner. Slits were first identified as genetic regulators of larval cuticle patterning in the Drosophila embryo and their role in directing commissural axons during the early development of central nervous system has been widely studied32. Our recent study found that osteoblast-secreted SLIT3 is able to mediate skeletal angiogenesis that in turn facilitates bone formation16,33. Slits are also reported to control global energy metabolism by enhancing energy expenditure. C-terminal SLIT2 fragments have been studied as a beige adipocyte-derived factor that acts locally to augment beige adipocyte development by activating canonical PKA signaling22. SLIT2 proteolytic cleavage is also noted to be highly conserved and likely central to SLIT2 function34,35. Similar to a previous study, we detected similar peptide signals in serum and osteoblast supernatant samples22.It is noted that this local secretion of SLIT2 within adipose tissue may play distinct functions or be complementary to osteoblast-secreted SLIT2, with osteoblasts here identified as the major source of circulating SLIT2. Moreover, macrophage derived-SLIT3 also drives sympathetic-dependent thermogenesis by elevating the phosphorylation of tyrosine hydroxylase (TH) through PKA/CaMKII signaling36. In this study, we provided evidence that SHN3-regulated skeletal SLIT2 secretion is a critical thermogenic regulator that ameliorates body weight gain and benefits glucose homeostasis, further expanding the potential therapeutic value of the Slit family in treating obesity and related metabolic disorders.
In addition, the evidence for skeletal regulation of adipose tissue raises an important question—does bone mass positively or negatively correlate with fat mass and systemic energy balance? Previous studies utilizing different genetic models with either high bone mass models or osteoporotic animals have offered conflicting results on the relationship between bone and systemic energy metabolism7,9,10,13,37,38. Interestingly, elimination of osteoblastic SLIT2 did not alter in vivo bone quality, implying that the systemic energy regulation functions of osteoblasts are separate from their canonical role to mediate bone matrix production.
These findings both support and expand upon prior work identifying that adipocytes secrete a SLIT2 C-terminal fragment22, here finding that the same fragment is produced by osteoblasts (SLIT2-C) and that indeed osteoblasts are a major source of the total circulating serum SLIT2-C. Similarly, SLIT2-C here is seen to exert a WAT browning effect consistent with previously reported adenoviral gain-of-function studies22. These findings suggest a complimentary role for bone versus adipose-derived SLIT2-C. Transcriptional regulation of bone and adipose Slit2 appears to be independent of the skeletal SHN3/SLIT2 axis (Supplementary Fig 4b and 4j, k), raising the possibility that bone and adipose derived SLIT2 could be differentially regulated and respond to distinct induction conditions. Ultimately, directly parallel studies of bone and adipose conditional Slit2 deletion, ideally also including dual deletion at bone sties, will be needed to understand how these two sources of SLIT2 work together to regulate adipose browning and systemic energy metabolism.
Notably, we still did not eliminate the possibility that additional osteoblast-secreted factors in addition to SLIT2 contribute to the energy metabolism phenotype of Shn3-deficient mice. Apart from the Slit family, we also observed additional potential Shn3-regulated osteokines that may be relevant to regulation of energy metabolism such as Isthmin1 and BMPs, though these other factors showed both more modest alterations in expression after Shn3-depletion than SLIT2, and their basal expression levels in osteoblasts were much lower than that of SLIT239,40,41,42. This study also does not eliminate the possibility that there are other osteokines with thermogenic potential or regulating energy metabolism via distinct mechanisms. The identification of SLIT2 may facilitate the identification of additional such osteokines, such as through examination of mice with compound deletion of SHN3 and SLIT2 in bone for evidence of additional residual skeletal metabolic regulatory activity that could then be attributed to an alternative mediator. Alternatively, the search for additional osteokines could focus on mediators impacting effector mechanisms other than white adipose browning. Additionally, these findings also highlight the potential of osteoanabolic agents to have multiple desirable clinical effects, including simultaneously improving bone mass while also reducing weight gain or the manifestations of metabolic syndrome. Consideration of secreted SLIT2 levels in response in clinical studies of different osteoanabolic agents may represent an important next step towards clinically harnessing the bone to adipose crosstalk pathway identified here.
The lean phenotype driven by Shn3-deficiency displays distinct metabolic features from those seen in other studies of skeletal regulation of metabolism, indicating that bone is likely to engage multiple mechanisms to control energy metabolism. For example, SOST predominantly exerts its metabolic effects on mature white adipocytes and progenitors by accelerating fatty acid oxidization, impairing lipid synthesis and blocking adipogenic progenitor differentiation13,43. Lipocalin-2 attenuated food intake, causing a negative energy balance44,45. In contrast, Shn3-deficient mice exhibit increased food intake and lean body mass with increased energy expenditure. This indicates that targeting Shn3 may have an additive or synergistic effect with other anti-obesity therapies in treating metabolic disorders, thereby justifying further study and development of means to therapeutically inhibit SHN3. When combined with past data on the functions of SHN3 in regulating bone formation, we here identify dual therapeutic value of SHN3 inhibition in treating both osteoporotic diseases and obesity-related disorders24,25,46.
Methods
All animal experiments were conducted in compliance with approved protocols by the Weill Cornell Medical College (IACUC) and Xiamen University animal care committees. All research complies with relevant ethical regulations.
Animals
Shn3−/− , Shn3f/f, Slit3f/fand Osx Cre mice were produced as previously described47. Slit2f/f mice were a generous gift from Dr. Marc-Tessier Lavigne. Adipoq-Cre mice were purchased from the Jackson Laboratory. Mice were maintained in a barrier facility, fed ad libitum chow (Teklad 2014 Diet, Teklad), and followed a standard day-night lighting cycle. 6-week-old mice were used and fed with 60% HFD (Research diet, D12492i) to establish diet-induced-obesity models. Animals for the control-fat-diet group were fed with 10% fat diet (Research diet, D12450J) ad libitum.
Osteoblast and SVF isolation, in vitro beige adipocyte induction and co-culture assays
Shn3Osx and Shn3f/f calvarial osteoblast (COB) precursors were isolated, seeded on the upper chamber of a co-culture system (Corning) and differentiated for 14 days as previously described48,49. SVF cells were isolated as previously described50. Shn3f/f iWAT tissues were used for isolation of SVF cells. In brief, iWAT harvested from 4-week-old male mice was digested with collagenase II (Worthington, 1 mg/mL) and dispase II (Roche, 2 mg/mL). SVF cells were seeded on a 6-well-plate and passaged when reaching 70–80% confluence. P2 SVF cells were used for differentiation by adding induction cocktail containing IBMX (Sigma), dexamethasone (Sigma), insulin (Eil Lilly) and rosiglitazone (Sigma)for 48 h and then switched to maintenance media. SVF cells were co-cultured with differentiated COBs for 6 days before RNA isolation.
Murine serum collection, processing and volumes for different assays
In brief, rodents were sacrificed by cervical dislocation performed by trained researchers and blood samples were collected via cardiac puncture in a collection tube (BD Biosciences) before blood clotting. Collection tubes were then stored in 4 °C for at least an hour followed by a 10000 g centrifugation for 5 min at 4 °C. Serum was transferred to a clean 2 mL microcentrifuge tube and stored in −80 °C before use. For SLIT2 sandwich ELISA, 80uL of freshly isolated murine serum was used for ELISA assays. For C-peptide analysis, 10 uL of stored serum was used for C-peptide analysis. For Mass Spectrometry analysis, 100 uL of serum was processed utilizing High-SelectTM Top14 Abundant Protein Depletion Midi Spin Columns (A36371, ThermoScientific) according to the manufacturer’s handbook. For Western Blots, 1 uL murine serum was used for each sample and processed as below.
Quantitative PCR and Western Blot analysis
Quantitative polymerase chain reaction (qPCR) and Western Blotting (WB) were performed as previously described49,51,52. For Western Blotting of murine serum, 1uL of murine serum sample was prepared containing 4X NuPAGETM LDS sample buffer (NP0008, Thermo Scientific) with reducing agent and boiled before Western Blotting. Primary antibodies specific to UCP1 (1:500, ab10983, Millipore), SLIT2 (1:500, ab134166, Abcam), β-Catenin (1:1000, 9562 L, Cell signaling), β-Actin (1:5000, 4967 L, Cell signaling), Transferrin (1:5000, ab82411, Abcam) and HSP90 (1:5000, 4874 S, Cell signaling) were used for immunoblotting. Of note, the use of Slit2 germline null serum would be desirable for validation of immunoblotting. However, due to the perinatal lethal phenotype of Slit2-/- mice53, such validation is challenging and only examination of conditional knockout mouse serum displaying a partial ablation of SLIT2 levels is feasible. In light of this, we considered the approximately 50 kDa as representing the SLIT2-C fragment and the approximately 180 kDa as full length SLIT2. The primers (Origene, US) used for qPCR are listed in Supplementary Table 1. Samples were assessed for differences in the transcript levels of thermogenic genes, Hivep3 and Slit2.
Paraffin tissue processing, H&E staining and immunohistochemistry
Liver, pancreas, white adipose tissue and brown adipose tissue were harvested, dehydrated and embedded in paraffin as previously described54,55. Samples were sectioned at 5μm thickness followed by a common H&E staining protocol16. UCP1 antibody (1:500, ab10983, Abcam) was used for immunohistochemistry. In brief, adipose tissue sections were deparaffinized, rehydrated in serial ethanol solution and blocked by hydrogen peroxide and BSA solution. Primary UCP1 antibody was incubated overnight and consequently followed by goat anti-rabbit HRP conjugated secondary antibody (1:5000, 31460, Invitrogen) incubation. After each step of antibody incubation, sections were washed with PBS at least 3 times to remove excess antibody. DAB substrate (DAB-M, Sigma) was dropped on the surface of the specimen to generate signals and mounted with DPX (06522, Sigma) before analysis.
Micro-CT imaging and data analysis
Micro-computed tomography (µCT) was performed by using a Scanco Medical Micro-CT 35 system (Scanco Medical, Switzerland) as previously described15,56. In brief, femurs were scanned at 7 micrometer resolution. For analysis of femoral bone mass, a region of trabecular bone 2.1 mm wide was contoured, starting 300 microns from the proximal end of the distal femoral growth plate. Femoral trabecular bone was thresholded at 211 permille. Femoral cortical bone was thresholded at 350 permille. Calvarium was thresholded at 260 permille. A Gaussian noise filter optimized for murine bone was applied to reduce noise in the thresholded 2-dimensional image. Cortical and trabecular thickness (Cort. Th, Tb. Th), trabecular number (Tb. N), trabecular spacing (Tb. Sp) and bone volume/total volume (BV/TV) were calculated by contouring the corresponding regions of the scan slices. 3D reconstruction images were generated by stacking the contours of 2D images. µCT analysis was conducted by an individual blinded to the genotype of each mouse.
Intraperitoneal glucose tolerance tests and intraperitoneal insulin tolerance tests
For intraperitoneal glucose tolerance tests (IGTTs) and intraperitoneal insulin tolerance tests (ITTs), mice on HFD for 8 (thermoneutral housed) or 11 weeks (room temperature housed) were fasted for at least 6 h before receiving an intraperitoneal injection of glucose solution (1.5 g/kg body weight for IGTTs, Sigma) or insulin solution (1U/kg body weight for ITTs, Lilly). Blood samples were collected at corresponding time point and glucose levels were evaluated through blood glucose test strips (Roche). Results were analyzed by Two-way ANOVA with repeated measures.
Subcutaneous fat graft transplantation
Fresh subcutaneous fat grafts were harvested from the inguinal region of 8-week-old male C57BL6/J donor mice. Host mice were anesthetized with isoflurane and shaved to expose the entire dorsal region. A 5-8 mm vertical incision was made alongside the spine. A 400 mg fat graft was implanted into secondary hosts for 4 weeks. Grafts were then fixed in 4% PFA for histological analysis or homogenized for gene expression studies.
C-peptide and SLIT2 sandwich ELISA
For in vivo circulating SLIT2 level assessment, SLIT2 sandwich ELISA was performed utilizing mouse serum collected from HFD-fed Shn3−/−, WT, Slit2f/f and Slit2Osx mice. For in vitro osteoblast supernatant Slit2 quantification, osteoblasts were isolated, differentiated as described above. Conditioned media was collected for consequent ELISA assay.
Sandwich ELISA assays were performed according to manufacturers’ protocols (C-peptide ELISA kit, Cat No. 80-CPTHU-E01.1, ALPCO; Mouse Slit2 ELISA kit, Cat No. NBP2-82435, Novus Biologicals). Preliminary experiments were performed before formal assays to evaluate the range of sample dilution. For linearity validation of SLIT2 sandwich ELISA, the recombinant SLIT2-C terminal fragment (aa1114-1521, 9430-SL-050, R&D systems) and the recombinant SLIT2-N fragment (aa26-900, 5444-SL, R&D systems) were utilized and prepared at concentration of 20, 10, 5, 2.5, 1.25, 0.63, 0.31 ng/mL in sample diluent. Serum samples collected from 18-week-old HFD-fed murine model were serially diluted by sample diluent (undiluted, 1:1 diluted, 1:2 diluted, etc.). Samples were freshly prepared and all reagents were equilibrated to room temperature prior to ELISA assays. 80uL SLIT2-C, SLIT2-N, neat and serially diluted serum were added to respective wells of a ready-to-use microplate according to manufacturers’ guidance and incubated for 2 h at room temperature on a microplate shaker. The contents of the wells were decanted and washed with 1X wash buffer for at least 3 times. Conjugated detection antibodies were added and incubated for 60 min before another washing process. Substrates were added to each well and incubated for 10 min before adding stop solution. A microplate reader was used for reading the absorbance at 450 nm.
For formal experiments of SLIT2 sandwich ELISA of murine serum, serum samples were diluted at 1:1 ratio by sample diluents provided by ELISA kit manufacturer before an ELISA assay. 80uL of standards, controls and samples were pipetted into respective wells of the ready-to-use microplate and the following ELISA steps were similar to the ELISA validation above.
Analysis of mitochondrial stress test through Seahorse XFe96
Oxygen consumption rates of primary inguinal cells were determined through an XFe96 Extracellular Flux Analyzer (Seahorse Biosciences). Primary inguinal cells from HFD-fed WT and Shn3−/− mice were harvested as previously described50. Cells were seeded at 10,000 cells per well on a 96-well-plate (within 103680-100, Agilent) for mitochondrial stress test. Mitochondrial stress testing was performed according to manufacturer’s guidelines (103680-100, Agilent). In brief, on the day of analysis, cells were washed twice, equilibrated with basal respiration media (10 mmol/L glucose, 1 mmol/L sodium pyruvate, 2 mmol/L glutamine in DMEM basal media) and incubated for 45 min in CO2-free incubator before testing. Port injection solutions were prepared as follows (final concentration): 1.5 μM oligomycin (Port A), 1 μM FCCP (Port B) and 0.5 μM Rotenone/Antimycin A (Port C). Each cycle consisted of 4 min of mixture and 2 min of measurement. Data were analyzed through Seahorse Wave Desktop software (ver 2.6.3, Agilent) and presented as mean ± standard deviation (SD).
Analysis of osteoblast bulk RNA sequencing
The expression data matrix of bulk RNA sequencing data was obtained and analyzed as previously described16. In brief, reads were aligned to the mm9 mouse transcripts through STAR (ver 2.3.0e) with default parameters. Bam files were sorted and indexed by samtools. Gene counts were obtained by applying feature counts (ver 1.4.3) to sorted bam files. Reads without unique-mapping were abandoned. Genes with no expression counts were discarded. DESeq2 (version 1.4.5) R package was employed to normalize gene counts. Differentially expressed genes (DEGs) between KO and WT groups were detected (FDR < 0.1 and log2 fold-change>0.5). Thermogenic cytokines were screened through literature review6,57,58. Genes that were differentially expressed were rank ordered based of the degree of differential expression and then manually screened for secreted factors. The top 7 differentially expressed genes encoding for secreted factors were then visualized by a heatmap and then further scrutinized for additional study based 1) deprioritizing matrix proteins that are unlikely to reach systemic circulation other than short propeptide fragments, 2) prioritizing genes with a robust absolute degree of expression in osteoblasts, and 3) prioritizing factors known to have a metabolic regulatory activity in other contexts.
Sample preparation for mass spectrometry and in-gel digestion
For secreted proteins of differentiated osteoblasts isolated from 14-week-old HFD-fed wild type mice were concentrated through basic methanol chloroform precipitation (300 mL of serum free media after an 18-hour incubation) with PMSF (HYB0496, MCE) and protease inhibitor cocktail (HYK0010, MCE) added. For serum samples, 100uL serum were processed utilizing High-SelectTM Top14 Abundant Protein Depletion Midi Spin Columns (A36371, ThermoScientific) according to the handbook by manufacturer. MS samples were further prepared using SCASP method before processing mass spectrometry analysis59.
Orbitrap Fusion Lumos
Peptide samples (n = 2) were resolved in 2% ACN/0.1%FA for LC-MS analysis. The LC used was an EASY-nLC 1200 system (Thermo Fisher Scientific, San Jose, CA) harboring a home-pulled emitter integrative 75 µm by 35 cm C18 column packed with 1.8 µm 120 Å C18 material (Welch, Shanghai, China). A 135-min LC separation was configured on the basis of the mix of buffer (0.1% formic acid in H2O) and buffer B (80% acetonitrile containing 0.1% formic acid). Buffer B was made to increase from 4 to 34% in 120 min, then to surge to 100% in 3 min, and then kept at 100% for 8 min. The LC-MS flow rate was kept at 300 nl/min. The Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) instrument coupled to a nanoelectrospray ion source (NanoFlex, Thermo Fisher Scientific) was used. In brief, following MS settings were applied: 1 MS1 scan at 60 k resolution with an AGC target of 4 × 105 and max injection time of 50 ms in the range of 350–1200 Th, followed by 40 DIA scans with segment widths adjusted to the precursor density. The scan resolution in the Orbitrap was set to 30 k with an AGC target of 1 × 106 and max injection time of 54 ms. The HCD collision energy was set to 32%.
Predicted library by DIA-NN
The protein sequence files from Uniprot/Swissprot human were input to DIA-NN (v1.8.1)60. “FASTA digest for library-free search” and “Deep learning-based spectra, RTs and IMs predication” were enabled. Protease “Trypsin/P” was selected. Missed cleavages of “1” and Maximum number of variable modification of “2” were selected to generate the predicted libraries. “N-term M excision”, “C carbamidomethylation”, “Ox(M)”, “Ac(N-term)” were enabled. The other parameters were set as followed: Peptide length range “7–30”, Precursor charge range “2–4”, Precursor m/z range “300–1800” and Fragment ion m/z range “200–1800”.
Targeted analysis of DIA data by DIA-NN
DIA files (raw) files were loaded into DIA-NN (v1.8.1). Libraries (tsv) generated by DDA or DIA data were selected, and the predicted library (speclib) was selected. FASTA files which were used for DDA database searches were added. “FASTA digest for library-free search” and “Deep learning-based spectra, RTs and IMs prediction” were not enabled. “Generate spectral library” was enabled. “Protein inference” was set to “protein name (from FASTA)”. Other parameters were left as default. The protein groups and precursors lists were filtered at 1% FDR, using global q-values for protein groups and both global and run-specific q-values for precursors.
Mass spectrometry by in-gel digestion
Protein samples to be assessed (n = 2) were reconstituted (supernatant protein pellet) or stored (purified serum samples) in 10 mM PBS and 0.02% sodium azide, pH 7.4. After mixed with 5X SDS and denaturation, a SDS-PAGE was performed and followed by a Commassie Blue staining to isolate 50–75 kDa peptides. Gels were washed 10 times in pure water, twice in destain buffer (25 mM NH4HCO3 and 50% ACN in water) and incubated in distilled water overnight at 4°C. After dehydration, gels were lyophilized, incubated in reduction buffer (10 mM DTT/25 mM NH4HCO3) and alkylation buffer (50 mM IAA/25 mM NH4HCO3) for 1 h. Samples were then digested with 0.02ug/uL trypsin (Promega), desalted and lyophilized before mass spectrometry.
The lyophilized peptide fractions were resuspended in ddH2O containing 0.1% formic acid, and 2ul aliquots of which was loaded into a nanoViper C18 (Acclaim PepMap 100, 75 μm ×2 cm) trap column. The online Chromatography separation was performed on the Easy nLC 1200 system (ThermoFisher). The trapping and desalting procedure was carried out in 20 μL 100% solvent A (0.1% formic acid). Then, an elution gradient of 5-38% solvent B (80% acetonitrile, 0.1% formic acid) in 60 min was used on an analytical column (Acclaim PepMap RSLC, 75 μm × 25 cm C18-2 μm 100 Å). DDA (data-dependent acquisition) mass spectrum techniques were used to acquire tandem MS data on a ThermoFisher Q Exactive mass spectrometer (ThermoFisher, USA) fitted with a Nano Flex ion source. Data was acquired using an ion spray voltage of 1.9 kV, and an interface heater temperature of 275°C. For a full mass spectrometry survey scan, the target value was 3 × 106 and the scan ranged from 350 to 2000 m/z at a resolution of 70,000 and a maximum injection time of 100 ms. For the MS2 scan, only spectra with a charge state of 2–5 were selected for fragmentation by higher-energy collision dissociation with a normalized collision energy of 28. The MS2 spectra were acquired in the ion trap in rapid mode with an AGC target of 8000 and a maximum injection time of 50 ms. Dynamic exclusion was set for 25 s.
Metabolic cage methods and data analysis
Metabolic cage experiments were performed in the XMU animal facility with technical support from the Metabolic Phenotyping Core of Weill Cornell Medicine. Mice were individually housed within the Promethion High-Definition Multiplexed Respirometry System for Mice (Sable Systems International, North Las Vegas, NV, USA). Metabolic cages were located within ambient temperature and light-controlled environmental enclosures, DB034-LT Laboratory Incubator (Darwin Chambers Company, St Louis, MO, USA). Mice were exposed to a 12 h light/dark cycle and ambient temperature of 22 °C, unless otherwise specified. Mice were housed on pine chip bedding, without nestled, and provided ad libitum access to food and acidified water. Energy expenditure was assessed via indirect calorimetry and calculated using the Weir equation (3.941 kcal/L × VO2 + 1.106 kcal/L × VCO2)61. Respirometry values were determined every 5 min; the dwell time for each cage is 30 s, with baseline cage sampling frequency of 30 s occurring every four cages. Rates of oxygen consumption and carbon dioxide production were acquired with a sampling frequency of 1 s. Rates of oxygen consumption and carbon dioxide production were acquired with a sampling frequency of 1 s. Respiratory exchange ratios (RER) were calculated as VCO2/VO2. Food and water intake, and body mass, were assessed gravimetrically. Distance traveled (activity) was determined by XY position displacements that are represented by beam breaks (PedMeters and AllMeters). Data were analyzed through CalR. Two groups template was used for data analysis. Two-way repeated-measures ANOVA was used for whole-body energy expenditure results, respiratory exchange ratio, food consumption and locomotor activities. Correlations were examined with non-parametric Spearman correlation test62.
rAAV9-mediated in vivo silencing of Shn3
Artificial miRNA-containing plasmids targeting murine Shn3 were generous gifts from Dr. Guangping Gao. Replication-deficient recombinant AAV (rAAV) vector design and production were performed as previously described24. In brief, engineered amiR cassettes targeting Shn3 (amiR-Shn3) or control (amiR-Ctrl) were constructed within vector plasmids between CB promoter and the reporter gene Egfp which enable visual tracking of transduced cells or tissues. Plasmids containing amiR cassettes, AAV2/9 and helper plasmids were mixed and transfected into HEK293 cells (CRL-1573TM, ATCC) using PEI-MAX 40000 (Polysciences, 24765-1) to generate rAAV for experimental use. These rAAV batches were then collected and purified following a traditional CsCl sedimentation protocol. The concentration of rAAV9-amiR-Shn3 and rAAV9-amiR-Ctrl was then titered as previously described24.
For systemic delivery of rAAV, intravenous injections were performed when male mice reached 6 weeks of age. A total volume of 100 uL containing 4 × 1011 GC rAAV9-amiR-Shn3 or rAAV9-amiR-Ctrl were injected into the tail vein. Rodents administered with rAAV9 were subsequently exposed to HFD feeding for 12 weeks to induce DIO. Body weights were collected weekly. 10 weeks after injection, IGTT and ITT tests were performed to determine glucose tolerance and insulin sensitivity. At the endpoint, DIO murine models were sacrificed and their organs were subjected to IVIS optical imaging and downstream analysis.
Statistical analysis
Replicates are described in the figure legends. All statistical calculations and graphs were created using GraphPad Prism Ver 9.0. The results shown were reported as mean ± standard deviation (SD). When applicable, Student’s t-test, One-Way analysis of variance (ANOVA) followed by Tukey’s test, Two-way ANOVA with Bonferroni’s multiple comparison test were used to evaluate the statistical significance. Correlations were examined with non-parametric Spearman correlation test. Statistical significance was accepted as a p-value less than 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided with this paper. All relevant data are available from the corresponding authors. Uncropped blots and processed mass spectrometry data are provided within source data files in this paper. Bulk RNA Seq of murine osteoblasts can be accessed through following link: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA395701. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD05425863,64. Source data are provided with this paper.
References
Bartelt, A. & Heeren, J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 10, 24–36 (2014).
Bartelt, A. et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 17, 200–205 (2011).
Bartelt, A. & Heeren, J. The holy grail of metabolic disease: brown adipose tissue. Curr. Opin. Lipidol. 23, 190–195 (2012).
Zhang, Y. et al. The starvation hormone, fibroblast growth factor-21, extends lifespan in mice. Elife 1, e00065 (2012).
Rao, R. R. et al. Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 157, 1279–1291 (2014).
Zhou, R. et al. Endocrine role of bone in the regulation of energy metabolism. Bone Res 9, 25 (2021).
Lee, N. K. et al. Endocrine regulation of energy metabolism by the skeleton. Cell 130, 456–469 (2007).
Wei, J., Hanna, T., Suda, N., Karsenty, G. & Ducy, P. Osteocalcin promotes β-cell proliferation during development and adulthood through Gprc6a. Diabetes 63, 1021–1031 (2014).
Tangseefa, P. et al. The mTORC1 complex in pre-osteoblasts regulates whole-body energy metabolism independently of osteocalcin. Bone Res 9, 10 (2021).
Tangseefa, P. et al. Deletion of Rptor in preosteoblasts reveals a role for the mammalian Target of rapamycin complex 1 (mTORC1) complex in dietary-induced changes to bone mass and glucose homeostasis in female mice. JBMR 5, e10486 (2021).
Wei, J. & Karsenty, G. An overview of the metabolic functions of osteocalcin. Rev. Endocr. Metab. Disord. 16, 93–98 (2015).
Zhang, X., Cheng, Q., Wang, Y., Leung, P. S. & Mak, K. K. Hedgehog signaling in bone regulates whole-body energy metabolism through a bone-adipose endocrine relay mediated by PTHrP and adiponectin. Cell Death Differ. 24, 225–237 (2017).
Kim, S. P. et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc. Natl Acad. Sci. USA. 114, E11238–E11247 (2017).
Jones, D. C. et al. Regulation of adult bone mass by the zinc finger adapter protein Schnurri-3. Science 312, 1223–1227 (2006).
Shim, J.-H. et al. Schnurri-3 regulates ERK downstream of WNT signaling in osteoblasts. J. Clin. Invest. 123, 4010–4022 (2013).
Xu, R. et al. Targeting skeletal endothelium to ameliorate bone loss. Nat. Med. 24, 823–833 (2018).
Taylor, R., Al-Mrabeh, A. & Sattar, N. Understanding the mechanisms of reversal of type 2 diabetes. lancet Diab Endocrinol. 7, 726–736 (2019).
Wagner, J. D., Cann, J. A., Zhang, L. & Harwood, H. J. Chapter 14 - Diabetes and Obesity Research using Nonhuman Primates. in Nonhuman Primates in Biomedical Research (Second Edition) (eds. Abee, C. R., Mansfield, K., Tardif, S. & Morris, T.) 699–732 (Academic Press, 2012).
Hu, F., Qiu, X. & Bu, S. Pancreatic islet dysfunction in type 2 diabetes mellitus. Arch. Physiol. Biochem. 126, 235–241 (2020).
Zhong, L. et al. Single cell transcriptomics identifies a unique adipose lineage cell population that regulates bone marrow environment. Elife 9, e54695 (2020).
Huang, W. & Olsen, B. R. Skeletal defects in osterix-cre transgenic mice. Transgenic Res. 24, 167–172 (2015).
Svensson, K. J. et al. A secreted Slit2 fragment regulates adipose tissue thermogenesis and metabolic function. Cell Metab. 23, 454–466 (2016).
Lattin, J. E. et al. Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res. 4, 5 (2008).
Yang, Y.-S. et al. Bone-targeting AAV-mediated silencing of Schnurri-3 prevents bone loss in osteoporosis. Nat. Commun. 10, 2958 (2019).
Oh, W.-T. et al. WNT-modulating gene silencers as a gene therapy for osteoporosis, bone fracture, and critical-sized bone defects. Mol. Ther. 31, 435–453 (2023).
Hill, J. O., Wyatt, H. R. & Peters, J. C. Energy balance and obesity. Circulation 126, 126–132 (2012).
Borén, J., Taskinen, M.-R., Björnson, E. & Packard, C. J. Metabolism of triglyceride-rich lipoproteins in health and dyslipidaemia. Nat. Rev. Cardiol. 19, 577–592 (2022).
De Laet, C. et al. Body mass index as a predictor of fracture risk: a meta-analysis. Osteoporos. Int. a J. Establ. result Coop. Eur. Found. Osteoporos. Natl Osteoporos. Found. USA 16, 1330–1338 (2005).
Fan, Z. et al. Comparison of OSTA, FRAX and BMI for predicting postmenopausal osteoporosis in a han population in Beijing: a cross sectional study. Clin. Interv. Aging 15, 1171–1180 (2020).
Yeap, B. B. et al. Reduced serum total osteocalcin is associated with metabolic syndrome in older men via waist circumference, hyperglycemia, and triglyceride levels. Eur. J. Endocrinol. 163, 265–272 (2010).
Fiore, C. E., Pennisi, P., Pulvirenti, I. & Francucci, C. M. Bisphosphonates and atherosclerosis. J. Endocrinol. Invest. 32, 38–43 (2009).
Blockus, H. & Chédotal, A. Slit-Robo signaling. Development 143, 3037–3044 (2016).
Li, N. et al. Osteoclasts are not a source of SLIT3. Bone Res. 8, 11 (2020).
Wang, K. H. et al. Biochemical purification of a mammalian slit protein as a positive regulator of sensory axon elongation and branching. Cell 96, 771–784 (1999).
Brose, K. et al. Slit proteins bind robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 96, 795–806 (1999).
Wang, Y.-N. et al. Slit3 secreted from M2-like macrophages increases sympathetic activity and thermogenesis in adipose tissue. Nat. Metab. 3, 1536–1551 (2021).
Zoch, M. L., Abou, D. S., Clemens, T. L., Thorek, D. L. J. & Riddle, R. C. In vivo radiometric analysis of glucose uptake and distribution in mouse bone. Bone Res 4, 16004 (2016).
Kim, S. P. et al. Lrp4 expression by adipocytes and osteoblasts differentially impacts sclerostin’s endocrine effects on body composition and glucose metabolism. J. Biol. Chem. 294, 6899–6911 (2019).
Tseng, Y.-H. et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 454, 1000–1004 (2008).
Chen, Y. et al. Potential functions of the BMP family in bone, obesity, and glucose metabolism. J. Diab Res. 2021, 6707464 (2021).
Baboota, R. K., Blüher, M. & Smith, U. Emerging role of bone morphogenetic protein 4 in metabolic disorders. Diabetes 70, 303–312 (2021).
Jiang, Z. et al. Isthmin-1 is an adipokine that promotes glucose uptake and improves glucose tolerance and hepatic steatosis. Cell Metab. 33, 1836–1852.e11 (2021).
Kim, S. P. et al. Bone-derived sclerostin and Wnt/β-catenin signaling regulate PDGFRα(+) adipoprogenitor cell differentiation. FASEB J. Publ. Fed. Am. Soc. Exp. Biol. 35, e21957 (2021).
Mosialou, I. et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543, 385–390 (2017).
Mosialou, I. et al. Lipocalin-2 counteracts metabolic dysregulation in obesity and diabetes. J. Exp. Med. 217, e20191261 (2020).
Yallowitz, A. R., Shim, J.-H., Xu, R. & Greenblatt, M. B. An angiogenic approach to osteoanabolic therapy targeting the SHN3-SLIT3 pathway. Bone 172, 116761 (2023).
Hammad, H. et al. Transitional B cells commit to marginal zone B cell fate by Taok3-mediated surface expression of ADAM10. Nat. Immunol. 18, 313–320 (2017).
Debnath, S. et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139 (2018).
Li, Z. et al. TAOK3 is a MAP3K contributing to osteoblast differentiation and skeletal mineralization. Biochem. Biophys. Res. Commun. https://doi.org/10.1016/j.bbrc.2020.07.060 (2020).
Yu, H., Emont, M., Jun, H. & Wu, J. Isolation and differentiation of murine primary brown/beige preadipocytes. Methods Mol. Biol. 1773, 273–282 (2018).
Kim, J. M. et al. The ERK MAPK pathway is essential for skeletal development and homeostasis. Int. J. Mol. Sci. 20, 1803 (2019).
Greenblatt, M. B. et al. CHMP5 controls bone turnover rates by dampening NF-κB activity in osteoclasts. J. Exp. Med. 212, 1283–1301 (2015).
Plump, A. S. et al. Slit1 and Slit2 cooperate to prevent premature midline crossing of retinal axons in the mouse visual system. Neuron 33, 219–232 (2002).
Cohen, P. et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156, 304–316 (2014).
Seale, P. et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Invest. 121, 96–105 (2011).
Zou, W. et al. MLK3 regulates bone development downstream of the faciogenital dysplasia protein FGD1 in mice. J. Clin. Invest. 121, 4383–4392 (2011).
Priest, C. & Tontonoz, P. Inter-organ cross-talk in metabolic syndrome. Nat. Metab. 1, 1177–1188 (2019).
Cohen, P. & Kajimura, S. The cellular and functional complexity of thermogenic fat. Nat. Rev. Mol. Cell Biol. 22, 393–409 (2021).
Gan, G. et al. SCASP: a simple and robust SDS-aided sample preparation method for proteomic research. Mol. Cell. Proteom. 20, 100051 (2021).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
WEIR, J. B. D. E. B. New methods for calculating metabolic rate with special reference to protein metabolism. J. Physiol. 109, 1–9 (1949).
Mina, A. I. et al. CalR: a web-based analysis tool for indirect calorimetry experiments. Cell Metab. 28, 656–666.e1 (2018).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Chen, T. et al. iProX in 2021: connecting proteomics data sharing with big data. Nucleic Acids Res. 50, D1522–D1527 (2022).
Acknowledgements
We appreciate Dr. Marc-Tessier Lavigne for generously sharing Slit2f/f mice. We thank Dr. Guangping Gao for generously sharing artificial miRNA-containing plasmids targeting murine Shn3.We thank all the members in the Animal Facility and Confocal Microscopic Core facility at Xiamen University for animal maintenance metabolic cage study and imaging studies. We also thank Dr. Shuyong Lin, Dr. Chensong Zhang, all the staff in Metabolic Phenotyping Core and Department of Pathology of Weill Cornell Medicine for their technical support on metabolic cage and histological assessment. We thank Dr. Francois Marchildon for technical support on indirect calorimetry studies. We thank Shuangquan Su and Biorender in helping us generating the schematic diagram of in vitro and in vivo assays. We also thank all the members in the Xu Lab and Greenblatt Lab for their critical review and helpful discussion of this manuscript. RX is supported by National Natural Science Foundation (NSFC) of China (81972034, 92068104 and 82372362), Natural Science Foundation of Fujian Province (2022J06003) and Project of Xiamen Cell Therapy Research (Grant No. 3502Z20214001). MBG is supported by Pershing Square Sohn Cancer Research Alliance and Pershing Square MIND Prize awards, the NIH under R01AR075585, R01HD115274, R01CA282815, and a Career Award for Medical Scientists from the Burroughs Welcome Foundation. HL is supported by NSFC (82230085, 82272572) and Science and Technology Major Project of Changsha (kh2102015). NL is supported by NSFC of China (82002262). JS is supported by a Young Investigator Award from the Children’s Tumor Foundation. SB is supported by the Arthritis National Research Foundation (1065843), a Weill Cornell Jumpstart Award, and an award from the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2021R1A6A3A14038667). PC is supported by NIH RC2DK129961.
Author information
Authors and Affiliations
Contributions
R.X., M.B.G., and Z.Li. designed, conducted, and analyzed the majority of experiments. R.X., M.B.G., and H.L. supervised the project. R.X. and M.B.G. conceived the project. Z.Li., B.S., N.L., X.Z., and R.H. performed all mouse studies. Z.Li., B.S., N.L., J.S., X.Z., R.H., and X.C. maintained and genotyped all mice. Z.Li conducted metabolic cage studies with the assistance of M.K. and P.C. Z.Li, B.S., N.L., J.S., X.Z., X.C., and J.H. analyzed the results. J.S., S.B., A.R.Y, S.D., M.C., and Z.Ling. assisted specimen acquisition. Y.H. and G.L. assisted AAV-related experiments. M.K. and P.C. assisted with thermoneutral experiments and offered critical support in study design. Z.Li and C-Q.Z. performed sample preparation, LC-MS and analyzed data. X.S. offered immunoblot antibodies and provided critical support for micro-CT analysis. R.X., M.B.G., and Z.Li. prepared the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Grant Mastick, Toshio Matsumoto and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Shi, B., Li, N. et al. Bone controls browning of white adipose tissue and protects from diet-induced obesity through Schnurri-3-regulated SLIT2 secretion. Nat Commun 15, 6697 (2024). https://doi.org/10.1038/s41467-024-51155-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51155-6
- Springer Nature Limited