

Abstract
Whether small nucleolar RNAs (snoRNAs) are involved in the regulation of liver cancer stem cells (CSCs) self-renewal and serve as therapeutic targets remains largely unclear. Here we show that a functional snoRNA (SNORD88B) is robustly expressed in Hepatocellular carcinoma (HCC) tumors and liver CSCs. SNORD88B deficiency abolishes the self-renewal of liver CSCs and hepatocarcinogenesis. Mechanistically, SNORD88B anchors WRN in the nucleolus, promoting XRCC5 interacts with STK4 promoter to suppress its transcription, leading to inactivation of Hippo signaling. Moreover, low expression of STK4 and high expression of XRCC5 are positively correlated with HCC poor prognosis. Additionally, snord88b knockout suppresses mouse liver tumorigenesis. Notably, co-administration of SNORD88B antisense oligonucleotides (ASOs) with MST1 agonist adapalene (ADA) exert synergistic antitumor effects and increase overall murine survival. Our findings delineate that SNORD88B drives self-renewal of liver CSCs and accelerates HCC tumorigenesis via non-canonical mechanism, providing potential targets for liver cancer therapy by eliminating liver CSCs.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC), accounting for approximately 90% of liver cancer, is one of the most prevalent malignancies with poor prognosis globally1. Due to HCC heterogeneity and high recurrence, current treatments including surgical excision, chemoradiotherapy, and targeted drugs have modest effects on HCC2,3. New treatments come from an improved understanding of hepatocarcinogenesis. Cancer-initiating cells or cancer stem cells (CSCs), located in the top layer of tumors with high initiation potential, are mainly responsible for the heterogeneity and high recurrence of tumors4,5. Plenty of cell surface markers have been identified to sort liver CSCs, including CD13, CD133, CD44, CD90, EpCAM, and so on4. Using up-to-date techniques, researchers have proposed that liver CSCs may originate from differentiated hepatocytes or EpCAM+ ductal cells6,7. One of these clinical trials found that CAR-modified T cell targeting CD133 ameliorates progression-free survival in late-stage metastatic malignancies8. However, due to the plasticity of CSCs, targeting therapy solely based on surface markers is insufficient. It is urgent to explore targets based on the intrinsic characteristics of liver CSCs, which requires a deeper understanding of the mechanisms underlying their self-renewal.
Like embryonic or normal stem cells, CSCs depend on sustained activation of stemness signaling, including Hippo/YAP1, Wnt/β-catenin, Notch, and Hedgehog pathways9. Of note, aberrant activation of these pathways in CSCs may evoke uncontrolled cell proliferation and abnormal differentiation, leading to tumor initiation. While the Hippo signaling is inactivated, YAP1 enters the nucleus, which accounts for approximately 60% of HCC cases10, and acts as a transcription-regulatory factor to promote cell stemness and proliferation. Clearly, the Hippo pathway is regulated by a kinase cascade, in which large tumor suppressors 1 and 2 (LATS1/2) are phosphorylated and activated by autophosphorylation of the mammalian STE20-like protein kinase 1 and 2 (MST1/2, gene named STK4/3), resulting in YAP1 phosphorylation and degradation in the cytoplasm11. Interestingly, SNORA26 co-expression genes can be apparently enriched into the Hippo signaling pathway in cancer12. However, it remains unclear how the Hippo signaling is linked to small non-coding RNAs to affect the stemness in liver CSCs.
Small nucleolar RNAs (snoRNAs) are a class of single-stranded, small non-coding RNAs with a length of 60~300 nt13. Most of the well-characterized snoRNAs have been collected and analyzed by SNORic and snOPY databases. Consistent with its unique nucleolar localization, the canonical functions of snoRNAs are post-transcriptional modification and maturation control of ribosomal RNA (rRNA) by binding proteins to form small nucleolar ribonucleoproteins (snoRNPs)14. However, this paradigm limits the elucidation of other functions of snoRNAs. Emerging evidence indicates that dysregulated snoRNAs are involved in human malignancies in a nucleolar-independent manner. For instance, SNORD50A/B specifically located in the cytoplasm directly binds and degrades K-Ras to prevent human cancer progression15. Interestingly, SNORD113-1 has been reported as a tumor suppressor in HCC16 and SNORD17 promotes HCC progression via non-canonical functions17. These studies imply the critical roles of snoRNAs in HCC progression. However, their precise roles in liver CSCs remain largely unclear. Here we identify a conserved snoRNA SNORD88B (originated from C19orf48 gene transcript, NCBI ID: 692203) that is highly expressed in liver CSCs. Elevated SNORD88B anchors WRN in the nucleolus to inhibit STK4 (codes MST1 protein) transcription via recruitment of XRCC5, leading to the augmentation of self-renewal of liver CSCs. Administration of MST1 agonist adapalene (ADA) and ASOs against SNORD88B exerts synergistic therapeutic effects on mouse HCC tumor models.
Results
SNORD88B is highly expressed in liver CSCs
To explore the role of snoRNAs in the oncogenesis of HCC, we isolated CSCs (CD13+CD133+) and non-CSCs (CD13−CD133−) from human HCC tumor tissues based on our sorting strategy18,19 and conducted snoRNA transcriptome sequencing. The top 10 snoRNAs with high expression in CSCs were clustered (Fig. 1A). We screened out 5 snoRNAs with high abundance and validated their up-regulation in liver CSCs through qRT-PCR (Fig. S1A). We then depleted these snoRNAs using short hairpin RNA (shRNA) in HCC primary cells, and performed sphere-formation assays to detect their functions on stemness of liver CSCs. Among these five snoRNAs, we found that SNORD88B depletion most dramatically suppressed oncosphere formation (Fig. 1B). Thus, we focused on SNORD88B in the self-renewal of liver CSCs.

A 5 × 106 liver CSCs (CD13+CD133+) cells and non-CSCs (CD13−CD133−) were respectively enriched from human HCC tumor tissues via flow cytometry, followed by snoRNA sequencing (left panel). Differentially expressed snoRNAs (fold change >2) in CSCs were shown in the right panel, and the top 10 upregulated snoRNAs were denoted. B 5 upregulated snoRNAs with high abundance were depleted in HCC primary cells via shRNA. Spheres more than 100 μm in diameter were counted. HCC #1 and HCC #2 denoted HCC sample numbers. sh#1 and sh#2 indicate two effective shRNAs targeting snoRNAs. n = 3 independent experiments. Data are shown as means ± SD. Exact P values from left to right: 4.9E−08, 2.4E−07. C Schematic representation of human SNORD88B. The position of the arrowheads indicates SNORD88A/B/C position in linear gene locus. SNORD88B was denoted in red arrowheads. E1, exon #1. D, E Northern blot of SNORD88B expression levels in oncospheres (S) and non-spheres (N), or in CD13+CD133+ CSCs (C) and CD13−CD133− non-CSCs (N). 18S rRNA served as a loading control. F Total RNA from HCC oncospheres was extracted from the cytoplasm, nucleoplasm, nucleolus, and followed by qRT-PCR (left panel) and Western blot (right panel). U1 RNA served as a positive control for nuclear location. β-actin, NUP62, and Nucleolin are protein markers for cytoplasm, nucleoplasm, and nucleolus, respectively. n = 3 biologically independent experiments. Data are shown as means ± SD. G Representative FISH staining of SNORD88B and nucleolar marker Nucleolin (NCL) in liver CSCs or non-CSCs originated from primary HCC cells. Nuclei are annotated by white dotted lines. Scale bar, 15 μm. H Violin Plot of SNORD88B expression in HCC tumor tissues compared with normal tissues using TCGA database. Mapping by R software. Exact P value: 1.7E−08. I Kaplan–Meier survival analysis of HCC patients, which were from the TCGA database and grouped into two subsets according to SNORD88B expression levels. Kaplan–Meier method with the log-rank test was used. Mapping by R software. Survival curves with a 95% confidence interval were shown. J Violin plot of SNORD88B expression in HCC primary tumors with different sizes using TCGA database. Exact P values from upper to lower: 0.0163, 0.036, 0.0018. T1 (n = 92), T2 (n = 57), T3 (n = 28), T4 (n = 11). K Box plot of SNORD88B expression in different stages of HCC using TCGA database. Boxes represent the interquartile range (IQR); upper and lower lines correspond to the 75th and 25th percentiles, respectively. Whiskers above and below boxes extend to the 5th and 95th percentiles, respectively. Horizontal lines within boxes represent median values. Stage I (n = 84), Stage II (n = 51), Stage III (n = 48). Exact P value: 0.0225 *P < 0.05; **P < 0.01; ***P < 0.001 by two-tailed Student’s T-test. Data are representative of at least three independent experiments.
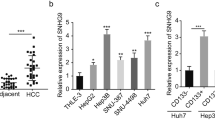
SNORD88B is located in the intron region between the fourth and fifth exons in the C19orf48 gene locus of human chromosome 19, with a length of 91 nt (Fig. 1C). Based on NCBI and snOPY databases, the C19orf48 gene contained other two homologous transcripts (SNORD88A and SNORD88C). Compared with other homologous transcripts, SNORD88B showed the highest abundance in HCC samples (Fig. S1B), which was further confirmed by Immunofluorescence staining in human HCC tumor tissues (Fig. S1C). Moreover, we found SNORD88B, but not its linear gene C19orf48, was highly expressed in HCC primary tumors (Fig. S1D, E), oncospheres (Fig. S1F), and liver CSC marker positive cell subtypes (Fig. S1G, H) via qRT-PCR, and further validated by Northern blot (Fig. 1D, E). In addition, an absolute quantitative assay showed that SNORD88B contained ~95 or 100 copies per cell in liver CSCs (Fig. S1I).
Subcellular fractionation showed that SNORD88B mainly resided in the nucleolus (Fig. 1F). Consistently, the location of SNORD88B was verified in liver CSCs (Fig. 1G) via RNA fluorescence in situ hybridization (FISH). Furthermore, the up-regulation of SNORD88B in HCC samples was further confirmed using The Cancer Genome Atlas (TCGA) dataset (Fig. 1H), and its expression levels were also increased with HCC progression (Fig. S1J). Kaplan–Meier analyses showed that SNORD88B was negatively related to the prognosis of HCC patients, but not SNORD88A or SNORD88C (Fig. 1I and Fig. S1K, L). Additionally, SNORD88B was positively correlated with larger tumor sizes and advanced stages in HCC (Fig. 1J, K). These data indicate that high expression of SNORD88B in liver CSCs and liver cancer is associated with poor prognosis.
SNORD88B promotes the self-renewal of liver CSCs
To investigate the function of SNORD88B in the regulation of liver CSC self-renewal, we generated SNORD88B-depleted cells using lentivirus-mediated shRNA and confirmed knockdown efficiency by Northern blot and qRT-PCR (Fig. 2A and Fig. S2A). Notably, serial sphere-formation assays showed that SNORD88B depletion remarkably reduced the self-renewal capacity of liver CSCs, which could be rescued by overexpression of SNORD88B (Fig. 2B). SNORD88B knockdown did not affect intracellular levels of its linear gene C19orf48 (Fig. S2B, C). It has been reported that C19orf48 can function as a drug-resistance gene in lung cancer20, suggesting C19orf48 could be involved in the pathogenesis of CSCs. To validate the role of its linear gene in the regulation of liver CSCs, we constructed C19orf48 silenced cell lines. We observed that SNORD88B displayed comparable levels in C19orf48-depleted and control cells, indicating that the upregulated expression of SNORD88B is independent of the transcription of its linear gene (Fig. S2D). Moreover, C19orf48 depletion did not affect the formation of oncospheres, which could be reduced by SNORD88B ASO treatment (Fig. S2E). We observed that SNORD88B depletion decreased the proportion of CD13+CD133+ cells (Fig. S2G). Finally, CD13 and CD133 expression levels were positively correlated with expression levels of SNORD88B in oncospheres (Fig. S2H). These data suggest that SNORD88B promotes liver CSC self-renewal in vitro.

A Northern blot showed SNORD88B depletion efficiency in HCC cell lines and HCC primary cells. Overexpression of SNORD88B (oe88B) rescued the expression of SNORD88B. shVec, shRNA empty vector; sh88B, depletion of SNORD88B. B Sphere-formation assays of depleted or rescued SNORD88B HCC cells. Statistical results of sphere-formation ratios were calculated by serial sphere-formation assays. Scale bar, 500 μm. n = 3 biologically independent experiments. Exact P values from left to right: 0.001044, 1.12E−05, 0.000877, 5.62E−05, 1.86E−05, 0.000109. Data are shown as means ± SD. C Gradient numbers of SNORD88B depletion and control cells were subcutaneously injected into BALB/c nude mice (8 W) for 3 months to detect tumor initiation. The typical tumor images were displayed in the left panel and the ratios of tumor-free mice were shown in the right panel. n = 7 for each group. Scale bar, 1 cm. Exact P value: 0.0139. D 1 × 106 SNORD88B depleted or control cells were subcutaneously injected into BALB/c nude mice (8W), followed by measurement of tumor progression every 4 days. n = 5 for each group. Results are shown as means ± SD. Exact P values from left to right: 0.00177, 7.88E−05, 0.00037, 1.43E−05. E Orthotopic liver tumor growth was imaged via luciferase signals. Representative images are shown in the left panel, and statistical results are shown as means ± SD (right panel). n = 5 for each group. Exact P values from left to right: 0.0006, 0.0004. F The effect of silencing SNORD88B expression through ASOs was verified in Huh7 cells. n = 3 independent experiments. Data are shown as means ± SD. Exact P values from left to right: 0.00045, 1.74E−05. G, H Primary HCC cells were subcutaneously injected into BALB/c nude mice. 25 mg/kg ASOs were injected around the tumors on days 24, 26, 28, 30 and 32. Mice were sacrificed on the 40th day after injection, and the tumors were excised (G) and weighed (H). Representative images were shown. Data are shown as means ± SD. n = 5 mice per group. Scale bar, 1 cm. Exact P value: 1.41E−05. I In vivo tumorigenic cell frequency assay. Tumor-free mice are counted (right panel) and representative tumor images are shown (left panel). n = 7 for each group. Exact P value: 2.19E−05. J 1 × 106 SNORD88B-overexpressing or control cells were subcutaneously injected into BALB/c nude mice, followed by measurement of tumor progression every 4 days. n = 5 for each group. Results are shown as means ± SD. Exact P values from left to right: 0.0163, 0.0011, 0.00016, 0.0021. K Orthotopic liver tumor growth was imaged via luciferase signals. Representative images are shown in the left panel, and statistical results are shown as means ± SD (right panel). Immunodeficient NSG mice were used. n = 5 for each group. Exact P value: 0.000278. *P < 0.05; **P < 0.01; ***P < 0.001 by two-tailed Student’s T-test. Data are representative of at least three independent experiments.
The limiting dilution assay is a gold standard to assess CSC potential21. SNORD88B depletion and control cells were serially transplanted into immunodeficient mice to analyze rates of secondary tumor formation. We found that SNORD88B depletion suppressed tumor initiation capacity (Fig. 2C and Fig. S2I). Moreover, SNORD88B depletion but not its linear gene remarkably reduced tumor propagation (Fig. 2D and Fig. S2F). We next injected luciferase-labeled HCC cells into mouse livers to assess tumor growth in situ. We observed that SNORD88B deficiency dramatically suppressed orthotopical tumor propagation (Fig. 2E). To further investigate its therapeutic roles, we designed two effective ASOs to silence SNORD88B expression (Fig. 2F). SNORD88B ASO administration remarkably inhibited tumor growth in patient-derived xenograft (PDX) HCC models (Fig. 2G, H). These data suggest that SNORD88B depletion suppresses the self-renewal of liver CSCs.
We next established SNORD88B-overexpressing HCC cells (Fig. S2J) and found that its linear gene expression was not affected (Fig. S2K). Serial sphere-formation assays showed that SNORD88B overexpression promoted oncosphere formation (Fig. S2L). SNORD88B overexpression also elevated the ratio of liver CSCs (Fig. S2M). Moreover, SNORD88B overexpression could augment tumor initiation capacities (Fig. 2I), CSC frequency (Fig. S2N), and tumor propagation (Fig. 2J, K). Collectively, SNORD88B promotes liver CSCs self-renewal and HCC progression.
Snord88b knockout in mice prevents HCC oncogenesis and propagation
Given that SNORD88B was highly homologous in humans and mice, mouse ortholog snord88b also located in the intron region of the fourth and fifth exons on the 2410002F23Rik gene locus of chr 7, with a length of 93 nt (Fig. S3A). The key motif of mouse snord88b was the same as the human’s and was further verified by sequencing (Fig. S3B). We then generated snord88b knockout (KO) mice using the CRISPR/Cas9 technique to evaluate its in vivo functions (Fig. S3C). Interestingly, we found that only deleting the intronic segments of the snord88b locus resulted in successful knockout mice, while mutating the core motif region of snord88b may lead to embryonic lethality. Snord88b deletion in mice was confirmed by Northern blot and DNA sequencing (Fig. 3A and Fig. S3C, D). Compared with other homologous transcripts, snord88b was modestly expressed in normal mouse livers but high abundant in mouse tumors (Fig. S3E). Of note, the expression of its linear mRNA of 2410002F23Rik showed no difference between WT and KO mice (Fig. S3F).

A Northern blot analysis for snord88b expression in mouse livers. B Schematic diagram of mouse liver tumor. C Tumors were induced in snord88b+/+ and snord88b−/− mice by hydrodynamic tail-vein injection. After 14 days (D14) and 20 days (D20), mice were sacrificed and livers were shown. Black arrows indicate liver tumors. n = 10 for each group. Scale bars, 1 cm. D Representative H&E images of snord88b+/+ and snord88b−/− livers sections at D14 and D20 after hydrodynamic injection. Scale bar, 200 μm. E, F Quantitation of tumor foci numbers (E) and sizes (F) in the livers, n = 4 mice per group. Results are shown as means ± SD. Exact P values: 0.0215, 0.0054. G The ratios of liver weight vs. body weight were presented at D14 and D20 after hydrodynamic injection. n = 4 mice per group. Results are shown as means ± SD. Exact P values: 1.26E−05, 0.00088. H Expression profiles of CSC-related genes in WT or KO tumor tissues. I FACS analysis for mouse liver CSCs (Cd90+Cd44+) sorted from snord88b deleted and control tumor cells. n = 3 biologically independent experiments. Data are shown as means ± SD. Exact P value: 0.003. J Sphere-formation assays were performed with deleted or control snord88b HCC cells. The ratios of sphere area/total area were calculated (right panel). Scale bar, 500 μm. n = 3 independent experiments. Data are shown as means ± SD. Exact P values: 0.002641, 0.013216. K Schematic diagram of liver tumor induced in snord88b-overexpressing mice. L Representative tumor images of control and oe88b livers at D20 after hydrodynamic injection. Scale bar, 1 cm. M, N Quantitation of tumor foci sizes, n = 4 mice per group (M) and tumor area in the livers (N). n = 3 for each group. Results are shown as means ± SD. Exact P value: 0.000901.*P < 0.05; **P < 0.01; ***P < 0.001 by two-tailed Student’s T-test. Data are representative of at least three independent experiments.
Hydrodynamic injection is a classical and efficient mouse model for HCC induction22. We injected sleeping beauty transposition system plasmid HrasG12V plus shp53 into 8-week-old WT and KO mice. At the 14th and 20th days after tail-vein injection, we sacrificed mice for macroscopic examination (Fig. 3B). Interestingly, liver tumorigenesis was dramatically reduced in snord88b KO mice compared with littermate WT mice (Fig. 3C). HCC progression was more aggressive in WT mice, with larger nodular hyperplasia areas and more tumor foci (Fig. 3D–F). Ratios of liver weight vs. body weight are another important indicators of tumor malignancy23, which was much lower in KO than in WT mice (Fig. 3G). Consistent with the inhibition of tumor progression in snord88b KO mouse livers, snord88b KO mainly inhibited HCC cell proliferation rather than affecting apoptosis (Fig. S3G). Of note, snord88b knockout did not affect liver metabolism (Fig. S3G) and morphology of the nucleolus in hepatocytes (Fig. S3H).
We next determined the effect of snord88b on the self-renewal of liver CSCs in mice. We found that snord88b knockout suppressed the expression of CSC-related genes (Fig. 3H), proportion (Fig. 3I), and sphere formation of CSCs (Fig. 3J). In addition, a mouse model with specific high expression of snord88b in livers was established by adenovirus, followed by hydrodynamic tail-vein injection to induce liver cancer (Fig. 3K). We observed that snord88b overexpression dramatically increased liver tumor sizes and oncogenic areas (Fig. 3L–N). Taken together, we conclude that SNORD88B is an intrinsic factor in promoting HCC oncogenesis and propagation.
SNORD88B prevents WRN trafficking from the nucleolus to the nucleoplasm
We then explored the molecular mechanism of SNORD88B in the regulation of liver CSC self-renewal. We predicted that SNORD88B could mediate 2′-O-methylation of 28S rRNA at C3680 via snOPY web (Fig. S4A). We then tested rRNA 2′-O-methylation in control, SNORD88B-deficient, and oe88B in SNORD88B-deficient HCC cell lines via an approach based on RT with limiting dNTP concentration24 (Fig. S4B). We observed that similar amplification between SNORD88B-deficient and control cells in the high levels of dNTPs. In contrast, low dNTP concentrations led to a difference in amplification as an indication of the presence or absence of 2′-O-methylation (Fig. S4C). SNORD88B-deficiency reduced 2′-O-methylation at 28S-C3680, which could be rescued by oe88B (Fig. S4D). Finally, we observed deletion of the matching region (18–27 nt) with 28S RNA in SNORD88B (Δ88B) disrupted rRNA 2′-O-methylation modification. Loss of 2′-O-methylation at 28S-C3680 did not affect spheroid formation (Fig. S4E). These results indicate that SNORD88B modulates the self-renewal of liver CSCs via a non-canonical mechanism.
We then conducted an RNA pulldown assay to identify potentially associated proteins via SNORD88B biotin-labeled probes, followed by mass spectrometry. Of note, a non-canonical protein WRN, a nucleolar shuttle protein, was identified to be a SNORD88B associated protein candidate in oncospheres lysates (Fig. 4A and Fig. S5B). Their interaction was further validated by Western blot (Fig. 4B). RNA-binding sites in WRN (https://www.uniprot.org/uniprotkb/Q14191/feature-viewer) suggest that WRN might be an RNA-binding partner25. WRN could also specifically precipitate SNORD88B rather than other snoRNA in RNA immunoprecipitation (RIP) assay (Fig. 4C). In addition, 3D structure prediction indicated an interaction between SNORD88B and WRN through NPDock Web Servers (Fig. S5A). RNA truncation assay revealed that fragment 27–55 nt of SNORD88B was essential for binding to WRN (Fig. 4D). Based on our predictions, we mapped the region 27–55 into two mutational fragments, 27–34 (Mut#1), and 35–55 (Mut#2). We verified that the 27–34 region of SNORD88B was the binding site for WRN (Fig. S5C). Additionally, the deletion of the 28S-C3680 region did not affect the binding with WRN. Through domain mapping assays, we found that the RecQ domain of WRN protein was required for binding to SNORD88B (Fig. S5D). Furthermore, the co-localization of SNORD88B with WRN in the nucleolus of spheres was confirmed by immunofluorescence staining (Fig. S5E). These data indicate that SNORD88B interacts with WRN in the nucleolus.

A Oncospheres were lysed and incubated with biotinylated SNORD88B sense, antisense, and sepharose 4B beads control. Eluted fractions were resolved by SDS–PAGE followed by silver staining and mass spectrometry identification. The black arrow indicates the WRN band. B The interaction between SNORD88B and WRN was detected by Western blot. C Oncospheres were used for RIP assay. SNORA49 and ACTB were used as controls. n = 3 biologically independent experiments. Results are shown as means ± SD. Exact P value: 0.0399. D Schematic diagram of truncated segments in SNORD88B divided by its secondary structure (upper panel). SNORD88B truncated fragments were incubated with HCC oncosphere lysates, followed by RNA pulldown assay and Western blot (lower panel). E Total protein lysates of oncospheres were separated into cytoplasmic, nucleoplasmic, and nucleolar fractions. The protein levels of WRN were detected in these fractions by Western blot. Nucleolin and GAPDH served as internal references for the nucleolus and the cytoplasm. F Expression levels of the WRN in SNORD88B depleted, overexpressed, and control HCC cells were examined by qRT-PCR. n = 3 independent experiments. Results are shown as means ± SD. G Immunofluorescence staining analysis of GST-labeled WRN and NCL in WRN WT, ΔNoLS, and ΔRecQ CSCs. Representative images are shown. Scale bars, 10 μm. H WRN WT, ΔNoLS, ΔRecQ, and control were overexpressed in HCC cells, followed by an oncosphere formation assay. Representative images are shown in the left panel. Scale bar, 500 μm. Statistical results of sphere-formation ratios were shown in the right panel. n = 3 independent experiments. Data are shown as means ± SD. Exact P values from top to lower: 0.00012, 9.95E−05, 1.97E−05, 0.0076, 0.0023, 0.00072. I WRN WT, ΔNoLS, ΔRecQ, and empty vectors were overexpressed in WRN knockdown cells, followed by clone formation assay. n = 3 independent experiments. Data are shown as means ± SD. Exact P values from top to down: 0.0104, 0.004, 0.0022. J Co-IP assays detected the binding of endogenous WRN and XRCC5 in oncospheres. K GST-labeled full-length WRN (WRN) and ΔNoLS WRN (ΔNoLS) were overexpressed in SNORD88B overexpression cells, followed by immunoprecipitation and Western blot. L Immunofluorescence staining showed co-localization of WRN and XRCC5 in SNORD88B depleted and control CSCs. n = 3 independent experiments. Results are shown as means ± SD. Representative images were shown. Scale bars, 10 μm. Exact P value: 0.000186. *P < 0.05; **P < 0.01; ***P < 0.001 by two-tailed Student’s T-test. Data are representative of at least three independent experiments.
Unexpectedly, we observed that WRN was abundantly localized in the nucleoplasm in SNORD88B depleted oncospheres (Fig. S5E). Since SNORD88B was mainly located in the nucleolus, we proposed that SNORD88B could anchor WRN in the nucleolus. Similarly, subcellular fractionation assay verified that SNORD88B depletion promoted the translocation of WRN from the nucleolus to the nucleoplasm (Fig. 4E). Changing expression levels of SNORD88B did not affect the expression of WRN (Fig. 4F and Fig. S5F). The nucleolar localization signal (NoLS) at the C-terminal residues 1403~1432 of WRN is necessary for the nucleolar localization of WRN26. To further explore the effect of WRN translocation on the regulation of self-renewal of liver CSCs, we constructed GST-labeled WRN with the deletion of NoLS (ΔNoLS) or the SNORD88B interacting region RecQ domain (ΔRecQ) (Fig. S5G). NoLS or RecQ domain truncation disrupted nucleolar localization of WRN (Fig. 4G). Consistently, ΔNoLS or ΔRecQ expressed HCC cells almost abrogated oncosphere formation and tumor cell propagation (Fig. 4H, I). In addition, Actinomycin D (ActD) can promote WRN translocation from the nucleolus to the nucleoplasm27. Of note, ActD treatment remarkably reduced the proportions of CSCs (Fig. S5H). These observations suggest that the translocation of WRN modulates the self-renewal maintenance of liver CSCs.
We used an anti-WRN antibody to perform an immunoprecipitation assay through oncosphere lysates and identified a potential interacting protein candidate (Fig. S5I, J). XRCC5, also named Ku80, was verified to interact with the C-terminus of WRN (amino acid 940-1432) to stimulate the exonuclease activity of WRN28. The WRN and Ku heterodimer complex in nucleoplasm exists in physical and functional interaction in DNA metabolic pathways, such as double-stranded break repair and VDJ recombination29. We confirmed their interaction via co-immunoprecipitation (co-IP) assay (Fig. 4J). Furthermore, we expressed GST-labeled WT or NoLS-truncated WRN in SNORD88B-overexpressed HCC cells and found NoLS-truncated WRN (oe88B + ΔNoLS) bound more XRCC5 protein in the nucleoplasm (Fig. 4K). Parallelly, SNORD88B knockdown enhanced the interaction of WRN with XRCC5 in the nucleoplasm (Fig. 4L). Collectively, SNORD88B anchors WRN in the nucleolus to prevent its trafficking into the nucleoplasm.
XRCC5 is enriched onto the STK4 promoter to inhibit its transcription
XRCC5 acts as a negative transcription regulator to inhibit the transcription of downstream genes30,31. So we conducted transcriptome microarray to screen out upregulated genes in SNORD88B depleted oncospheres, and performed integrative gene set enrichment analysis (GSEA). We noticed that SNORD88B depletion dramatically activated Hippo signaling (Fig. 5A). Among Hippo signaling-related genes, SNORD88B depletion most increased STK4 expression, which is consistent with the most reduced STK4 expression in SNORD88B-overexpressing cells (Fig. 5B, C). Of note, we observed that STK4 expression levels were negatively related to SNORD88B expression levels (Fig. 5D). Moreover, SNORD88B depletion promoted expression and phosphorylation of MST1 (encoded by STK4 gene) (Fig. 5E). We depleted STK4 expression in HCC cells (Fig. S6A, B), and performed sphere-formation assays. We observed that STK4 knockdown remarkably promoted sphere formation (Fig. 5F). These results indicate that STK4 negatively regulates the self-renewal of liver CSCs via the SNORD88B-mediated Hippo signaling pathway.

A GSEA analysis revealed that differential genes between SNORD88B depletion and control cells were attributed to Hippo signaling. NES normalized enrichment score, FDR false discovery rate, FWER familywise error rate. B, C Expression levels of Hippo signaling target genes in SNORD88B depleted or overexpressed oncospheres were detected by qRT-PCR. n = 3 independent experiments. Results are shown as means ± SD. Exact P values from left to right B: 0.01578, 0.01268, 0.002940, 0.000967, 0.01274, 0.04938, 0.000106, 0.0010226, 0.0096286, 0.03333, 0.035386, 0.003267. Exact P values from left to right C: 0.00935, 0.038837, 0.0181, 0.01746, 5.24E−05, 0.05. D SNORD88B and STK4 mRNA levels were detected by qRT-PCR, followed by correlation analysis. Black dots represent HCC samples (n = 23). E Expression levels and phosphorylation of MST1 were examined by Western blot in SNORD88B depleted and control cells. F Sphere-formation assay for STK4 knockdown in HCC cells. Scale bar, 500 μm. n = 3 mice per group. Results are shown as means ± SD. Exact P values from top to down: 0.00136, 0.0009, 0.017, 0.00925. G Huh7 oncospheres were lysed and used for ChIP assay with XRCC5 or IgG antibodies, followed by qPCR. n = 3 biologically independent experiments. Results are shown as means ± SD. Exact P value: 0.000476. H The nuclear protein/DNA complexes in oncospheres were pulled down by biotinylated DNA probes from the STK4 promoter −3800 to −3600 region, followed by Western blot assays. NSP, non-specific probe. I ChIP-qPCR analysis of XRCC5 enrichment on the −3800 to −3600 bp region of the STK4 promoter in SNORD88B depletion, overexpression, and control cells. n = 3 independent experiments. Results are shown as means ± SD. Exact P values from top to down: 0.0055, 0.0031, 0.00144. J SNORD88B depletion enhanced the chromatin accessibility of the STK4 promoter by DNase I digestion assays. n = 3 independent experiments. Results are shown as means ± SD. Exact P values: 0.00322, 0.00324. K, L Expression levels of XRCC5 in HCC samples (K) and metastatic patients (L) provided by Wang’s cohort (GSE14520). M0 represents patients with low metastases and M1 represents patients with high metastases. M Expression levels of XRCC5 in HCC samples provided by TCGA. N Kaplan–Meier survival curves of HCC samples from TCGA. Based on the XRCC5 expression levels, patients were divided into two groups. Kaplan–Meier method with the log-rank test was used. *P < 0.05; **P < 0.01; ***P < 0.001 by two-tailed Student’s T-test. Data are representative of at least three independent experiments.
To further explore how XRCC5 regulates STK4 transcription, we performed chromatin immunoprecipitation (ChIP) assays with anti-XRCC5 antibody. 4 kb region upstream of the STK4 gene transcription start site (TSS) was mapped into 10 segments (Fig. S6C). We found that XRCC5 protein was enriched at −3800~−3600 bp region of the STK4 promoter (Fig. 5G). We pulled down the protein/DNA complex in oncosphere nuclear extract via a biotin-labeled DNA probe and validated by Western blot (Fig. 5H). We performed a dual luciferase reporter assay (DLR) to verify their interaction (Fig. S6D). We noticed that XRCC5 associated with the STK4 promoter at the −3000~−4000 region to suppress its expression in WRN-depleted cells (Fig. S6E, F). In addition, SNORD88B overexpression remarkably increased XRCC5 enrichment on the STK4 promoter (Fig. 5I). Moreover, SNORD88B depletion dramatically augmented DNase I sensibility on the STK4 promoter region (Fig. 5J). In contrast, SNORD88B overexpression apparently reduced DNase I sensibility in the STK4 promoter region (Fig. S6G). We next silenced XRCC5 expression by small interfering RNAs (siRNAs) and validated by qRT-PCR and Western blot (Fig. S6H). We observed that XRCC5 depletion remarkably increased the transcription of luciferase in DLR assay (Fig. S6I) and STK4 expression (Fig. S6J). We further analyzed expression levels of XRCC5 by GEO and TCGA databases. We noticed that XRCC5 was highly expressed in HCC tumor and metastasis patients (Fig. 5K–M). Additionally, XRCC5 expression was positively correlated with poor prognosis of HCC patients (Fig. 5N). Taken together, these results indicate that XRCC5 binds to the STK4 promoter to inhibit its transcription.
MST1 negatively regulates the self-renewal of liver CSCs as an upstream kinase of Hippo signaling
To further determine the function of STK4 in liver CSCs, we detected expression levels of STK4 in HCC tumors and peri-tumor tissues, oncospheres and non-spheres, and liver CSCs and non-CSCs. We found STK4 was lowly expressed in tumor tissues, oncospheres, and liver CSCs (Fig. 6A–C). These observations were further confirmed by immunohistochemical staining (Fig. 6D and Fig. S7A). Of note, low expression of STK4 was also validated by the GEO cohort (Fig. 6E). In addition, STK4 depletion accelerated tumor growth via subcutaneous tumor formation assay (Fig. 6F). Consistently, MST1 depletion dramatically promoted expression of Hippo signaling downstream target genes (Fig. S7B). MST1 is an upstream kinase in the Hippo signaling pathway that causes phosphorylation of the downstream effector YAP1 protein. We observed an increase in nuclear localization of YAP and downregulation of YAP phosphorylation in HCC tumor tissues, which was consistent with MST1 expression and phosphorylation (Fig. 6G, H and Fig. S7C). These data indicate that MST1 functions as a tumor suppressor.

A–C SKT4 expression was detected in HCC tumors and peri-tumors (A), oncospheres and non-spheres (B), as well as liver CSCs and non-CSCs (C). n = 3 independent experiments. Results are shown as means ± SD. Exact P values from left to right A: 0.0406, 0.0115, 0.0063, 0.0027, 0.036, 0.0104. Exact P values from left to right B: 0.0063, 0.0069, 0.0003, 0.013, 0.0025. Exact P values from left to right C: 0.022, 0.003, 0.031, 0.029, 0.0037. D Immunohistochemistry with anti-MST1 antibody was performed in HCC samples. Scale bar, 400 μm. E Expression levels of SKT4 in HCC samples provided by Wang’s cohort (GSE14520). F 1 × 106 STK4 depleted or control cells were subcutaneously injected into BALB/c nude mice, followed by measurement of tumor parameters every 4 days. n = 3 for each group. Results are shown as means ± SD. Exact P values from left to right: 0.00062, 0.00126, 0.027, 0.016. G Immunohistochemistry with YAP antibody was performed in human HCC samples. Scale bars, 75 μm. H Expression levels of MST1, pMST1, and pYAP1 were examined by Western blotting in HCC tumors and peri-tumors. I STK4 deficiency rescued the impaired sphere-formation capacity, which was caused by SNORD88B ASO2. Representative images (upper panel) and statistical results (lower panel) are shown. Scale bar, 500 μm. n = 3 independent experiments. Results are shown as means ± SD. Exact P values from left to right: 0.0005, 0.023, 2.82E−05, 0.002. J Western blot analysis showed that SNORD88B overexpression remarkably reduced YAP1 phosphorylation, which could be rescued by overexpression of MST1. K Localization of YAP1 in SNORD88B knockdown or control liver CSCs was analyzed by immunofluorescence staining. Scale bars, 40 μm. *P < 0.05; **P < 0.01; ***P < 0.001 by two-tailed Student’s T-test. Data are representative of at least three independent experiments.
We observed that MST1 depletion was able to rescue oncosphere formation in SNORD88B-depleted HCC cells (Fig. 6I). Of note, SNORD88B overexpression decreased MST1 expression (Fig. 6J). Moreover, MST1 overexpression in SNORD88B-overexpressed cells remarkably enhanced YAP1 phosphorylation (Fig. 6J). In addition, SNORD88B depletion prevented phosphorylated YAP1 into the nucleus (Fig. 6K). Moreover, ASO treatment also activated Hippo signaling (Fig. S7D). Parallelly, MST1 expression and cytoplasmic YAP1 were also upregulated in snord88b KO mouse tumors (Fig. S7E). These data indicate that SNORD88B-mediated MST1 downregulation promotes the self-renewal of liver CSCs and tumor growth through suppression of YAP1 phosphorylation.
Administration of MST1 agonist ADA with ASOs against SNORD88B displays synergistic antitumor effects
Adapalene (ADA) has been documented to be an agonist for MST132. We found that ADA could promote phosphorylation of MST1 and YAP1 (Fig. 7A). Moreover, ADA treatment abolished oncosphere formation of HCC cell lines and HCC primary cells (Fig. 7B). We nest induced HCC tumor in 8-week-old snord88b KO and WT mice by hydrodynamic injection, followed by twice oral ADA administration (Fig. 7C). We observed that ADA administration dramatically reduced tumor formation and tumor growth (Fig. 7D). Consequently, ADA treatment promoted survival rates of snord88b KO mice compared to those of WT mice (Fig. 7E). Of note, administration of ADA with ASOs against SNORD88B displayed apparent synergistic antitumor effects on mouse HCC tumor models (Fig. 7F–H), which therapy disrupted YAP activation (Fig. S7F). Finally, we injected luciferase-labeled human HCC cells into murine livers to establish orthotopic patient-derived tumor cell (PDC) models for validation of therapeutic effects. Consistently, administration of ADA plus ASOs against SNORD88B exerted a dramatic therapeutic effect on orthotopical PDC xenografts (Fig. 7I, J). Taken together, these results indicate that the administration of ADA and ASOs against SNORD88B has synergistic therapeutic effects on HCC tumors.

A The effect of ADA treatment on MST1 phosphorylation was detected by Western blot. B Sphere-formation assays for ADA treatment in HCC cells. Representative images (left panel) and statistical results (right panel) are shown. Scale bars, 500 μm. n = 3 independent experiments. Results are shown as means ± SD. Exact P values from left to right: 0.0018, 0.0033. C Schematic representation of the therapy method. WT and KO mice were used. D Mice were treated with ADA (10 mg/kg) and vehicle at days 14 and 20. The macroscopic appearance of snord88b+/+ and snord88b−/− livers are shown. n = 6 for each group. Scale bars, 200 μm. E Survival curve of indicated mice after treatment with hydrodynamic injection. n = 6 for each group. Exact P value: 0.00971. F Illustration of ASO/ADA treatment timeline in mice. G, H Treatment with ASO combined ADA improves synergistic therapeutic effect in mice bearing tumors after hydrodynamic injection. Representative H&E images (G) and survival analysis (H) are shown. n = 5 for each group. Scale bars, 200 μm. Exact P value G: 0.0132. I Schematic representation of the therapy method. Immunodeficient B-NSG mice were used. J Orthotopic human liver tumor growth was imaged via luciferase signals. Representative images are shown (left panel), and statistical results are shown as means ± SD (right panel). n = 3 for each group. Exact P values from upper to lower: 0.0003, 0.0011, 0.0062, 0.003. *P < 0.05; **P < 0.01; ***P < 0.001; by two-tailed Student’s T-test. Data are representative of at least three independent experiments.
Discussion
HCC progression is fueled by CSCs33. Thus targeting liver CSCs as therapeutic strategies will become a hope for the treatment of HCC patients. Epigenetic regulations act as a critical layer for the self-renewal of liver CSCs. Our lab previously defined some pivotal non-coding RNAs to modulate the self-renewal of liver CSCs18,34,35,36,37,38,39. In this study, we identified a category snoRNA SNORD88B which was highly expressed in liver CSCs and HCC tumors. SNORD88B initiated self-renewal of liver CSCs via a non-canonical fashion by anchoring WRN protein in the nucleolus, leading to suppression of Hippo signaling activation. Intriguingly, the administration of ADA plus ASOs against SNORD88B exerted dramatic synergistic therapeutic effects on HCC tumors.
In recent years, accumulative studies have revealed the important role of snoRNAs in tumorigenesis40. On the one hand, they function in a canonical manner by affecting the function of the nucleolus. snoRNAs activate PARP-1 to ADP-ribosylate DDX21 to promote rDNA transcription, ribosome biogenesis, and cell growth in human breast cancer41. SNORD42A accelerates the growth and survival of leukemia cells via directing 2′-O-methylation at U116 of 18S rRNA24. On the other hand, snoRNAs modulate tumor propagation in a nucleolar function-independent manner. SNORD17 binds NPM1 and MYBBP1A to inhibit p53 activation to drive HCC progression17. Here we identified a non-canonical function of SNORD88B by binding WRN and altering its nucleolar-nucleoplasmic distribution to regulate self-renewal of liver CSCs and HCC tumorigenesis.
WRN mutation-induced iPSCs can maintain long-term self-renewal42. Meanwhile, WRN can inhibit self-renewal of embryonic stem cells (ESCs) via de novo DNA methylation of OCT4 promoter43. These data suggest that abrogated WRN may participate in stem cell reprogramming and self-renewal. Mutation or deletion of WRN is related to increased cancer susceptibility in non-MSI (microsatellite instability) cancers44. Lower expression of WRN may serve as an independent factor for poor prognosis of early HCC45 and advanced HCC46. We further revealed a WRN shuttling mechanism based on snoRNA that may be involved in the regulation of liver CSCs self-renewal and hepatocarcinogenesis, adding a layer for WRN regulation.
Dysregulation of Hippo signaling is a critical character of many human tumors, and YAP has been reported to be critical for the maintenance of multiple cancer hallmarks. We previously showed that the accumulation of YAP in the nucleus exerts a critical role in the self-renewal of liver CSCs37. Interestingly, silencing WRN expression induces an accumulation of YAP protein47. In this study, we explained this phenotype was dependent on XRCC5-STK4 axis regulation. XRCC5, a negative transcription factor, directly binds negative regulatory element 1 (NRE1) to inhibit MMTV gene transcription in mice30. We found that XRCC5 interacted with the NRE1-like sequences (−3800 to −3600 bp region) existed in the STK4 promoter to suppress its transcription, providing a possible mechanistic explanation for the high expression of XRCC5 in HCC samples and its positive correlation with poor clinical prognosis of HCC.
Despite our limited understanding of the molecular mechanisms underlying functional snoRNAs in tumorigenesis, accumulating evidence has suggested that snoRNAs may be ideal candidates for tumor therapy. A combined treatment of rapamycin and adenovirus-delivered SNORD44 efficiently inhibits CRC growth in CDX models48. ASO-mediated silencing of SNORA23 hinders pancreatic ductal adenocarcinoma propagation49. Here we showed that ASOs against SNORD88B dramatically reduced tumor burden in PDX models, indicating that targeting SNORD88B by ASOs could be a potential therapeutic strategy for the treatment of HCC patients. Human clinical trials have validated the safety of ASO intrathecal administration50,51. However, no ASO drugs have been approved by the FDA yet. Efficient delivery of ASOs into tumors and prevention or mitigation of subsequent immune response are still required to be further investigated. With the advancement of non-viral delivery systems such as nano-delivery and liposome delivery systems, we believe that ASOs will be irreplaceable in the treatment of tumors in the future.
In summary, our findings reveal a non-canonical function of SNORD88B in liver CSCs by anchoring WRN in the nucleolus and inhibiting the transcription of STK4 via XRCC5. The SNORD88B-WRN-XRCC5-STK4-YAP1 axis facilitates the self-renewal of liver CSCs and enhances HCC tumorigenesis. Our work suggests that snoRNAs may be targets for HCC therapy, paving the way to develop more effective therapeutic strategies for HCC patients.
Methods
All experiments in this article comply with all relevant ethical regulations and are approved by the Institutional Committee of Institute of Biophysics, Chinese Academy of Sciences. All mice were housed under a specific pathogen-free environment, with 12 h dark/light cycle, 20 ± 2 °C, and 50% humidity conditions, and were used for experiments at the age of 6–8 weeks. The animal experimental protocols were approved by the Institutional Animal Care and Use Committee of Institute of Biophysics, Chinese Academy of Sciences. Mice were sacrificed when they developed tumors larger than 20 mm in diameter or a total tumor burden not exceeding 12% of the animal’s body weight or skin ulceration. In this study, we confirm that at no point during the study did any animal exceed the maximal permitted tumor size or burden. There are no biases in the selection of patients’ sex and gender. The materials used in this study are listed in Supplementary Data 1.
HCC samples
We selected surgically resected primary HCC tumor tissues and paired peri-tumor tissues from a cohort of 28 HCC patients who had not received previous chemotherapy or radiotherapy. Clinical-pathological features of HCC patients are shown in Table S1. Of these cases, all patients had HBV-infection background. Pathological diagnosis was made according to the histology of tumor specimens or biopsy and examined by experienced pathologists. All tissue samples were obtained from consenting patients and approved by the Institutional Review Board of the Institute of Biophysics, Chinese Academy of Sciences. All specimens were obtained from the partial hepatectomy series at the Department of Hepatobiliary Surgery, PLA General Hospital (Beijing, China).
HCC Cell lines, HCC primary cells, and sphere-formation assay
Human HCC cell lines Huh7, Hep3B, and PLC/PRF/5 (PLC) were provided by Dr. Zeguang Han (Shanghai Jiao Tong University School of Medicine, Shanghai, China). All these cell lines were maintained in a DMEM medium supplemented with 10% FBS, 100 μg/ml penicillin G, and 100 U/ml streptomycin. Tumor bulk was dissociated into 1 m3 with scissors in digestion buffer (0.1% collagenase IV, 0.01% DNase, 0.05% proteinase) at 37 °C for 45 min, with a shaker at 1–2 cycles/second. Then the supernatant was collected and passed through a 100 μm cell strainer. Centrifuge the filtrate at 50 × g for 1 min and collect the supernatant, further centrifuge at 150 × g for 8 min, and the cells were enriched in the pellet. After red cell elimination, HCC primary cells were obtained and passaged through serial sphere-formation assay.
A sphere-formation assay was performed with 2000 HCC cells, murine HCC cells, or 5000 primary HCC cells. These cells were seeded into Low Attachment 6-well plates (Corning) and cultured in DMEM/F12 serum-free medium supplemented with 20 ng/ml bFGF, 20 ng/ml EGF, N2, and B27. Sphere initiating ratio = (sphere number) /indicated cells × 100%.
shRNA and siRNA knockdown system
All shRNAs were designed on RNAi designer (Thermo). 3 shRNAs of each indicated gene were selected and cloned into pSicoR-Puro (catalog 12084, Addgene) lentivirus vector, and the primers were listed in Table S2. pSicoR-Puro vectors were co-transfected with packaging plasmids pVSVg (catalog 8454, Addgene) and psPAX2 (catalog 12,260, Addgene) plasmids into 293T cells. Lentivirus was collected after 2 days and filtered with a 0.45 μm strainer. After mixed with equal volume fresh DMEM, HCC cells were infected at 37 °C for 12 h, followed by puromycin selection. HCC cells were passaged and gene silencing efficiency was analyzed by qRT-PCR, and the primers are listed in Supplementary Data 2.
For the siRNA knockdown system, all siRNAs were designed on the DSIR website. HCC cells were cultured in 6-well plates. According to the manufacturer’s instructions, when cells reached 70% density, 100 pmol siRNAs and their negative control (Tsingke, China) were transfected with Lipofectamine 3000 (Life Technologies, US) per well, and the primers are listed in Table S3.
Generation of SNORD88B knockout mice by CRISPR/Cas9 technology
SNORD88B knockout mice on a C57BL6/J background were generated using a CRISPR-mediated approach52. A pair of sgRNAs targeting intron sequences flanking the SNORD88B sequences was designed. Corresponding sgRNAs and identified primers are shown in Table S4. Approximately 250 zygotes from C57BL/6 mice were injected with corresponding sgRNAs and subsequently transferred into the uterus of pseudo-pregnant ICR females from which viable F0 mice were obtained. Genomic DNA deletion was verified by PCR and DNA sequencing, followed by a Northern blot. Both male and female mice were induced for HCC tumorigenesis at 8 weeks. At least five mice per group were used in each experiment.
In the process of experiments and outcome assessment, researchers have no subjectivity about the allocation. BALB/c nude (CAnN.Cg-Foxn1nu/Crl) mice, developed through crosses and back-crosses between BALB/cABom-nu and BALB/CAnNCrj-nu, lack a thymus, are unable to produce T-cells. 7-week-old female BALB/c nude mice were obtained from Beijing Vital River Laboratory Animal Technology. B-NDG (NOD-PrkdcscidIL2rgtm1/Bcgen) mice lack mature T-cells, B cells, and functional NK cells, and display cytokine signaling deficiencies due to the deletion of the IL2rg gene from the NOD-scid genetic background. 7-week-old female Immunodeficient B-NSG mice were purchased from Biocytogen. These mice were used in follow-up experiments after a week of acclimatization in the animal room. For the limiting dilution assay, gradient numbers of HCC cells mixed with 100 µl Matrigel were subcutaneously injected into BALB/c nude mice (8 W) for 3 months to detect tumor initiation. For the subcutaneous tumor formation assay, 106 HCC cells were subcutaneously injected into BALB/c nude mice (8W), followed by measurement of tumor progression every 4 days. Tumor volume = (length × width2)/2. Mice were sacrificed when they developed tumors larger than 20 mm in diameter or skin ulceration. All mouse experiments were conducted in accordance with the relevant guidelines and approved by the Institutional Animal Care and Use Committees at the Institute of Biophysics, Chinese Academy of Sciences.
Hydrodynamic tail-vein injection
For intrahepatic delivery of the transposon system, Gender-randomized 8-week-old mice with C57BL/6 background were secured and hydrodynamically injected with 20 μg of pbabe-c-mycT58A+HRasG12V plasmid (catalog 11130, Addgene), 20 μg of pT2-shP53 (catalog 124261, Addgene) and 20 μg of luciferase and SB transposases expressing plasmid (catalog 20207, Addgene) via lateral tail vein. All plasmids were suspended in sterile-filtered 0.9% NaCl or 1% PBS solution. The supernatant solution was adjusted to a total volume of 10% (in ml) of the total body weight (in grams) and subsequently injected in less than 7 s. All injected mice were monitored through luciferase signals and sacrificed at appropriate time points.
Mouse model for overexpressing SNORD88B
The U6 promoter in the AAV-sgRNA (catalog 60231, Addgene) plasmid was replaced by the liver-specifically expressed promoter TGB (AAV-TBG-GFP). SNORD88B gene was cloned into an AAV-TBG-GFP adenovirus vector, which was co-transfected with packaging plasmids pHelper and pAnc plasmids into 293T cells. Two days later, the adenovirus was collected using a Universal Virus Concentration Kit (C2901S, Beyotime), dissolved in 1 ml of normal saline. 200 μl of virus was injected into mice via the tail vein using an insulin needle. Five days later, liver tumors were induced in mice by hydrodynamic tail-vein injection. Gender-randomized 8-week-old mice with C57BL/6 background were used.
Isolation of nucleoli
2 × 107 Huh7 cells were collected and suspended in 200 μl lysis buffer (10 mM Tris pH 7.4, 140 mM NaCl, 1.5 mM MgCl2, 1× protease inhibitor cocktail) for 10 min on ice. 20 μl lysates were taken out for total RNA extraction or Western blot. The remaining lysate was centrifuged at 300 × g for 5 min at 4 °C to pellet the nuclear fraction and the supernatant was the cytoplasmic fraction. To prepare nucleoplasm and nucleolar fractions, the nucleus pellet was resuspended by 20 μl sucrose solution (340 mM, 5 mM MgCl2) and broken by sonication until complete nuclei cannot be observed under the microscope. 200 μl sucrose solution (880 mM, 5 mM MgCl2) was gently added to the bottom of the sonicated nuclear fraction and then centrifuged at 500 g for 20 min at 4 °C to pellet nucleoli, and the supernatant was the nucleoplasm fraction. The pellet was resuspended in 200 μl sucrose buffer (340 mM) as a nucleolar fraction for analysis.
Immunohistochemistry assay
Paraffin sections of tumor tissue were deparaffinized in xylene and rehydrated in gradient alcohol, and treated with 3% hydrogen peroxide (H2O2) for 10 min to block the activity of endogenous peroxidase. The sections were incubated with boiling Tris-EDTA buffer for 15 min and blocked with 10% donkey serum for 30 min. Subsequently, the slides were incubated with primary antibodies at RT (room temperature) for 2 h. After incubation with HRP-conjugated secondary antibodies at RT for 1 h, the slides were performed with DAB, counterstained with hematoxylin, and then dehydrated and mounted.
Real-time PCR (qRT-PCR)
Total RNA was isolated from corresponding samples using standard Trizol methods. The complementary DNA (cDNA) template was synthesized using a 5× All-In-One RT Master Mix. SYBR Green reaction system was applied for quantitative real-time PCR analysis according to the manufacturer’s instruction on the CFX Connect™ Real-Time PCR Detection System (Bio-Rad, USA). The experiment was performed three times independently and the results were analyzed by 2−ΔΔCt method. ACTB or U6 was used as endogenous control. Primers used in this study are listed in Supplementary Data 2.
In Vivo Imaging system (IVIS)
1 × 106 luciferase transfected Huh7 cells were injected in situ into the liver of mice. After 2 weeks, 3 mg D-fluorescein potassium salt was injected intraperitoneally into the mice and waited for 10 min. Then the mice were anesthetized with isoflurane. Luciferase activity was detected by the lumina3 machine. Results are analyzed using Living Image 4.3 software (PerkinElmer).
Flow cytometry
Human liver CSCs were stained with FITC-conjugated CD13 and PE-conjugated CD133 antibodies or PE-conjugated CD44 antibodies and sorted via flow cytometry. According to the reported markers of murine liver CSCs53, cells were stained with PE-conjugated Cd90 and APC-conjugated Cd44 antibodies and sorted via flow cytometry. The sorting strategy of combined Cd90 with Cd44 has good crosstalk in mice. FlowJo 10.8.1 software was used for subsequent analysis. The gate strategies of FACS were all listed in Supplementary Data 3.
RNA fluorescence in situ hybridization (FISH)
Biotin-labeled SNORD88B probes (sequence: GUGCACCGUGUCCUCAGGGGCA) were synthesized and used for RNA FISH. Fluorescein TSA kit was used to couple 488 fluorescent signals to biotin, after which indicated antibodies or fluorescence-conjugated secondary antibodies were added. After sealing, treated samples were observed by confocal microscopy (Nikon, A1R+).
Generation of PDC models and ASO treatment
20 nt of ASOs are fully phosphorothioated and contain five 2′-O-methyl-modified ribonucleotides at both ends. 1 × 106 HCC primary cells obtained from patients were subcutaneously injected into 8-week-old female B-NSG mice. ASOs (25 mg/kg) were peri-tumor injected on days 24, 26, 28, 30, and 32. Mice were sacrificed on the 40th day after injection, and the tumors were excised and weighed. The hydrodynamic injection tumor model was established as described before. ASOs (50 mg/kg) were intraperitoneally injected on days 14 and 20. Survival rates of indicated mice were calculated after 40 days. ASO sequences are shown in Table S3.
Assay of 2′-O-methylation on 28S-C3680
2′-O-methylation of 28S-C3680 was analyzed according to a previously published protocol24. Reverse transcription was performed using RNase H minus Moloney leukemia virus reverse transcriptase, RNasin ribonuclease inhibitor, and 1 mM of reverse primer targeting the sequence downstream to 28S-C3680, with either 10 mM or 1 mM dNTPs in the reaction. The reaction was incubated for 5 min at 37 °C and stopped at 70 °C for 15 min. Amplification of cDNA was measured by qPCR and methylation levels were calculated with the formula 2(CT-low/CT-high).
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed according to the normative protocol (Uptate Biotechnology, Inc.). Briefly, oncosphere cells were fixed in 1% formaldehyde for 10 min at 37 °C, and then lysed by SDS buffer for 10 min on ice, followed by ultrasonic to shear DNA into fragments between 200 and 1000 bp. The samples were precleared with Protein A/G Agarose Beads (catalog G103-01, GenStar) for 0.5 h in rotor, and then incubated with the anti-XRCC5 or control antibodies. The eluted fractions were then subjected to DNA isolation and analysis by qPCR. Primer sequences are shown in the Table S5.
RNA pulldown and mass spectrometry assay
Biotin-labeled RNA probes (SNORD88B sense and antisense sequences) were in vitro transcribed with the T7 RNA polymerase (catalog 108817670010, Roche) and biotin RNA labeling mix (catalog 11685597910, Roche). Oncosphere lysate was incubated with a 3 mg probe overnight at 4 °C. Then the streptavidin-conjugated agarose CL-4B beads (catalog 85881, Sigma-Aldrich) were added to enrich RNA probes for 2 h. After washing with RIPA buffer, the retrieved proteins were resolved by SDS–PAGE and visualized by silver staining. Differential bands from three independent experiments were excised and mixed for mass spectrometry (LTQ Orbitrap XL, Thermo).
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis and database search: In-Gel Digestion, silver-stained gel bands were destained, reduced with DTT, and alkylated with iodoacetamide. Trypsin was then added for overnight digestion. Peptides were extracted using multiple steps with different concentrations of acetonitrile. The peptide mixtures obtained from the digestion were first analyzed by LC-MS/MS. Database searching and protein identification were performed using the SEQUEST HT search engine in Thermo Proteome Discoverer (version 1.4.0.288) against the Uniprot_human_2018 database. The search parameters were as follows: trypsin as the enzyme with up to two missed cleavages allowed, precursor ion mass tolerance of less than 10 ppm, and fragment ion mass tolerance of less than 20 mDa. Carbamidomethylation of cysteine was set as a fixed modification, while oxidation of methionine and lactation of lysine were set as variable modifications. Search results were filtered using Percolator with a Delta Cn cutoff of less than 0.1 and an FDR of 1%. Peptide filtering was based on peptide confidence set to high.
Domain mapping
According to WRN functional domain regions, WRN was divided into three fragments, which were constructed into pcDNA3-C-terminal GST plasmids. Lipo3000 was used to transfer plasmids into WRN knockdown cells. After 4 days, nuclear fractions were extracted and preformed via RNA pulldown and Western blotting.
SnoRNA and transcriptome sequencing
For snoRNA sequencing, total RNA was isolated with Trizol regent from liver CSCs (CD133+CD13+) and non-CSCs (CD133−CD13−) from human HCC samples. Then snoRNAs were sequenced using a human snoRNA Array (YINGBIO Tech, China). For identification of snoRNA downstream target genes, total RNA was isolated from SNORD88B-depleted or control oncosphere cells, followed by NimbleGen sequencing (BGI Tech Company) analysis.
Overexpression of SNORD88B and MST1
For SNORD88B overexpression, its full-length sequence was cloned into the pBPLV-GFP vector. For MST1 overexpression, pBPLV-GFP-MST1 plasmid was constructed. Then these plasmids were co-transfected with package plasmids (pBPLV: VSVG: pLp1: pLp2 = 5: 2.8: 4.2: 2 μg) into 293T cells for virus production. After the supernatant was filtered with a 0.45 μm strainer and mixed with an equal volume of fresh DMEM, HCC cells were infected for 12 h, followed by GFP sorting. The full-length or lacking domain sequence of WRN was cloned into pGEX-6P-1 plasmid and transiently transfected into Huh7 cells and then induced to express by IPTG.
DNase I sensibility assay
Nuclei were isolated according to the protocol of the nuclei-isolation kit (catalog 78833). Then cell nuclei were suspended in 200 μl DNase Digestion Buffer and digested with 2 U DNase I for 5 min at 37 °C. After stopping the digestion, total DNA was extracted and followed by qRT-PCR.
Bioinformatics analysis
R software (V.4.2.0) was used to analyze SNORD88 in liver cancer based on the TCGA database. Firstly, the RNA-sequencing data of liver cancer were selected, combined with the patient’s prognosis, and pathological stage information. After removing meaningless data, the violin maps and boxplots were drawn using the R package of ggplot2, pheatmap, VennDiagram, etc. Survival analyses of HCC patients with high or low SNORD88 expression was performed using the package of survival and survminer. and then drew a survival curve using ggsurvplot package.
Computational modeling of SNORD88B-WRN interaction
Predicting RNA and protein interactions has previously been reported (Liang Y, Mol Ther Nucleic Acids, 2020). The secondary structure of SNORD88B was predicted using the RNAfold web server (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi), as described by Zuker in Nucleic Acids Research, 2003. The minimum free energy (MFE) and partition function were checked, under the condition of avoiding isolated base pairs. Next, the 2D structure of SNORD88B was submitted to RNA Composer (https://rnacomposer.cs.put.poznan.pl/) to generate a 3D structure as described by Popenda et al. in Nucleic Acids Research, 2012. The crystal structure of WRN was retrieved from AlphaFold (AF-Q14191-F1). We then performed RNA-protein docking studies using PyMol software and HDock (http://hdock.phys.hust.edu.cn, default parameters). Hydrogen bond interactions with high confidence scores were selected. The detailed interactions of RNA-protein were performed using chimerax.
Statistics and reproducibility
Data were statistically analyzed by two-tailed Student’s T-test using the GraphPad Prism 8.0 and Excel 2019 software. P < 0.05 was considered significant (*P < 0.05; **P < 0.01; ***P < 0.001); NS, non-significant. At least three independent experiments were performed with similar results for each assay, and representative experiments are shown. Tumorigenic cell frequency was calculated according to extreme limiting dilution analysis website instructions (ELDA, https://bioinf.wehi.edu.au/software/elda/).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The source data for Figs. 1–7 and Supplementary Figs. 1–7 are provided as a Source Data file. SnoRNAs sequencing data that support the findings of this study have been deposited in the Gene Expression Omnibus under accession code GSE233521. SNORD88B deficiency sequencing data are available from the Gene Expression Omnibus database under accession code GSE233520. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD044847. The remaining data are available within the Article, Supplementary Information, or Source Data file. Source data are provided with this paper.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Huang, A., Yang, X. R., Chung, W. Y., Dennison, A. R. & Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target Ther. 5, 146 (2020).
Ji, J. et al. MicroRNA expression, survival, and response to interferon in liver cancer. N. Engl. J. Med. 361, 1437–1447 (2009).
Lee, T. K., Guan, X. Y. & Ma, S. Cancer stem cells in hepatocellular carcinoma - from origin to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 19, 26–44 (2022).
Meacham, C. E. & Morrison, S. J. Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337 (2013).
Matsumoto, T. et al. Proliferating EpCAM-positive ductal cells in the inflamed liver give rise to hepatocellular carcinoma. Cancer Res. 77, 6131–6143 (2017).
Shin, S. et al. Genetic lineage tracing analysis of the cell of origin of hepatotoxin-induced liver tumors in mice. Hepatology 64, 1163–1177 (2016).
Wang, Y. et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 7, e1440169 (2018).
Tsui, Y. M., Chan, L. K. & Ng, I. O. Cancer stemness in hepatocellular carcinoma: mechanisms and translational potential. Br. J. Cancer 122, 1428–1440 (2020).
Harvey, K. F., Zhang, X. & Thomas, D. M. The Hippo pathway and human cancer. Nat. Rev. Cancer 13, 246–257 (2013).
Ma, S., Meng, Z., Chen, R. & Guan, K. L. The hippo pathway: biology and pathophysiology. Annu. Rev. Biochem. 88, 577–604 (2019).
Liu, J., Liao, X., Zhu, X., Lv, P. & Li, R. Identification of potential prognostic small nucleolar RNA biomarkers for predicting overall survival in patients with sarcoma. Cancer Med. 9, 7018–7033 (2020).
Kiss, T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell 109, 145–148 (2002).
Lafontaine, D. L. & Tollervey, D. Birth of the snoRNPs: the evolution of the modification-guide snoRNAs. Trends Biochem. Sci. 23, 383–388 (1998).
Siprashvili, Z. et al. The noncoding RNAs SNORD50A and SNORD50B bind K-Ras and are recurrently deleted in human cancer. Nat. Genet. 48, 53–58 (2016).
Xu, G. et al. Small nucleolar RNA 113-1 suppresses tumorigenesis in hepatocellular carcinoma. Mol. Cancer 13, 216 (2014).
Liang, J. et al. Non-coding small nucleolar RNA SNORD17 promotes the progression of hepatocellular carcinoma through a positive feedback loop upon p53 inactivation. Cell Death Differ. 29, 988–1003 (2022).
Wang, Y. et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 16, 413–425 (2015).
Wu, J. et al. The long non-coding RNA LncHDAC2 drives the self-renewal of liver cancer stem cells via activation of Hedgehog signaling. J. Hepatol. 70, 918–929 (2019).
Li, S. et al. The human lung cancer drug resistance-related gene BC006151 regulates chemosensitivity in H446/CDDP cells. Biol. Pharm. Bull. 33, 1285–1290 (2010).
Boumahdi, S. et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature 511, 246–250 (2014).
Ju, H. L. et al. Investigation of oncogenic cooperation in simple liver-specific transgenic mouse models using noninvasive in vivo imaging. PLoS ONE 8, e59869 (2013).
Kondylis, V. et al. NEMO prevents steatohepatitis and hepatocellular carcinoma by inhibiting RIPK1 kinase activity-mediated hepatocyte apoptosis. Cancer Cell 28, 830 (2015).
Pauli, C. et al. Site-specific methylation of 18S ribosomal RNA by SNORD42A is required for acute myeloid leukemia cell proliferation. Blood 135, 2059–2070 (2020).
Yang, F. et al. Cis-acting circ-CTNNB1 promotes beta-catenin signaling and cancer progression via DDX3-mediated transactivation of YY1. Cancer Res. 79, 557–571 (2019).
Suzuki, T., Shiratori, M., Furuichi, Y. & Matsumoto, T. Diverged nuclear localization of Werner helicase in human and mouse cells. Oncogene 20, 2551–2558 (2001).
Shiratori, M. et al. WRN helicase accelerates the transcription of ribosomal RNA as a component of an RNA polymerase I-associated complex. Oncogene 21, 2447–2454 (2002).
Karmakar, P., Snowden, C. M., Ramsden, D. A. & Bohr, V. A. Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res. 30, 3583–3591 (2002).
Orren, D. K. et al. A functional interaction of Ku with Werner exonuclease facilitates digestion of damaged DNA. Nucleic Acids Res. 29, 1926–1934 (2001).
Giffin, W. et al. Sequence-specific DNA binding by Ku autoantigen and its effects on transcription. Nature 380, 265–268 (1996).
Chung, U. et al. The interaction between Ku antigen and REF1 protein mediates negative gene regulation by extracellular calcium. J. Biol. Chem. 271, 8593–8598 (1996).
Yadav, P., Bhatt, B. & Balaji, K. N. Selective activation of MST1/2 kinases by retinoid agonist adapalene abrogates AURKA-regulated septic arthritis. J. Immunol. 206, 2888–2899 (2021).
Yamashita, T. & Wang, X. W. Cancer stem cells in the development of liver cancer. J. Clin. Invest. 123, 1911–1918 (2013).
Gu, Y. et al. Circular RNA circIPO11 drives self-renewal of liver cancer initiating cells via Hedgehog signaling. Mol. Cancer 20, 132 (2021).
Wang, Y. et al. LncRNA HAND2-AS1 promotes liver cancer stem cell self-renewal via BMP signaling. EMBO J. 38, e101110 (2019).
Wu, J. Y. et al. The long non-coding RNA LncHDAC2 drives the self-renewal of liver cancer stem cells via activation of Hedgehog signaling. J. Hepatol. 70, 918–929 (2019).
Zhu, P. et al. LncBRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat. Commun. 7, 13608 (2016).
Chen, Z. et al. Circular RNA cia-MAF drives self-renewal and metastasis of liver tumor-initiating cells via transcription factor MAFF. J. Clin. Invest. 131, e148020 (2021).
Zhu, P. et al. lnc-beta-Catm elicits EZH2-dependent beta-catenin stabilization and sustains liver CSC self-renewal. Nat. Struct. Mol. Biol. 23, 631–639 (2016).
Liang, J. et al. Small nucleolar RNAs: insight into their function in cancer. Front. Oncol. 9, 587 (2019).
Kim, D. S. et al. Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol. Cell 75, 1270–1285.e14 (2019).
Shimamoto, A. et al. Reprogramming suppresses premature senescence phenotypes of Werner syndrome cells and maintains chromosomal stability over long-term culture. PLoS ONE 9, e112900 (2014).
Smith, J. A. et al. A role for the Werner syndrome protein in epigenetic inactivation of the pluripotency factor Oct4. Aging Cell 9, 580–591 (2010).
Chu, W. K. & Hickson, I. D. RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer 9, 644–654 (2009).
Yu, M. C. et al. Prediction of early-stage hepatocellular carcinoma using OncoScan chromosomal copy number aberration data. World J. Gastroenterol. 23, 7818–7829 (2017).
Goh, W. W. et al. Network-based pipeline for analyzing MS data: an application toward liver cancer. J. Proteome Res. 10, 2261–2272 (2011).
Fausti, F. et al. ATM kinase enables the functional axis of YAP, PML and p53 to ameliorate loss of Werner protein-mediated oncogenic senescence. Cell Death Differ. 20, 1498–1509 (2013).
Yuan, S., Wu, Y., Wang, Y., Chen, J. & Chu, L. An oncolytic adenovirus expressing SNORD44 and GAS5 exhibits antitumor effect in colorectal cancer cells. Hum. Gene Ther. 28, 690–700 (2017).
Cui, L. et al. Small nucleolar noncoding RNA SNORA23, up-regulated in human pancreatic ductal adenocarcinoma, regulates expression of spectrin repeat-containing nuclear envelope 2 to promote growth and metastasis of xenograft tumors in mice. Gastroenterology 153, 292–306.e2 (2017).
Kordasiewicz, H. B. et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 74, 1031–1044 (2012).
Miller, T. M. et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442 (2013).
Zhu, P. et al. IL-13 secreted by ILC2s promotes the self-renewal of intestinal stem cells through circular RNA circPan3. Nat. Immunol. 20, 183–194 (2019).
Wang, Q. et al. DNMT1-mediated methylation of BEX1 regulates stemness and tumorigenicity in liver cancer. J. Hepatol. 75, 1142–1153 (2021).
Acknowledgements
We thank Drs. Xiang Ding, Junying Jia, Yihui Xu, Xiang Shi, Shu Meng, Wei Xu, and Ge Xu (Institute of Biophysics) for technical support. We also thank Jing Li (Cnkingbio Company Ltd, Beijing, China) for technical support. This work was supported by the National Key R&D Program of China (2019YFA0508501, 2020YFA0803501); Strategic Priority Research Programs of the Chinese Academy of Sciences (XDB0570000); the National Natural Science Foundation of China (82130088, 81921003, 31930036, 82022058), Natural Science Foundation of Beijing (Z231100007223013).
Author information
Authors and Affiliations
Contributions
Y.G. designed the experiments, interpreted the results, and wrote the paper; Z.Y. and Z.Z. performed SNORD88B-related experiments; S.L. generated the cell line used in this study; J.W. performed hydrodynamic injection to induce HCC mice model; L.H. provided human HCC samples; N.L. and Y.X. helped with experimental design and identified CRISPR/Cas9-mediated mice model; P.Z. designed some experiments, provided experimental resources and technical support; Y.W. initiated and designed the study, wrote the paper; Z.F. initiated and acquired funding for the study, organized, designed, wrote the paper. All authors read and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Vincenzo Cardinale, Chunru Lin, and Shuguo SUN for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gu, Y., Yi, Z., Zhou, Z. et al. SNORD88B-mediated WRN nucleolar trafficking drives self-renewal in liver cancer initiating cells and hepatocarcinogenesis. Nat Commun 15, 6730 (2024). https://doi.org/10.1038/s41467-024-50987-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50987-6
- Springer Nature Limited