

Abstract
Shotgun proteomics analysis presents multifaceted challenges, demanding diverse tool integration for insights. Addressing this complexity, OmicScope emerges as an innovative solution for quantitative proteomics data analysis. Engineered to handle various data formats, it performs data pre-processing – including joining replicates, normalization, data imputation – and conducts differential proteomics analysis for both static and longitudinal experimental designs. Empowered by Enrichr with over 224 databases, OmicScope performs Over Representation Analysis (ORA) and Gene Set Enrichment Analysis (GSEA). Additionally, its Nebula module facilitates meta-analysis from independent datasets, providing a systems biology approach for enriched insights. Complete with a data visualization toolkit and accessible as Python package and a web application, OmicScope democratizes proteomics analysis, offering an efficient and high-quality pipeline for researchers.
Similar content being viewed by others


Introduction
Mass spectrometry-based proteomics has emerged as an indispensable tool for unraveling the intricate molecular mechanisms underlying complex diseases and biological phenomena. This technique enables simultaneous interrogation of thousands of proteins, allowing for the discovery of novel protein candidates without prior knowledge or defined targets. The flexibility provided by shotgun proteomics enables large-scale exploration of protein-protein interaction (PPI) networks1, subcellular processes2,3, protein thermal stability4,5, protein-drug interactions6,7, and signaling pathways8. Furthermore, due to the pivotal role of proteins in bridging genotypic information to phenotypic outcomes, proteomics complements other ‘omics disciplines, including genomics, transcriptomics, and metabolomics. Despite its widespread adoption and continuous technical advancements, proteomics continues to present substantial challenges, with data analysis complexity remaining a prominent issue9,10.
To achieve comprehensive information from proteomics data, the current approach requires the use of an array of software tools9,10,11. This encompasses raw data processing, protein identification, quantitation, differential proteomics, and enrichment analysis (e.g., MaxQuant12, PatternLab V13, DIA-NN14, Perseus15, PatternLab V, Progenesis QI for Proteomics, MSstats16; DAVID17, Enrichr18). While this conventional pipeline forms the core of proteomics data analysis, additional steps like exploring PPIs, conducting network analyses, and customizing data visualization are often necessary.
The complexity and number of computational tools present an initial barrier, particularly for non-programmers and newcomers to proteomics, as mastering the functions, capabilities, and limitations of each software tool demands a steep learning curve9. Furthermore, the challenge with tools for differential proteomics analysis involves accommodating the wide spectrum of data formats generated by search engines and quantitative proteomics software (Supplementary Data 1). Considering the distinctive features and necessities of each tool, encompassing data structure, normalization, and treatment of missing values, any new software tool must account for these varied input options. Additionally, a versatile and generic format is crucial to facilitate data importation from emerging proteomics tools, incorporate innovative statistical analyses, and integrate data from other ‘omics’ platforms, such as genomics and transcriptomics.
Statistical analysis for differential proteomics can exhibit substantial variations contingent upon the experimental design adopted by researchers. Typically, experimental designs can be broadly categorized into two main types: static and longitudinal19. Despite the common occurrence of both static and longitudinal analyses, many proteomics tools often lack comprehensive coverage of the wide spectrum of experimental designs (Supplementary Data 1).
Within the proteomics workflow, enrichment analysis assumes a pivotal role in aiding researchers to uncover system-level biological insights. One widely adopted approach is over-representation analysis (ORA), wherein experimentally derived entities are compared against annotated databases to ascertain whether biologically relevant properties are overrepresented in the experimental gene list20. Alternatively, gene-set enrichment analysis (GSEA) compares two conditions to find coordinated changes in gene expression in biologically relevant databases21. While enrichment analysis is widely utilized in proteomics, many existing proteomics tools have limitations in this regard (Supplementary Data 1), prompting researchers to explore web-based tools offering extensive datasets for conducting such analyses. Enrichr, for instance, offers access to over 224 distinct and regularly updated libraries18.
Despite the importance of enrichment analysis within the broader proteomics workflow, its power can be significantly enhanced by comparing individual experiments with independent results or data published by third-party research groups. This meta-analysis approach reduces false discovery rates and enables a more reliable assessment of molecular features associated with biological phenomena, as numerous sources contribute to a systems-level investigation9,22. While specific enrichment tools support the analysis of multiple gene lists, many existing proteomics tools frequently lack the capacity to conduct a broad analysis across multiple experiments and integrate data with other omics technologies (Supplementary Data 1). For researchers aiming to analyze multiple gene lists, several web-based tools are accessible, including Metascape9 and DAVID17. Nevertheless, these tools often overlook crucial protein statistics, such as protein fold changes, and offer a restricted array of figures that can integrate enrichment outcomes with respective protein attributes.
Considering the complexities inherent in the proteomics workflow, we introduce OmicScope—an integrative solution designed to streamline proteomics data analysis from differential expression to system-level integration. Here, we elucidate the features of OmicScope, available both as a Python package and a web application (https://omicscope.ib.unicamp.br/), providing a unique environment capable of handling and integrating quantitative proteomics data, enrichment outcomes, and comparing independent studies. OmicScope performs differential analysis across diverse platforms, conducts enrichment analysis using 224 Enrichr libraries, and integrates independent studies to enhance biological insights. Additionally, we offer a comprehensive suite of graphical outputs, encompassing gold-standard proteomics figures and a unique set of plots tailored to enhance comprehension in individual experiments and systems-level analyses. OmicScope also provides a versatile array of output files for integration with third-party software, including tables, vectorized images, network files, and its own output format. Together, OmicScope stands as a user-friendly tool engineered to facilitate proteomics data analysis, integration, and interpretation for the entire research community.
Results
Overview
To develop OmicScope, we conducted a survey of computational tools designed to conduct proteomics downstream analysis, with a particular focus on those capable of performing differential analysis. Our survey identified 15 computational environments, which were evaluated based on criteria such as tool distribution (package, web tool, desktop application), input formats, features for conducting differential proteomics analysis, capacity for enrichment analysis, data integration capabilities, meta-analysis functionalities, export options, and code availability. The details of all evaluated tools are provided in Supplementary Data 1, which served as the basis for defining the features of OmicScope.
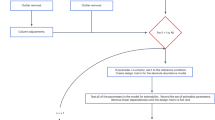
OmicScope was designed to be an integrative pipeline for proteomics data analysis, encompassing differential proteomics, enrichment analysis, and meta-analysis. Developed as Python package, OmicScope pipeline includes three primary components: OmicScope, EnrichmentScope, and Nebula (Fig. 1). Once quantitative data is inserted into the workflow, the OmicScope determines differentially regulated proteins (DRPs). These DRPs are then subjected to enrichment analysis using the EnrichmentScope algorithm, aiming to elucidate key biological features. Additionally, individual studies analyzed using the OmicScope and/or EnrichmentScope algorithms can be exported and used as input for Nebula. In Nebula, users can analyze multiple studies collectively, establishing correlations and identifying shared features across independent results. Each component, when activated, generates a set of figures and tables, streamlining user interactions for both the package and web application.

The OmicScope workflow begins with the import of data from various sources, including outputs from proteomics tools and generic formats. Once imported, the OmicScope module determines differentially regulated proteins. These proteins are then directed to the EnrichmentScope module, which facilitates over-representation and gene-set enrichment analyses. Data derived from both OmicScope and EnrichmentScope can be seamlessly used as input for Nebula, a module that integrates results from multiple studies using a systems biology approach. Each module within OmicScope is equipped with its own visualization toolset and allows for the export of tables, vectorized images, and graphML files.
To facilitate the access of non-programmers to OmicScope’s pipeline, we implemented the whole package’s functionalities in a user-friendly and highly interactive web application (See details in Appendix and Supplementary Figs. 13–15). OmicScope Web allows users to extract proteome information from dynamic plots, including bar plots, dot plots, and networks. In addition to providing explanations for each plot and its corresponding parameters, the web application enables users to customize the OmicScope workflow to meet their specific requirements. To enhance user experience by minimizing clicks and simplifying data handling, the OmicScope web application automatically generates all results and figures based on user input, which are accessible throughout the analysis process.
Furthermore, OmicScope workflow prioritizes the reporting of proteomics results to the scientific community, providing broad range of export methods, including tables, figures, and networks. Our tool exports figures in a vectorized and high-definition manner, tables containing data used for plots, and networks using the universal graphML file format.
Input methods
Proteomics research exhibits substantial diversity in experimental workflows, including mass spectrometer selection, acquisition modes, fragmentation methods, and quantitative approaches. This inherent diversity requests a wide array of software tools for protein identification and quantitation, each with its strengths and limitations, leading to interoperability challenges10,23.
To address these challenges, OmicScope offers eight data import methods (See Methods, Appendix, and Fig. 1), including six tailored to widely adopted proteomic software: MaxQuant, PatternLab V, DIA-NN, Proteome Discoverer, FragPipe, and Progenesis QI for Proteomics. These methods import outputs from respective software considering their unique characteristics. For software not yet integrated into OmicScope, the “General” method allows users to create custom spreadsheets for input into the OmicScope pipeline. This method accepts generic expression files, making OmicScope compatible with data from various omics platforms, such as genomics and transcriptomics. “General” method is able to perform differential proteomics analysis or import existing statistical analyses based on imported spreadsheets.
Aiming to provide an import method that joins succinctness, simplicity, and speed, we implemented “Snapshot” method, in which the users can import proteomics results containing assessed proteins, along with their associated fold changes and statistical outcomes. While Snapshot presents certain limitations concerning the number of plots that can be generated (refer to Supplementary Data 2), this method substantially improves interoperability across studies, especially given that many studies typically provide restricted information from their analyses, as demonstrated in the cases of Nie 2021 and Wang 2021. By integrating all of these input methods, OmicScope stands out as the platform capable of handling the widest variety of files (see Supplementary Data 1).
OmicScope: the core module
The central module of OmicScope shares the same name as the algorithm described herein. This module plays a pivotal role in organizing data, performing normalization and data imputation, filtering proteins, and carrying out differential proteomics analysis. It identifies differentially regulated entities and generates ready-to-publish figures (Fig. 2A).

A OmicScope offers various data import methods, including established software and generic approaches. Once data is successfully imported, OmicScope defines data architecture, performs or import differential proteomics analysis, filters data, identifies differentially regulated proteins (DRPs), and generates tables, figures, and exports. In Crunfli study (provided as a Source Data file), the two-tail t-test was performed followed by multiple hypothesis correction using BH approach (B–E) Illustrative figures generated by OmicScope: (B) Bar plot displaying the count of identified proteins and DRPs. C Volcano plot with accompanying density plot highlighting the top 10 DRPs based on Adjusted p value. D Heatmap of DRPs with Adjusted p value less than 0.002, with colors representing z-score. COVID-19 patients and controls are denoted as dark cyan and purple, respectively. E Boxplot depicting the abundance of proteins identified from the MAPK. For this import method, boxplot considers 38 MS-runs coming from 19 subjects. Data are presented as median (center), quartiles (bound box), whiskers with 1.5*Interquartile Range, and outliers according to inter-quartile range. F Protein-protein interaction network generated by OmicScope with DRPs having an Adjusted p value less than 0.005. In the left graph, proteins are colored based on log2(fold change), while the right graph represents proteins colored according to their communities identified using Louvain algorithm.
To provide maximum versatility, pre-processing and statistical steps are optional within the OmicScope pipeline. When no statistical results are provided, OmicScope autonomously conducts statistical analysis, filtering data based on pre-specified parameters and selecting the most suitable statistical tests based on the data architecture (see Appendix section for details). This flexible architecture accommodates various experimental designs, including static and longitudinal approaches. In static cases, comparisons between independent groups are typically made using t-tests for binary comparisons or One-way ANOVA for more than two independent conditions. In longitudinal analyses, OmicScope employs the Storey approach19, considering that differentially regulated genes vary over time based on natural cubic splines. In this longitudinal approach, statistical evaluations consider both within-group and between-group comparisons. Once nominal p value is calculated, OmicScope performs Benjamini-Hochberg multiple hypothesis correction24. By default, OmicScope designates proteins as differentially regulated if their adjusted p value is below 0.05, although users can define other parameters, such as fold-change and nominal p value cutoffs (Fig. 2A).
OmicScope module offers a visualization toolkit for data overviewing, clustering, and protein-specific features (Supplementary Fig. 1). In the overview category, users can generate bar plots, volcano plots, MA-plots, and dynamic range plots, facilitating the visualization of data distribution and normalization, providing initial insights into the dataset. The clustering category includes functions for hierarchical clustering, principal component analysis, and K-means clustering, allowing users to compare samples based on protein abundances and assess sample clustering. In this category, users can select various metrics and calculation methods to perform clustering analysis for static and longitudinal experimental designs. Lastly, protein-specific category aims to extract deeper insights about selected proteins, using bar plots and box plots. In this category, OmicScope also includes an integration with STRING API, providing a PPI network of DRPs, being one of unique environments to couple quantitative proteomics to PPI survey (Supplementary Data 1).
To demonstrate the capabilities of OmicScope, we employed previously published COVID-19 studies as illustrative examples (refer to the Methods section for details). These studies employed quantitative proteomics and transcriptomics to investigate SARS-CoV-2’s effects on various tissues. Specifically, we conducted a single analysis example, showcasing both differential proteomics and enrichment analysis, using proteins quantified by Crunfli25 in the brain tissue of patients who succumbed to SARS-CoV-2 complications. In this study, the authors meticulously detailed the processing parameters and furnished quantitative outputs from the analysis, ensuring reproducibility, and enabling result comparisons.
Crunfli’s dataset was imported into OmicScope with default parameters, filtering out contaminants26, and resulting in the identification of 721 DRPs (Fig. 2B). After OmicScope defines the DRPs, proteomics figures can be generated using a dedicated function for each plot type. For scatter plots and heatmaps, users can specify gene names as arguments to highlight specific target proteins (as demonstrated in Fig. 2C). Additionally, for clustering analyses, users optionally can set a p value cutoff to filter proteins and conduct analyses based on statistical significance (Fig. 2D).
In Crunfli’s dataset, for instance, we selected the MAPK family, including MAPK1, MAPK14, and MAPK9, all of which showed upregulation in SARS-CoV-2 infection compared to the control group (Fig. 2E). Moreover, the protein-specific category includes a function for exploring PPIs by querying the STRING database27. In this network analysis, users can identify communities based on the Louvain algorithm28 and filter data based on protein p values and/or specific proteins. In our analysis, we filtered proteins based on a p value threshold (pAdjusted < 0.005), applied the Louvain algorithm to conduct modularity analysis, and exported the data to facilitate data visualization (Fig. 2F).
While the Crunfli dataset offers advantages for our pivotal analysis, it does pose a technical limitation due to the relatively small number of evaluated proteins in the study. To address this, we challenged OmicScope against a benchmark dataset provided by Meier29 and Demichev30. Meier spiked-in two distinct concentrations of Yeast digest into HeLa digest, while Demichev employed Frag-Pipe and DIA-NN workflows, resulting in the evaluation of over 12,000 proteins, specifically identifying DRPs from the yeast digest. Using OmicScope, we identified two distinct expression profiles highlighting differential abundance among yeast protein concentrations, as demonstrated by Meier and Demichev (Supplementary Fig. 2). These outcomes highlight OmicScope’s capacity to handle varying data formats and sizes, performing a reproducible analysis of differential expressions.
To illustrate OmicScope’s capabilities in conducting longitudinal analysis, we analyzed data provided by Grossegesse31, wherein they investigated proteome changes induced by SARS-CoV-2 in CaLu cell lines across four time points: 2, 6, 10, and 24 h post-infection (Supplementary Fig. 3A). In this analysis, OmicScope identified 614 proteins that were differentially regulated (p < 0.05) between the SARS-CoV-2 and Mock groups. Examination of the K-means plots revealed three protein clusters, wherein SARS-CoV-2 induced a distinct protein pattern compared to the Mock group (Supplementary Fig. 3B). Further analysis of the PPI network derived from proteins assigned to cluster 0, which exhibited the highest fold-change variation, demonstrated up-regulation of proteins associated with interferon signaling during SARS-CoV-2 infection, consistent with previous findings31,32. These results underscore OmicScope’s integrative feature, wherein proteins identified through K-means clustering can be leveraged to explore PPIs and elucidate molecular mechanisms underlying biological phenomena.
EnrichmentScope: enhancing biological insights
One of the critical and challenging aspects of omics studies is extracting meaningful biological insights from hundreds or even thousands of differentially regulated entities. A commonly applied method for this purpose is enrichment analysis, wherein experimental gene or protein sets are compared against pre-established datasets, which may encompass biological pathways, molecular functions, kinase-associated genes, and other relevant categories. EnrichmentScope addresses this challenge by furnishing specialized enrichment analysis capabilities.
After executing the OmicScope module, users can proceed to perform enrichment analysis on EnrichmentScope module, specifying between two approaches: Over-Representation Analysis (ORA, conventional enrichment) or GSEA. Then, users must select specific databases, choosing between the 224 libraries offered by Enrichr18. Optionally, EnrichmentScope also can consider all proteins evaluated in the study as background for enrichment analysis. Once the analysis is performed, the module provides a result table and a toolkit of visualization functions, including the ability to export quantitative and enrichment data (Fig. 3A, Supplementary Fig. 4).

A EnrichmentScope performs Over-represented analysis (ORA) or Gene Set Enrichment Analysis (GSEA) using Enrichr libraries. For Crunfli’s dataset (provided as a Source Data file), we applied ORA workflow, performing a Fisher’s exact test with multiple hypothesis correction using BH approach. B–F Depiction of figures generated using the EnrichmentScope module. B Dot plot illustrating the top 10 enriched terms in the analysis. C Dot plot showcasing the count of differentially regulated proteins in terms related to neurodegenerative diseases. D Heatmap of differentially regulated proteins associated with terms from (C). E Network connecting enriched terms with their respective proteins, colored based on foldchange. The labeled proteins are shared among all processes. F EnrichmentMap displaying all enriched pathways, colored by modules defined using the Louvain algorithm. Term labels were determined based on intra-module connectivity and p value.
EnrichmentScope offers visualization tools like dot plots, facilitating the assessment of enrichment statistics and the number of proteins considered for enrichment (Fig. 3B, C). Users can select top enriched terms based on adjusted p values to identify relevant biological processes (Fig. 3B). Another dot plot option allows users to explore protein regulation in depth, illustrating the number of DRPs in each enriched term (Fig. 3C). In Crunfli’s study, for instance, the top 10 enriched terms using KEGG Database were filtered, and pathways related to neurodegenerative diseases were selected, showing the ratio of up- and down-regulated proteins (Fig. 3B, C).
EnrichmentScope also generates heatmaps and network graphs linking enriched terms to respective proteins (Fig. 3D, E). These visualizations reveal protein fold changes and proteins overlap among groups, shedding light on key factors in biological events. In the previously chosen pathways, proteins related to processes such as the proteasome, electron transport chain, and cytoskeleton were shared across all neurodegenerative processes, offering insights into the effects of SARS-CoV-2 on COVID-19 patients. Following this analysis, users can further investigate proteins of interest within the OmicScope module using functions like box plots, PPI networks, and more.
A challenge encountered in enrichment analysis is dealing with data redundancy, particularly prevalent in hierarchical databases such as Reactome33 and Gene Ontology34, which can lead to an overwhelming amount of information, as many pathways indicate a similar biological function (Supplementary Fig. 5). To address this limitation, EnrichmentScope apply systems biology approach similar to what is proposed by EnrichmentMap, wherein enrichment terms are represented as nodes within a network35 (Fig. 3F). Besides providing a simplified network representation, this strategy also simplifies information extraction, reduces data redundancy without omitting data and aids in selecting targets for further experimental validation. To connect each enriched term in the network, the algorithm calculates the pairwise Jaccard similarity indices, considering genes/proteins overlapped between target terms (See Appendix). By default, EnrichmentScope establishes links when the Jaccard Similarity Index exceeds 0.25, enabling graph construction. Additionally, EnrichmentScope automatically searches for communities within the enrichment map, labeling nodes (terms) that present highest intra-module degree (Fig. 3F). In addition to integrating quantitative and enrichment data, our implementation offers a wide selection of libraries, two enrichment approaches, and network visualization capabilities, distinguishing it as a notable feature compared to other platforms (see Supplementary Data 1).
Nebula: from singular studies to meta-analysis
The advent of omics platforms has exponentially increased the accumulation of data over the years, driving scientists to develop tools capable of comparing independent studies or even integrating experiments in a multi-omics fashion. Therefore, OmicScope introduces the Nebula module, designed to enhance data integration, interpretability, and comparison between studies. Although evaluating multiple independent proteomes simultaneously is a common approach, our software survey revealed that meta-analysis is a rare feature among computational proteomics tools (see Supplementary Data 1)9,36,37.
The Nebula workflow utilizes the outputs of OmicScope/EnrichmentScope for data integration and visualization. These outputs have the extension “.omics” and can be generated by running the OmicScope module, which returns quantitative data, or the EnrichmentScope module, which provides both quantitative and enrichment results. For each independent analysis, one of these previously described modules must be executed, and Nebula will read each output file to compile them into a unified object. Once the files are imported into Nebula, a set of visualization functions becomes available for conducting studies comparisons at the protein and/or enrichment levels (Fig. 4A, Supplementary Fig. 6).

A Nebula facilitates comparative analysis of independent studies based on OmicScope and EnrichmentScope outputs (provided as a Source Data file). B–H Figures generated using Nebula. B Bar plot depicting the count of whole (gray) and differentially regulated proteins/genes (colored) across various studies, as well as the combined count. C Dot plot showing the count of up-regulated and down-regulated entities. D Top 10 enriched pathways according to the KEGG database for all organs. Upset plots for (E) proteins and (F) enrichment terms, illustrating overlapping sizes among conditions. G Circular plot displaying all differentially regulated proteins and their shared relationships among evaluated groups (cyan links), along with shared enrichment terms among groups (black links). Each protein is annotated with its respective foldchange. H Circular plot depicting proteins differentially regulated in Oxidative phosphorylation among studies, with accompanying foldchange values. Source data are provided as a Source Data file.
To demonstrate Nebula’s capabilities, we used data from Crunfli 2022, Nie 2021, and Wang 2021. These selected studies assessed the effects of SARS-CoV-2 on patients’ tissues, with Crunfli examining proteomic signatures in the brain, Wang evaluating proteomics and transcriptomics effects in the lungs, and Nie reporting the liver as the most affected organ in proteomics terms. In Nie’s and Wang’s studies, the authors just provided DRPs and genes, enabling the application of the Snapshot method for ORA (Fig. 4B).
Nebula’s pipeline supports various plots that facilitate the simultaneous comparison of all target groups. Bar plots and dot plots offer an initial overview of the groups by comparing the number of proteins and pathways evaluated in each condition, serving as initial steps in establishing associations between studies (Fig. 4B–D). In the selected datasets, the lungs exhibited the highest number of DRPs and genes, followed by the liver and brain (Fig. 4B). Utilizing the Nebula integrative analysis approach, we noteworthy all examined tissues presents a consistent elevated number of up-regulated entities when compared to down-regulated counterparts (Fig. 4C). When filtering enrichment terms to highlight the top 10 pathways identified in each condition, Nebula can pinpoint several potential pathways worthy of further investigation (Fig. 4D).
To delve deeper into comparisons, Nebula offers tools for examining overlaps at both protein and enrichment levels. While Venn diagrams are commonly used for visualizing overlaps, they have limitations when comparing more than four conditions, producing illegible plots (Supplementary Fig. 7). To overcome this limitation, Nebula includes circus plots and upset plots in its pipeline (Fig. 4E, F). In the Upset plot38, each condition is depicted in a row, while columns illustrate non-zero intersections exclusively among the labeled groups specified in the frame (Fig. 4E, F). The advantage of the Upset plot lies in its readability and the absence of limitations regarding the number of groups analyzed. In the example datasets, only 19 proteins and genes exhibited dysregulation in all tissues, whereas the largest overlap encompassed 467 DRPs between the lung and liver proteomes (Fig. 4E). On the other hand, when examining overlaps in enrichment terms, the highest overlap was found between brain and lung proteomes, with 34 terms exclusively shared between these two tissues.
In addition to the Upset plot, Nebula can also perform comparisons across groups using circular plots. In this plot, Nebula links each group with lines, with each link representing a protein that overlaps between those conditions. Each protein also displays its respective fold change in the respective study, generating a circular heatmap. This circular plot complements the Upset plot by providing a view of the proportion of up- and down-regulated proteins shared among groups. As expected, in the studies under evaluation, the major shared proteins were up-regulated (Fig. 4G).
Nebula also offers a three-dimensional interpretation of data, considering groups, proteins, and enrichment terms simultaneously. Our circular diagram allows user to specify an enrichment term to be searched in all datasets, followed by the filtering of proteins associated with those terms in each study. Nebula then generates a circular plot that connects study and proteins, color-coding them based on their respective fold changes per group. In the example datasets, “oxidative phosphorylation”, enriched in all studies, was chosen to demonstrate that major proteins in this pathway were indeed up-regulated in all organs (Fig. 4H).
To provide systems-level information about multiple studies, Nebula’s array of visual representations also comprises network and statistical analyses. Similar to the methodology employed in EnrichmentScope, Nebula generates a graphical representation that establishes connections between studies and their corresponding DRPs, which also can be exported to third-party software tools (Fig. 5A).

A Nebula employs a systems biology approach, presenting proteins differentially regulated for each study as networks, enabling a detailed exploration of shared proteins among groups (provided as a Source Data file). B Differentially regulated proteins can be compared pairwise using statistical tests or similarity indices, with a heatmap displaying each pairwise result. Users have the option to define background size or utilize all identified genes in all conditions as background for Fisher’s Test. Here, we performed Fisher’s exact test to compare groups using human proteome background length (20,423). C Based on results from similarity indices or Fisher’s Test, users can generate a network, establishing links between studies according to parameter thresholds.
Two other systems biology strategies employed by Nebula to assess the similarity between studies are similarity analysis and statistical tests. In pairwise similarity analysis, Nebula computes similarity indices using the Jaccard algorithm by default across the target studies35. Nebula is also capable of using alternative metrics, like Pearson, Euclidean, and others, to calculate the similarity index using protein fold change. On the other hand, while performing statistical tests, Nebula applies Fisher’s Exact test to compare the overlap between studies by considering the entire set of imported proteins as the background, which results in pairwise p values. Similar to conventional enrichment analysis, users can optionally specify alternative background sizes, such as the number of reviewed proteins in a specific organism according to the Uniprot database. Alternatively, Nebula also encompasses other statistical analysis, such as t-test, Wilcoxon, or Kolmogorov-Smirnov test, using the fold-change distribution to compare studies. The results from the similarity and statistical analysis can be visualized using heatmaps and graphs. In the network representation, each node represents a group, while links are depicted according to pre-defined thresholds for similarity indices or p values(Fig. 5 B, C).
In the example discussed here, DRPs from the four groups were compared using the Fisher’s Exact Test approach, utilizing the reviewed human proteome database as the background (proteome size: 20,423 proteins). The heatmap showcases all pairwise p values generated in this analysis (Fig. 5B), whereas the network representation filters p values below 0.05 and connects each group accordingly (Fig. 5C). This analysis illustrates that the effect triggered by SARS-CoV-2 exhibits a stronger relationship at the protein level, particularly between the liver and lung proteomes, as previously suggested by other Nebula plots.
Discussion
Alongside the maturation of the MS-based Proteomics, the field has witnessed the evolution of mass spectrometers, leading to enhanced resolution, sensitivity, and consequently, the capability to identify thousands of proteins. In response to these advancements, computational analysis has progressed in parallel with technological advancements, offering several methods for protein identification and quantification. This study evaluated 15 pipelines commonly used for downstream proteomics analysis, comparing their acceptance of input formats, methods for conducting differential proteomics analysis, capacity for enrichment analysis, generation of figures, capabilities for meta-analysis, and available export formats. In this context, OmicScope was not designed to replace existing tools but to function as a versatile pipeline capable of exploiting the powerful features of various methodologies. Its primary objective is to facilitate effective, integrative, and snapshot downstream analysis of proteomics datasets, including differential analysis, enrichment analysis, and meta-analysis.
In our software survey, we observed a high variability in the number of input formats accepted by proteomics tools, with 10 tools capable of handling only one or two input formats. Noteworthy, PatternLabV13 and Progenesis QI for Proteomics were included in our comparison due to their capacity for differential expression analysis and related visualizations, however, their primary function revolves around protein identification and quantitation from raw MS files. Our results also highlighted MSstats16 for its inclusion of nine distinct input formats based on proteomics tools. Nevertheless, we did not find a generic file format that facilitates the easy integration of data from alternative sources into the MSstats pipeline. Despite offering a limited number of input file formats compared to MSStats, OmicScope provides two generic methods to handle a variety of data formats, in addition to six import methods based on well-established proteomics tools. This flexibility also allows integration and analysis of data from alternative omics platforms, including genomics and transcriptomics, and facilitates the straightforward incorporation of previously published data often presented in a more concise format.
While analyzing statistical pipelines utilized for differential proteomics, our findings revealed significant variations among tools concerning the supported experimental designs, applied statistical approaches, and user interface features. In terms of experimental designs, most software platforms include longitudinal analysis in their workflow; however, this strategy often requires programming skills or substantial computational resources, particularly with desktop applications. Additionally, we observed several applications offering either frequentist (e.g., Perseus, PatternLabV, Progenesis) or probabilistic (e.g., MSPypeline, DEP, Protti) approaches. In the OmicScope statistical pipeline, we provide methods for normalization and data imputation; however, our tool currently lacks methods for outlier rejection, inclusion of additional filtering steps, and assessment of quantitative accuracy, which will be the focus of future implementations. While evaluating to experimental designs, OmicScope integrates both frequentist and probabilistic approaches to accommodate static and longitudinal experimental designs, this functionality being accessed either through Python scripting or via web interface.
In static analyses, OmicScope employs t-tests or one-way ANOVA to effectively identify DRPs among two or more conditions, ensuring consistent outcomes in various scenarios. In longitudinal analysis, our tool employs the Storey methodology, incorporating a generalized linear model and considering gene variability over time through natural cubic splines. This approach allows OmicScope to investigate within- and between-group variations, encompassing more biological inquiries. Additionally, OmicScope employs the Benjamini-Hochberg method to correct p values, effectively controlling false discovery rates.
While other software may support both static and longitudinal experimental designs, OmicScope offers the advantage of automating all statistical analyses, thereby reducing the number of clicks and interactions needed to obtain initial results. Despite our tool facilitating data analysis and adhering to stringent parameters, this approach may not fully meet user demands or introduce some bias, requiring optimization of certain parameters (e.g., K-values in k-means plots, degrees of freedom for longitudinal analysis). To address this limitation, OmicScope provides users with the capability to adjust statistical parameters straightforwardly. Additionally, to allow proper reporting of the statistical pipeline, our tool also records all steps performed, which can be obtained via command line or downloading respective information in web app.
Regarding enrichment analysis, we observed that nine applications incorporate enrichment analysis within their workflows. Perseus, MsPypeline, ProStar39, ProteoSign40, Tidyproteomics41, protti42, and AlphaPeptStat support ORA, though Perseus requests the download of target databases and other tools present a restricted number of databases. On the other hand, DEP and TidyProteomics primarily focus on GSEA, with DEP offering the most extensive array of libraries by coupling enrichment functions to Enrichr. In line with DEP, OmicScope harnesses the vast resource of 224 databases available in Enrichr to perform ORA and Gene Set Enrichment Analysis (GSEA) algorithms. Beyond the analysis itself, OmicScope offers a set of innovative visualization features, including network analysis, heatmaps, and dot plots. These visualization tools serve a dual purpose: they enhance the presentation of enrichment analysis statistics and incorporate protein fold-change data, hereby integrating proteomics with enrichment results and providing system-level information.
In addition to the conventional proteomics workflow, the integration of results with independent and orthogonal datasets has emerged as a valuable strategy to mitigate false discovery rates22. While the Perseus workflow stands out for its capacity to facilitate multi-study and multi-omics comparisons, other evaluated tools often require the use of third-party software and packages to perform additional analyses. OmicScope introduces the Nebula module, specifically designed for multi-study comparisons. Nebula presents innovative visualization strategies, facilitating an overall view and pairwise comparisons of target studies. By introducing UpSet plots and circular plots, Nebula addresses limitations of traditional methods, such as Venn diagrams, providing insights into overlapping proteins between datasets and the corresponding fold changes observed in each study. Additionally, Nebula workflow extends to network analysis for group comparisons, featuring statistical analysis and similarity indices that highlight relationships between target studies. Despite being designed to handle proteomics datasets, we showed that Nebula also takes advantage of using other omics strategies, such as transcriptomics.
To cater to the diverse community of proteomics researchers, several software tools incorporate a Graphical User Interface (GUI) alongside their primary packages, ensuring accessibility for users across a range of technical backgrounds. Similarly, OmicScope provides both a Python package and a web application for running OmicScope, EnrichmentScope, and Nebula modules. While working with Python package, OmicScope provides flexibility by facilitating integration with other pipelines and allowing developers to contribute updates to input formats and algorithms. On the other hand, the OmicScope web application assists newcomers in proteomics and non-programmers by providing the interactivity necessary for data analysis and interpretation, utilizing a clean graphical interface to execute proteomics workflows. Furthermore, both the web application and Python package support data export in various formats, including tables containing all information required for generating figures and graphML files that enable network visualization in third-party software, such as Cytoscape43.
In conclusion, OmicScope has been carefully designed to assist experimentalists in analyzing and interpreting proteomics datasets. Future updates may focus on addressing specific quantitative inquiries, such as co-expression networks analysis and dose-response experiments. Our tool aims to support proteomics researchers in tackling complex biological questions and understanding molecular pathways associated with various disorders. OmicScope is accessible as an open-access resource at https://OmicScope.ib.unicamp.br/ and can be downloaded from the PyPI repository for convenience.
Methods
OmicScope architecture and user interface
OmicScope is developed in Python (v. 3.11) and distributed in Pypi repository (https://pypi.org/project/omicscope/) under MIT license. The OmicScope pipeline comprises three modules: OmicScope, EnrichmentScope, and Nebula (Fig. 1), designed to handle differential expression analysis, enrichment analysis, and multi-study comparisons, respectively. Documentation and step-by-step usage can be found in https://omicscope.readthedocs.io/en/latest/.
The web application was developed using the Streamlit framework, leveraging the OmicScope package in the background. Interactive figures were developed separately using Altair v.444 and the Vega visualization grammar. Step-by-step instructions for using the web application are provided in the Supplementary material and https://omicscope.ib.unicamp.br/, covering both OmicScope and Nebula workflows.
Study design and data collection
For our study, we utilized previously published COVID-19 datasets to run the OmicScope pipeline. Crunfli et al.25 (PXD023781) performed label-free quantitative proteomics on Progenesis QI for Proteomics, evaluating 2,278 proteins in post-mortem brain tissue from COVID-19 patients. They identified 735 proteins as differentially regulated (pAdjusted <0.05, Source Data). This biological relevant dataset served as a demonstration for a single analysis in OmicScope, showcasing its ability to handle quantitative proteomics data, generate publication-ready figures, and perform enrichment analysis. To evaluate OmicScope’s performance on larger datasets, we applied our workflow to a benchmark dataset supplied by Demichev, and the corresponding results can be found in Supplementary Fig. 2. Demichev applied Frag-Pipe and DIA-NN methodologies to analyze data initially published by Meier. Meier’s study involved spiking 15 ng and 45 ng of Yeast digest into Hela background, aiming to find DRPs coming from Yeast proteome (PXD017703). Finally, we evaluated our longitudinal analysis workflow using a dataset provided by Grossegesse, which examines the impact of SARS-CoV-2 at four distinct time points31 (PXD024883).
To showcase the capabilities of the Nebula module, we combined Crunfli’s dataset with results from refs. 45,46. Nie and colleagues evaluated autopsied tissues from seven organs of COVID-19 patients, of which we focused on the liver, the most affected organ according to their findings. Additionally, Wang and colleagues assessed protein and RNA levels in lungs from COVID-19 patients, providing differentially regulated genes and proteins. Files related to single-study analyses or Nebula workflows are available as supplementary files.
Input and data structure
OmicScope offers eight distinct modules for integrating external data into its pipeline, six of which rely on widely-used proteomic software for protein identification and quantitation: Progenesis Qi for Proteomic® PatternLab V13, MaxQuant12, Proteome Discoverer®, FragPipe, and DIA-NN14. For alternative sources, the General and Snapshot methods support data importing, with Snapshot offering a more concise file format with limited information. Additionally, our import methods categorize raw data into three primary matrices: assay (protein abundances), phenotype data (metadata or pdata), and raw data (protein information). Users also have the option to independently import pdata into the workflow, replacing raw pdata when necessary, to tailor statistical analysis to specific needs. Detailed information on input data, method specifications, and data structure can be found in the appendix and Supplementary Data 2.
Data pre-processing and differential proteomics analysis—OmicScope module
As part of the pre-processing steps, OmicScope can perform both data normalization and data imputation. During normalization, our workflow supports quantile, median, or average-based approaches. For data imputation, we handle missing values using the median, average, or KNN methods. Additionally, differential proteomics analysis within the OmicScope workflow is optional, contingent on input data. In cases where p value information is absent, OmicScope prior calculates average protein abundances among biological replicates and filters proteins detected in all conditions. By default, OmicScope applies log2-transformation for statistical tests.
OmicScope conducts two data analysis pipelines based on experimental design: static and longitudinal. In the static approach, it performs t-tests or Analysis of Variance (ANOVA) for two or more conditions, respectively. For longitudinal analysis, OmicScope adapted the workflow proposed by Storey in 2005, employing a natural cubic spline in a generalized linear model to model gene expression over time19. After obtaining nominal protein p values, OmicScope corrects for multiple hypotheses using the Benjamini-Hochberg method24. Further details on the statistical workflow are provided in the Appendix.
Enrichment analysis—EnrichmentScope module
Enrichment analysis is an integral part of the OmicScope workflow, handled within the EnrichmentScope module. This module employs the GSEApy package47 to conduct two distinct methods of enrichment analysis: ORA and GSEA. EnrichmentScope leverages data from the OmicScope object to query databases provided by the Enrichr API. OmicScope currently supports Human, Mouse, Yeast, Fly, Fish, and Worm as target organisms due to its dependency on Enrichr18.
For ORA, EnrichmentScope uses the proteins differentially regulated in the OmicScope object as input, employing a hypergeometric test against target databases. In contrast, GSEA utilizes assay data and phenotype data to determine whether a database term is significantly enriched at the top or bottom of a ranked list of genes based on their differential regulation. In both cases, EnrichmentScope generates a table containing evaluated terms, Benjamini-Hochberg-adjusted p values, Combined Score (ORA) or Normalized Enrichment Score (GSEA), log10-transformed p values, proteins related to each term, and the respective regulation of each protein.
Multi-studies integration—Nebula module
The Nebula module within OmicScope facilitates the integration of data from independent studies, allowing a meta-analysis approach to directly compare multiple proteomics studies. Input data for Nebula is provided by the OmicScope and EnrichmentScope modules. While OmicScope exports quantitative analysis, EnrichmentScope exports both quantitative and enrichment results. The output is a text file with omics extension, including conditions and statistical parameters applied in the respective study. For each analysis conducted in OmicScope, one file can be exported and later imported into Nebula.
To import data into the Nebula workflow, users must place all exports in the same directory and import them collectively. Once imported, Nebula divides each file into quantitative and, where applicable, enrichment data. DRPs are defined based on a user-defined p value cutoff, consistently applied to all imported data. To ensure reproducibility and consistency between OmicScope and EnrichmentScope outputs, Nebula remaps genes reported in EnrichmentScope to match the naming and format used in OmicScope data. To compare the independent studies, Nebula uses “gene name” column to match the information among datasets. Despite this feature being focused primarily on proteomics data, Nebula also works with other omics disciplines, such as genomics and transcriptomics.
Outputs
Each module described above offers a unique set of visualization tools tailored to specific analyses (see Appendix for details). In the OmicScope package, figures are primarily generated using matplotlib, seaborn, networkx, and pycirclize48 packages. In the web application, plots are generated based on Altair and Vega libraries. This setup allows for the export of figures in vectorized formats (e.g., SVG) and high-resolution images. Additionally, data used to construct networks can be exported as graphML files, compatible with network-specific tools like Cytoscape43 and Gephi49.
Statistics and reproducibility
Our study aims to introduce a new computational tool for statistical analyses in proteomics studies, using benchmarking datasets for evaluation. As such, traditional experimental design elements such as sample size determination, exclusion criteria, randomization, and blinding are not applicable to our study.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Crunfli and benchmark datasets are deposited in the ProteomeXchange Consortium via the PRIDE50 partner repository (accession no. PXD023781, PXD017703, and PXD024883). Nie and Wang datasets can be found in supplemental material in respective publications. The raw files used to test OmicScope, EnrichmentScope, and Nebula have been deposited in the FigShare repository under accession code https://doi.org/10.6084/m9.figshare.2605342951. All other data included in the article are provided in the Supplementary Information and Source Data file. Source data are provided with this paper.
Code availability
The OmicScope package can be accessed on the GitHub repository at https://github.com/guireiso/omicscope/52.
References
Liu, F., Lössl, P., Rabbitts, B. M., Balaban, R. S. & Heck, A. J. R. The interactome of intact mitochondria by cross-linking mass spectrometry provides evidence for coexisting respiratory supercomplexes. Mol. Cell. Proteom. 17, 216–232 (2018).
Mulvey, C. M. et al. Spatiotemporal proteomic profiling of the pro-inflammatory response to lipopolysaccharide in the THP-1 human leukaemia cell line. Nat. Commun. 12, 5773 (2021).
Thul, P. J. et al. A subcellular map of the human proteome. Science 356, eaal3321 (2017).
Kurzawa, N. et al. Deep thermal profiling for detection of functional proteoform groups. Nat. Chem. Biol. 19, 962–971 (2023).
Savitski, M. M. et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784 (2014).
Bantscheff, M. et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044 (2007).
Lechner, S. et al. Chemoproteomic target deconvolution reveals Histone Deacetylases as targets of (R)-lipoic acid. Nat. Commun. 14, 3548 (2023).
Needham, E. J. et al. Personalized phosphoproteomics identifies functional signaling. Nat. Biotechnol. 40, 576–584 (2022).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Chen, C., Hou, J., Tanner, J. J. & Cheng, J. Bioinformatics methods for mass spectrometry-based proteomics data analysis. Int. J. Mol. Sci. 21, 2873 (2020).
Noor, Z., Ahn, S. B., Baker, M. S., Ranganathan, S. & Mohamedali, A. Mass spectrometry–based protein identification in proteomics—a review. Brief. Bioinform. 22, 1620–1638 (2021).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Santos, M. D. M. et al. Simple, efficient and thorough shotgun proteomic analysis with PatternLab V. Nat. Protoc. 17, 1553–1578 (2022).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Kohler, D. et al. MSstats Version 4.0: statistical analyses of quantitative mass spectrometry-based proteomic experiments with chromatography-based quantification scale. J. Proteome Res. 22, 1466–1482 (2023).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 14, 128 (2013).
Storey, J. D., Xiao, W., Leek, J. T., Tompkins, R. G. & Davis, R. W. Significance analysis of time course microarray experiments. Proc. Natl Acad. Sci. USA 102, 12837–12842 (2005).
Boyle, E. I. et al. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20, 3710–3715 (2004).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Tripathi, S. et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735 (2015).
Bantscheff, M., Lemeer, S., Savitski, M. M. & Kuster, B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal. Bioanal. Chem. 404, 939–965 (2012).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Crunfli, F. et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc. Natl Acad. Sci. USA 119, e2200960119 (2022).
Frankenfield, A. M., Ni, J., Ahmed, M. & Hao, L. Protein contaminants matter: building universal protein contaminant libraries for DDA and DIA proteomics. J. Proteome Res. 21, 2104–2113 (2022).
Szklarczyk, D. et al. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612 (2021).
Traag, V. A., Waltman, L. & van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
Meier, F. et al. diaPASEF: parallel accumulation–serial fragmentation combined with data-independent acquisition. Nat. Methods 17, 1229–1236 (2020).
Demichev, V. et al. dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 13, 3944 (2022).
Grossegesse, M. et al. Deep time course proteomics of SARS-CoV- and SARS-CoV-2-infected human lung epithelial cells (Calu-3) reveals strong induction of interferon-stimulated gene expression by SARS-CoV-2 in contrast to SARS-CoV. J. Proteome Res. 21, 459–469 (2022).
Kim, Y.-M. & Shin, E.-C. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 53, 750–760 (2021).
Gillespie, M. et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 50, D687–D692 (2022).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Merico, D., Isserlin, R., Stueker, O., Emili, A. & Bader, G. D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 5, e13984 (2010).
Reis-de-Oliveira, G. et al. Digging deeper in the proteome of different regions from schizophrenia brains. J. Proteom. 223, 103814 (2020).
Nascimento, J. M. et al. Proteomic signatures of schizophrenia-sourced iPSC-derived neural cells and brain organoids are similar to patients’ postmortem brains. Cell Biosci. 12, 189 (2022).
Lex, A., Gehlenborg, N., Strobelt, H., Vuillemot, R. & Pfister, H. UpSet: visualization of intersecting sets. IEEE Trans. Vis. Comput. Graph. 20, 1983–1992 (2014).
Wieczorek, S. et al. DAPAR & ProStaR: software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 33, 135–136 (2017).
Theodorakis, E. et al. ProteoSign v2: a faster and evolved user-friendly online tool for statistical analyses of differential proteomics. Nucleic Acids Res. 49, W573–W577 (2021).
Jones, J. et al. Tidyproteomics: an open-source R package and data object for quantitative proteomics post analysis and visualization. BMC Bioinform. 24, 239 (2023).
Quast, J.-P., Schuster, D. & Picotti, P. protti: an R package for comprehensive data analysis of peptide- and protein-centric bottom-up proteomics data. Bioinform. Adv. 2, vbab041 (2022).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
VanderPlas, J. et al. Altair: interactive statistical visualizations for Python. J. Open Source Softw. 3, 1057 (2018).
Nie, X. et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell 184, 775–791.e14 (2021).
Wang, S. et al. A single-cell transcriptomic landscape of the lungs of patients with COVID-19. Nat. Cell Biol. 23, 1314–1328 (2021).
Fang, Z., Liu, X. & Peltz, G. GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 39, btac757 (2023).
Shimoyama, Y. pyCirclize: circular visualization in Python. (2022).
Bastian, M., Heymann, S. & Jacomy, M. Gephi: an open source software for exploring and manipulating networks. Proc. Int. AAAI Conf. Web Soc. Media 3, 361–362 (2009).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Reis-de-Oliveira, G., Carregari, V. C., Sousa, G. R., de dos R. & Martins-de-Souza, D. OmicScope unravels systems-level insights from quantitative proteomics data. https://doi.org/10.6084/m9.figshare.26053429.v1 (2024).
Reis-de-Oliveira, G., Carregari, V. C., Sousa, G. R., de dos R. & Martins-de-Souza, D. OmicScope unravels systems-level insights from quantitative proteomics data. Zenodo https://doi.org/10.5281/zenodo.12030956 (2024).
Acknowledgements
This work was supported by the São Paulo Research Foundation (FAPESP) (grant numbers, 2018/01410-1, 2017/25588-1, 2019/05155-9, 2023/04767-6) and by the Brazilian National Council for Scientific and Technological Development (CNPq). The OmicScope web application is hosted by the Institute of Biology at the State University of Campinas (Unicamp). Part of Supplementary Fig. 3A is created using templates from Servier Medical Art (http://smart.servier.com/), licensed under a CC BY 4.0 license. We acknowledge Caroline Brandão-Teles for her use of early OmicScope versions and contribution to design and functionality opinions. We also recognize Marcos Akira for his technical support in the OmicScope web application.
Author information
Authors and Affiliations
Contributions
G.R.O. and G.R.R.S. developed OmicScope. G.R.O. analyzed all data. G.R.O. and V.C.C. tested the software and assisted selection and optimization of features. D.M.S. supervised the research and provided funding. G.R.O., V.C.C., and D.M.S. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Michael Bartl, Xiaojun Ding, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Reis-de-Oliveira, G., Carregari, V.C., Sousa, G.R.d.R.d. et al. OmicScope unravels systems-level insights from quantitative proteomics data. Nat Commun 15, 6510 (2024). https://doi.org/10.1038/s41467-024-50875-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50875-z
- Springer Nature Limited