

Abstract
The cardiac conduction system (CCS) is a network of specialized cardiomyocytes that coordinates electrical impulse generation and propagation for synchronized heart contractions. Although the components of the CCS, including the sinoatrial node, atrioventricular node, His bundle, bundle branches, and Purkinje fibers, were anatomically discovered more than 100 years ago, their molecular constituents and regulatory mechanisms remain incompletely understood. Here, we demonstrate the transcriptomic landscape of the postnatal mouse CCS at a single-cell resolution with spatial information. Integration of single-cell and spatial transcriptomics uncover region-specific markers and zonation patterns of expression. Network inference shows heterogeneous gene regulatory networks across the CCS. Notably, region-specific gene regulation is recapitulated in vitro using neonatal mouse atrial and ventricular myocytes overexpressing CCS-specific transcription factors, Tbx3 and/or Irx3. This finding is supported by ATAC-seq of different CCS regions, Tbx3 ChIP-seq, and Irx motifs. Overall, this study provides comprehensive molecular profiles of the postnatal CCS and elucidates gene regulatory mechanisms contributing to its heterogeneity.
Similar content being viewed by others

Introduction
For every heartbeat, electrical impulses are initiated by the pacemaker cells in the sinoatrial node (SAN) and spread throughout the atrial myocardium to reach the atrioventricular node (AVN) and the fast-conducting network of the ventricular conduction system (VCS), including the His bundle, bundle branches and Purkinje fibers. Disturbances in the CCS can manifest in various types of cardiac rhythm abnormalities, such as heart blocks, ventricular fibrillation, and ventricular tachycardias, potentially leading to sudden cardiac death1,2,3,4,5,6.
Following the anatomical discoveries of the SAN, AVN, the His bundle, bundle branches, and the Purkinje fibers more than 100 years ago by Arthur Keith and Martin Flack7, Wilhelm His Jr8, Sunao Tawara9, and Johann Evangelist Purkinje10, respectively, electrophysiological and morphological differences between CCS components, such as automaticity11, conduction velocity12, and cell-cell electrical coupling13,14, have been demonstrated. In addition, the developmental processes of the CCS and its origin have been studied using genetic model organisms15,16,17. These studies have revealed specific expression patterns and/or functional implications of individual key cardiac transcription factors within the CCS. For instance, T-box 3 transcription factor 3 (Tbx3) is found in the SAN and AVN, as well as the proximal part of the VCS18. On the other hand, Iroquois homeobox 3 (Irx3) is expressed exclusively in the VCS, while being absent in the SAN and AVN19,20. Etv1, a member of the E-twenty-six (ETS) transcription factor family, is also expressed in the VCS as well as atrial cardiomyocytes at a high level but not in the SAN and AVN21. Inhibitor of DNA binding 2 (Id2), an inhibitor of the helix-loop-helix transcription factors, is expressed in all components of the CCS, except the SAN22,23. Interestingly, loss of many of these CCS transcription factors (e.g., Tbx3, Irx3, Etv1, and Nkx2-5) often results in similar developmental and functional phenotypes, such as CCS hypoplasia and conduction defects, only after birth20,24,25. Collectively, these studies suggest that the CCS is developmentally dynamic through the pre-to-postnatal stages, potentially arising through complex interactions among transcription factors. Nevertheless, due to the low number of CCS cells and technical difficulties in isolating healthy CCS cells from the postnatal heart, our understanding of the molecular heterogeneity of the CCS and its regulatory mechanisms at a cellular level has been comparatively limited. In particular, molecular diversities within the AVN, which is anatomically subdivided into the compact AVN and lower nodal bundle (LNB, also known as lower nodal cells)26,27,28, as well as those within the VCS, consisting of morphologically distinct structures like the His bundle, left and right bundle branches, and Purkinje fibers, are not yet fully understood.
Leveraging the recent advancements in single-cell RNA-sequencing (scRNA-seq) techniques, a few studies have highlighted the cellular heterogeneity within the CCS. Goodyer et al. have demonstrated the molecular heterogeneity of different components of the CCS using micro-dissected embryonic day 16.5 (E16.5) mouse hearts29. Bhattacharyya et al. have shown heterogeneity in the atrioventricular region using fluorescence-guided micro-dissection in Gjd3-CreEGFP;Rosa26tdTomato mice at postnatal day 0 (P0)30. Additionally, Liang et al. have reported the cellular diversity within the micro-dissected SAN of adult mouse hearts31. While these studies elaborately demonstrate distinct CCS cell populations and their unique expression profiles, further research on the transcriptomic landscape across the postnatal CCS and comparative profiling of different CCS components would enhance our understanding of the molecular, anatomical, and functional diversities within the CCS. In addition, the mechanisms governing the establishment of these CCS heterogeneities and the potential molecular transitions during pre-to-postnatal developmental stages remain to be elucidated.
Here, we utilize the integration of scRNA-seq and spatial transcriptomic analyses in a postnatal CCS-reporter mouse model (Cntn2Cre/+;Rosa26tdTomato/+) to provide a comprehensive view of the cellular heterogeneity within the postnatal CCS at a high-resolution with spatial information. Our study identifies CCS regional markers with unique and overlapping patterns and compares the expression of functional genes (i.e., ion channels and sarcomeres) within the CCS. By integrating with embryonic CCS scRNA-seq data29, we examine the molecular similarities and differences between prenatal and postnatal stages of the CCS. Furthermore, we provide insights into the gene regulatory mechanisms of the CCS and further investigate regulatory relationships between two CCS-specific transcription factors, Tbx3 and Irx3. This involves functional validation of CCS marker regulation using isolated neonatal mouse atrial and ventricular myocytes overexpressing Tbx3 and/or Irx3. Our analysis explores region-specific transcriptional regulation that contributes to the heterogeneity within the CCS.
Results
Single-cell and spatial transcriptomic analyses of the CCS
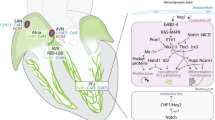
To understand the cellular and molecular constituents of the postnatal CCS, we simultaneously performed single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics (ST) using the CCS reporter mouse model, Cntn2Cre/+;Rosa26tdTomato/+ (CCS-tdT)32 (Fig. 1a). Consistent with the previous studies that utilized Contactin-2 (Cntn2)-driven reporters and Cntn2-targeting antibody to visualize the entire CCS system in mice21,33,34, all CCS components were reliably marked in the postnatal CCS-tdT mouse heart, which was validated by endogenous Cntn2 co-immunostaining (Fig. 1a and Supplementary Fig. 1).

a Representative epifluorescence image of a P4 CCS-reporter mouse (Cntn2Cre/+;Rosa26tdTomato/+) heart showing tdTomato-marked sinoatrial node, His bundle, bundle branches and Purkinje fibers. Scale bar = 1 mm. Imaging was performed on more than five hearts. b (top) Schematic overview of scRNA-seq experiment for the CCS profiling. Neonatal mouse cardiomyocytes were isolated from postnatal days 1, 2, and 4 (P1, P2, and P4) CCS-reporter mouse hearts, and tdTomato-positive CCS cells were purified using fluorescence-activated cell sorting (FACS). P1, P2, and P4 cells were barcoded separately using MULTI-seq, followed by 10x Genomics scRNA-seq. (bottom) Schematic overview of spatial transcriptomics of the CCS using GeoMx Digital Spatial Profiler (DSP). Cryosections of P4 CCS-reporter mouse hearts were collected on a slide, followed by the selection of regions of interest using segmentation and sequencing. Results from scRNA-seq and ST were integrated for analysis. Created with BioRender.com. c Uniform Manifold Approximation and Projection (UMAP) of scRNA-seq showing 8 CCS and 1 non-CCS clusters. A pie chart (top, left) shows the number of cells in each cluster. d Principal component analysis (PCA) of the CCS and non-CCS regions profiled by spatial transcriptomics (ST). e Expression profiles of the established CCS and non-CCS marker genes in each cluster of scRNA-seq data. f Expression profiles of the established CCS and non-CCS marker genes in each collected region of ST data. g Correlation analysis of CCS components between scRNA-seq and ST data. SAN sinoatrial node, cAVN compact atrioventricular node, LNB lower nodal bundle, HIS His bundle, PBB proximal bundle branch, PLBB proximal left bundle branch, PRBB proximal right bundle branch, DBB distal bundle branch, DLBB distal left bundle branch, DRBB distal right bundle branch, PF Purkinje fibers, RPF right Purkinje fibers, LPF left Purkinje fibers, RAA right atrial appendage, LENDO left ventricular endo-myocardium, LEPI left ventricular epi-myocardium, IVS-B interventricular septum (Base), IVS-A interventricular septum (Apex), RV right ventricle.
For scRNA-seq, cardiomyocytes were isolated from CCS-tdT mouse hearts at postnatal day 1, 2, and 4 (P1, P2, and P4). Subsequently, fluorescence-activated cell sorting (FACS)-purified CCS cells, which made up 0.1 − 0.2% of the total cardiomyocyte population (Supplementary Fig. 2), were subjected to multiplexed scRNA-seq (MULTI-seq)35 (Fig. 1b, Top). After pre-processing which excluded low-quality cells (Supplementary Fig. 3) and non-cardiomyocytes (1.03% of all cells) (Supplementary Fig. 4), a total of 6855 cardiomyocytes expressing an average number of 3388 genes per cell were identified. Through a series of unsupervised dimensional reduction analyses using uniform manifold approximation and projection (UMAP) and based on the expression of known CCS marker genes (see below), we identified 8 CCS and 1 non-CCS populations, including the sinoatrial node (SAN), compact atrioventricular node (cAVN), lower nodal bundle (LNB), His bundle (HIS), proximal bundle branch (PBB) and distal bundle branch (DBB), Purkinje fibers (PF), proliferative CCS (Prolif. CCS) and contractile myocytes (CM) (Fig. 1c and Supplementary Fig. 5).
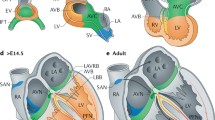
In parallel, ST was performed on cryosections of the P4 CCS-tdT mouse heart using the GeoMx Digital Spatial Profiler (Fig. 1b, Bottom). A segmentation approach allowed the precise capture of all tdTomato-positive cells in the CCS regions, such as SAN, cAVN, LNB, HIS, left and right proximal bundle branches (PLBB/PRBB), left and right distal bundle branches (DLBB/DRBB), left and right Purkinje fibers (LPF/RPF) (Supplementary Fig. 6; see the “Methods” section). Of note, our ST analysis included two sub-compartments of the AVN26,27,28, cAVN and LNB, which are anatomically situated adjacent to each other within the atrioventricular junction (Supplementary Fig. 6b and 7a). As described previously26,27,28, trichrome staining of the atrioventricular region showed that the LNB was surrounded by insulating fibrous tissue and basally located on the plane of atrioventricular insulation, in comparison to the apically located HIS (Supplementary Fig. 7a). Additionally, non-CCS regions adjacent to the selected CCS components, including the right atrial appendage (RAA), basal and apical interventricular septum (IVS-B, IVS-A), left ventricular endomyocardium (LENDO), left ventricular epimyocardium (LEPI), and right ventricle (RV) were collected for comparison. Among the 12,638 transcripts detected in ST, differentially expressed genes (4708 transcripts) showing |FC | > 3 and P < 0.05 in any possible comparison of two CCS components, were used to perform principal component analysis (PCA). PCA of ST data demonstrated that CCS components exhibited a globally linear clustering pattern reflecting the sequence of cardiac electrical activation, from impulse generation in SAN to propagation through the VCS (Fig. 1d). Moreover, these CCS components and their non-CCS counterparts were closely positioned. For instance, the RAA was grouped with the SAN, cAVN and LNB; IVS-B was grouped with the proximal VCS components (HIS, PLBB, and PRBB); and the IVS-A and the LENDO were grouped with distal VCS components (DLBB/DRBB and LPF/RPF).
In both scRNA-seq and ST analyses, all CCS regions highly expressed known pan-CCS markers (Hcn4, Cpne5 and Cacna2d2)22,29,36,37, whereas the non-CCS regions, such as the ‘Contractile myocytes’ cluster in scRNA-seq, and LENDO, LEPI, IVS-B, IVS-A and RV in ST, expressed higher levels of contraction genes (Tnnt2, Tnni1, Mb) with low expression of conduction genes (Fig. 1e, f). Also, both scRNA-seq and ST results corroborate the expression patterns of the known regional CCS markers, including Shox2 in the SAN38,39; Tbx3 in the SAN, AVN, and HIS40,41; Irx3 and Gja5 in the VCS19,20; Robo1 in the proximal VCS42,43; Slit2 and Sema3a in the distal VCS29,42; Gja1 in distal VCS and CM44; as well as atrial (Myl4, Myl7) and ventricular (Myl2, Myl3) sarcomere genes. Based on these observations, we characterized the CCS components as follows: SAN (Myl2low/Myl3low/Myl4high/Myl7high/Hcn4high/Tbx3high/Nkx2.5+/Shox2high/Irx3−/Robo1−); cAVN (Myl2mid/Myl3mid/Myl4high/Myl7high/Hcn4high/Tbx3high/Nkx2.5+/Robo1low/Shox2low); LNB (Myl2high/Myl3high/ Myl4low/Myl7low/Hcn4+/Gja5low/Gja1−/Irx3+/Tbx3+); HIS and PBB (Myl2+/Myl3+/Myl4−/Myl7−/Irx3+/Tbx3+/Robo1+); DBB and PF (Myl2+/Myl3+/Myl4−/Myl7−/Irx3+/Gja5+/Gja1+/Slit2+/Sema3a+); VCS (Myl2+/Myl3+/Myl4−/Myl7−/Irx3+/Gja5+/Sema3a+/Slit2+) and non-CCS cardiomyocytes (Hcn4−/Cpne5−/Cacna2d2−/Gja5−/Sema3a−/Tnnt2high/Tnni1high/Mbhigh). Notably, despite the anatomical proximity between the cAVN and LNB, a direct comparison between cAVN, LNB and HIS revealed that the LNB expressed ventricular genes (Myl2, Myl3) and VCS markers (Irx3, Etv1, Gja5), which were characteristics more similar to HIS than to cAVN (Supplementary Fig. 7b). This is consistent with previous studies demonstrating that the LNB is structurally continuous with the HIS and is derived from a ventricular lineage26,27,28,30.
Our scRNA-seq showed a cluster of cells (222 out of 6855 cells, 3.24%) annotated as ‘Proliferative CCS’, as these cells expressed CCS genes (Cntn2, Etv1, Irx3, Gja5) as well as cell cycle markers of G1/S phase (Mcm4, Mcm5) and G2/M phase (Fam83d, Cdc25b) (Supplementary Fig. 8). This suggests the presence of committed conduction cells undergoing proliferation in the early postnatal heart. Additionally, we compared P1, P2, and P4 MULTI-seq data to examine potential transcriptomic changes during early postnatal development in the CCS. Unlike our expectation, no differences in the proportion of P1, P2, and P4 cells per cluster, or in clusters per development stage were observed (Supplementary Fig. 9). Furthermore, no significant changes in gene expression were found, including the commonly known molecular signatures for cardiomyocyte maturation, such as a transition in myosin heavy chain isoforms from Myh6 to Myh7 and an increase in mitochondrial gene content.
Next, we conducted a comparison between the transcriptomic profiles of corresponding regions detected by both technologies (Fig. 1g). Correlation analysis showed strong concordances between scRNA-seq and ST datasets within nodal cells (SAN and cAVN); LNB and proximal VCS (HIS and PBB), and distal VCS (DBB and PF) components. Also, all ventricular non-CCS components in ST (LENDO, LEPI, IVS-B, IVS-A and RV) were highly correlated to the CM cluster in scRNA-seq, while the RAA in ST showed high similarity to the SAN and cAVN in scRNA-seq. Collectively, these results suggest that our scRNA-seq and ST methods credibly captured all CCS and non-CCS regions.
Transcriptional profiling of the postnatal CCS components
Next, we sought to identify regional CCS markers by integrating scRNA-seq and ST data. First, enriched genes of each CCS region were identified from scRNA-seq and ST datasets separately, by comparing each CCS region to other CCS and non-CCS clusters/regions (|FC| > 1.5, P < 0.05) (Fig. 2a and Supplementary Data 1 and 2). Subsequently, genes that were identified as common between scRNA-seq and ST were considered as specific regional markers with high confidence.

a CCS-specific enriched genes for each CCS component identified by both scRNA-seq and spatial transcriptomics. b Molecular gradients within the CCS components showing zonated enrichment of genes in Nodal cells (SA node, Compact AV node), Lower nodal bundle & Proximal VCS (His bundle, Proximal bundle branch), and Distal VCS (Distal bundle branch, Purkinje fibers). LBB left bundle branch, RBB right bundle branch, RAA right atrial appendage, LENDO left ventricular endo-myocardium, LEPI left ventricular epi-myocardium, IVS-B interventricular septum (Base), IVS-A interventricular septum (Apex), RV right ventricle, VCS ventricular conduction system.
In the SAN, as expected, Shox238,39 and Tbx340,41, well-known transcription factors in SAN development, were observed. In addition, we identified other potential markers, including Gnao1, which encodes G protein subunit alpha O1 and has been found to be involved in heart development45, as well as Ptchd4, Cdh12, and Nid2. The cAVN showed enrichment of known genes, such as Rspo3 and Bmp2, both of which are expressed in the atrioventricular canal of the developing mouse heart (E10.5) from which the AVN originates46,47, as well as Cacna1d, which encodes CaV1.3 channel known to regulate AVN automaticity48. Newly identified markers of the cAVN included Prdm6, which encodes PR/SET domain 6 and has been shown to be expressed in the developing cardiovascular49,50,51 and nervous system52. Interestingly, a genetic variant of Prdm6 has been reported to be associated with heart rate recovery during exercise53. The LNB showed enrichment of genes, including Ppp1r17, Hs3st3a1, and Lcp1. A previous study has shown that Ppp1r17, encoding protein phosphatase 1 regulatory subunit 17, is expressed in the interventricular septum during embryonic heart development, and is mostly lost in later developmental stages54. Like many other cardiac conduction genes, Ppp1r17 is also expressed in neurons55, yet its expression and function in the CCS are unknown. The HIS showed enrichment of Pcp4, Cdh6, and Cyp4b1. Although our analysis showed that these genes are most highly expressed in the His bundle, Pcp4 is known to be expressed throughout the VCS56 with critical roles in cardiac electrophysiology, and Cdh6 is known to be enriched in the CCS compared to non-CCS cardiomyocytes57. This suggests that there may be regional heterogeneity in the expression levels of these genes within the CCS. The PBB highly expressed Igfbp7 and Arxes1, while the DBB showed high expression of genes, such as Nppa, Gal, Ecm1, Cygb, Slc16a2, Pde10a, and Mbp. Nppa, encoding atrial natriuretic peptide58,59, is known to be expressed in embryonic trabecular cardiomyocytes60,61, and its expression in the ventricle is strongly reduced after birth. However, we observed low but specific expression of Nppa in the DBB, suggesting a possibility of persisting Nppa expression in the distal VCS postnatally. The PF showed enrichments of genes, including Mpped2, Corin, and Mylk4. In particular, Mpped2, encoding Metallophosphodiesterase domain-containing protein 2, has previously been reported to be expressed in neurons and embryonic ventricles47, although its functional involvement in the CCS has not yet been described.
The CCS is an anatomically continuous structure with a highly specialized function of generating and propagating electrical signals. Thus, while the integrated analytic approach above effectively uncovers the enrichment of gene expression in each individual CCS component, it may not be effective in capturing the gradual changes in gene expression along the CCS, in which case, genes would be enriched in more than one defined component of the CCS. Therefore, according to the PCA (Fig. 1d) and the correlation heatmap (Fig. 1g), we divided CCS components into three zones: ‘Nodal cells’ (SAN, cAVN), ‘LNB & Proximal VCS’ (HIS, PBB) and ‘Distal VCS’ (DBB, PF), and identified markers enriched in each zone by integrating scRNA-seq and ST datasets (Fig. 2b). ‘Nodal cells’ showed enrichment of genes, including Gcgr, which encodes glucagon receptor62 with roles in heart rate regulation63, Ctdspl, which encodes CTD Small Phosphatase Like, associated with atrial fibrillation through miR-26a-164, Enpep, and Bmp2. The ‘Proximal VCS’ zone showed high expression patterns of S100a6 encoding a Ca2+ binding protein Calcyclin65, Col4a4, Alcam, Cdh6, and Atp1b2. Also, in addition to the established PF markers, Sema3a66 and Gja567, the ‘Distal VCS’ zone showed enrichment of Pdzrn3, involved in heart maturation68, Irx1 and Irx269 encoding Iroquois homeobox (Irx) transcription factors, Mgp encoding Matrix Gla protein, Nsg2, and Sbk2. Interestingly, several PF and distal VCS genes, Mylk4, Corin, Sbk2, and Mgp, are also known to be expressed in the atria70,71. Together, these results demonstrate the zonated and gradient expression patterns of CCS marker genes, which reflect its highly specialized and integrated conductive structure and functions.
To further gain molecular insights into the distinctive physiological and electrophysiological properties of each CCS component, including automaticity and conduction velocity, we profiled the expression patterns of genes functionally associated with ion channels, receptors, gap junctions, and sarcomeres using our scRNA-seq and ST datasets (Supplementary Figs. 10 and 11). For example, Kcnj3 and Kcnj5, which form the acetylcholine-activated potassium channel (IKACh) Kir3.1 and Kir3.4, respectively, and are associated with bradyarrhythmia and sporadic atrial fibrillation72, as well as Cacna1g and Cacna1h, which encode CaV3.1 and CaV3.2, respectively, known to contribute to pacemaker activity73, were highly expressed in the CCS with strong enrichment in SAN and cAVN. Also, consistent with a previous study74, Kcnk3, encoding TASK-1 channel, showed enrichment in the VCS. It was notable that Kcnj12, encoding Kir2.2 responsible for cardiac inwardly rectifying potassium current (IK1), Gria3 encoding glutamate ionotropic receptor, Grik5 encoding glutamate ionotropic receptor kainite, and Ank3, encoding a cytoskeletal element ankyrin-G that regulates cardiac excitability75, were found to be enriched in the CCS compared to contractile myocytes. However, their functional implications in the CCS are not well understood. On the other hand, we found several CCS genes identified from scRNA-seq, which were not detected in ST, likely due to the limitation of the probe-based approach used in ST. Among those genes, we noted strong and CCS-specific expression of Scn10a (NaV1.8), known to modulate Scn5a-encoded NaV1.5 currents76, in the PF. In line with a recent study showing that only a short form of Scn10a mRNA is expressed in the VCS76, we observed a predominant expression of the short Scn10a transcript in the postnatal CCS. Mapping sequenced reads to the Scn10a gene in our scRNA-seq data revealed higher sequencing coverage in the last seven exons of the gene (exons 22 to 28), compared to exons 1 to 21 (Supplementary Fig. 12).
Next, we validated the identified CCS markers by performing immunostaining and fluorescence in situ hybridization (FISH) in the P4 CCS-tdT mouse heart. An SAN-enriched marker, Gnao1 was expressed in tdTomato-positive pacemaker cells (Fig. 3a), where Shox2 and another recently discovered SAN marker, Vsnl131, were expressed (Supplementary Fig. 13). Gnao1 expression was also detected in the cAVN and atrioventricular (AV) ring, but not in LNB. However, its expression was not specific to nodal cells, as it was observed in the right atrial appendage (Supplementary Fig. 14). We performed FISH to examine cAVN-enriched Prdm6, showing that it was primarily expressed in the cAVN and AV ring, as well as in surrounding non-CCS within the atrioventricular junction, but not in the LNB and other CCS regions (Fig. 3b and Supplementary Fig. 15). On the other hand, Ppp1r17 was expressed in the LNB and HIS, but not in cAVN (Fig. 3c and Supplementary Fig. 16), supporting our finding that LNB is molecularly more similar to HIS than to cAVN. We also examined proximal VCS-enriched markers. Consistent with scRNA-seq data, S100a6 was detected in the HIS, but not in any other CCS regions (Fig. 3d and Supplementary Fig. 17). However, its expression only partially overlapped with tdTomato-positive cells in the HIS and was also found in tdTomato-negative cells around the HIS and in the IVS. Previous studies have reported S100a6 expression in other cell types, including neurons77, fibroblasts and epithelial cells78, suggesting that the role of S100a6 may not be restricted to the CCS. Igfbp7, identified to be enriched in PBB in our analysis, displayed strong expression in both HIS and PBB, but not in PF (Fig. 3e and Supplementary Fig. 18). This suggests that Igfbp7 could serve as a proximal VCS marker. FISH staining showed that an established VCS marker79, Pcp4, was indeed expressed throughout the VCS, but showed stronger expression in the HIS and BB than in the PF, confirming our scRNA-seq and ST data (Supplementary Fig. 19). In addition, the expression of Nppa-encoded atrial natriuretic peptide (ANP) was primarily found in the distal VCS, including DLBB (Fig. 3f and Supplementary Fig. 20). Scn10a-encoded NaV1.8 was expressed highly in the PF, compared to the HIS (Supplementary Fig. 21). Moreover, another PF-enriched gene, Mpped2, was strongly and specifically expressed throughout the VCS, albeit with a lower level of expression in the HIS and PBB (Fig. 3g and Supplementary Fig. 22). Interestingly, Mpped2 and NaV1.8 exhibited a slightly broader expression pattern showing enrichment along the endomyocardium of the ventricular chambers, suggesting their potential roles within and around the VCS. Collectively, the simultaneous examination of both scRNA-seq and ST followed by histological validation demonstrates that the CCS consists of both specific and gradational molecular profiles.

Immunofluorescence (IF) and fluorescence in situ hybridization (FISH) staining were performed in P4 Cntn2Cre/+; Rosa26tdTomato/+ heart sections. a Gnao1 IF staining in tdTomato-positive SAN and cAVN, but not LNB; b Prdm6 FISH staining in the cAVN, but not LNB; c Ppp1r17 IF staining in the LNB and HIS; d S100a6 IF staining in the HIS; e Igfbp7 IF staining in the HIS and proximal LBB; f Nppa IF staining in the DBB; g Mpped2 FISH staining in the VCS. scale bars = 50 μm. The figures shown are representative of at least three independently repeated IF experiments or two FISH experiments. SAN sinoatrial node, cAVN compact atrioventricular node, LNB lower nodal bundle, HIS His bundle, LBB left bundle branch, RBB right bundle branch, LPF left Purkinje fibers, RPF right Purkinje fibers.
Transcriptomic comparison of the embryonic and postnatal CCS
The CCS forms through a tightly coordinated process of cell differentiation and specification during embryonic development, and maturation during early postnatal development20,25. As a recent study has characterized the embryonic (E16.5) CCS through scRNA-seq29, we sought to understand the transcriptional changes of the major CCS components during late embryonic and postnatal development. Embryonic CCS cells were first extracted from the original scRNA-seq datasets (GSE132658 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132658]) based on cellular identification profiles29, and then were integrated with our postnatal CCS scRNA-seq data with minimization of potential batch effects (Fig. 4a). This approach enabled a direct comparison of the molecular profiles of CCS components between embryonic and early postnatal stages.

a Integration of embryonic day 16.5 (E16.5) and postnatal (P1-P4) CCS scRNA-seq datasets shows a close overlay of the CCS components from different developmental stages. P: postnatal. b Correlation analysis between embryonic and postnatal CCS components exhibits a high degree of concordance between respective groups. c Expression patterns of the identified CCS component and regional markers in E16.5 CCS clusters, showing molecular similarities between embryonic and postnatal CCS. d Pairwise differential gene expression analysis between embryonic and postnatal CCS components, using the Wilcoxon Rank-Sum Test. *, adjusted P-value < 0.05. Source data are provided in Supplementary Data 3. SAN sinoatrial node, cAVN compact atrioventricular node, LNB lower nodal bundle, HIS His bundle, PBB proximal bundle branch, DBB distal bundle branch, PF Purkinje fibers, Prolif. CCS proliferative CCS, CM contractile myocytes, Tz-SAN transitional SAN, AVR AV ring, Atrial TZ atrial transitional zone, Ventricular TZ ventricular transitional cells, Tz-PF transitional PF.
Unbiased clustering of integrated cells displayed aligned patterns between individual CCS components from E16.5 and postnatal stages. For instance, the SAN, cAVN, HIS and PF cells of E16.5 and postnatal stages were well overlapped in the UMAP (Fig. 4a). Consistently, correlation analysis between E16.5 and postnatal CCS components demonstrated high concordances between the respective CCS regions, with significant clustering of CCS regions within each cardiac chamber (i.e., the atria and the ventricles) (Fig. 4b). We further examined whether the CCS marker genes identified in the postnatal CCS displayed similar expression patterns in the E16.5 CCS (Fig. 4c). Notably, SAN-enriched genes (Gnao1, Nid2, Bmp2), cAVN-enriched genes (Bambi, Gsg1, Rd3, Prdm6), nodal cells-enriched genes (Gcgr, Csrp2, Enpep, Ctdspl), HIS/proximal VCS-enriched genes (S100a6, Robo1, Scube2, Cdh6), and PF/distal VCS-enriched genes (Mpped2, Corin, Nsg2, Sema3a, Sbk2, Tsc22d3, Irx1 and Irx2) were consistently observed in the corresponding regions of the E16.5 CCS. This finding not only affirms the specificity of the newly identified CCS markers, but also indicates a high degree of transcriptomic consistency between these developmental stages, suggesting early establishment of molecular heterogeneity within the CCS.
Subsequently, we conducted pairwise differential gene expression analysis to identify the molecular differences between E16.5 and postnatal CCS components (|FC| > 1.5; P < 0.05; minimum % of expressing cells in either compared group > 50%) (Fig. 4d and Supplementary Data 3). We noted significant differences in the expression of known CCS marker genes, including higher Cpne5 and lower Igfbp5, Sln, and Vsnl1 expression in the postnatal SAN. Also, lower expression levels of Id2, Sln, Cav1, and Vsnl1, along with higher Rspo3 levels, were observed in postnatal cAVN, compared to the embryonic cAVN. Compared to the embryonic HIS, the postnatal HIS exhibited higher levels of Pcp4, Hcn4, and Igfbp7, yet lower levels of Tagln and Cnn1, encoding Transgelin (also known as SM22α) and Calponin 1, respectively, both of which are smooth muscle cell markers80. Given that Cnn1 is expressed in the embryonic mouse heart (E13.5) but becomes restricted to smooth muscle cells in adult mouse tissues81, this reduction may reflect postnatal CCS maturation. Similarly, the postnatal PF showed significant decreases in Bmp1082 and Corin83, genes that are expressed in the ventricular trabecular myocardium during development.
These observations collectively suggest that despite the high similarity in the overall molecular profiles of CCS components between E16.5 and early postnatal stages (P1-P4), there are discernible molecular changes that indicate the dynamic maturation process in the CCS during late embryonic and early postnatal development.
Heterogeneous transcriptional regulation of the CCS
To gain mechanistic insights into how distinct molecular patterning across the CCS is transcriptionally established, we performed network inference using SCENIC (Single-Cell Regulatory Network Inference and Clustering)84, which identifies regulons by predicting the transcription factors (TFs) and their candidate target genes based on co-expression and motif enrichment analysis. TF activity in each individual cell was calculated using AUCell and subjected to unbiased clustering. This analysis revealed TFs that potentially contributed to the establishment of the regional differences within the CCS (Fig. 5a and Supplementary Data 4). Transcriptional activities by Tbx3, Klf7, Creb5, Rara, and Sp3 were enriched in the nodes and proximal VCS; Shox2, Nr2f1, Nr2f2, and Gata6 in the nodes only; E2f6 in the nodes and distal VCS; as well as Mef2c and Gtf3a in the nodes and contractile cardiomyocytes. In addition, enrichments of Tcf4, Tbx5, Cebpb, Cebpd, Jun, Ets2, Fos, and Irf1 activities were observed across all CCS components, except contractile myocytes, whereas Irx2, Tcf7l2, Etv1, Srebf2, and Max displayed VCS-specific enrichment. These findings corroborated a large body of the literature highlighting the regulatory functions of Shox2 and Gata6 in the SAN85, Tbx3 in the SAN/cAVN/HIS24,25,41,86, and Tbx522,41,87 and Etv121,88 in the VCS. Gene regulatory networks of SAN, cAVN, HIS and PF were also constructed, elucidating the regulation of the regional marker genes, such as Gnao1, Gcgr, Bmp2 and Vsnl1 in the SAN; Prdm6, Rspo3, and Gde1 in the cAVN; Igfbp7, Cpne5, Scube2, and Cdh6 in the HIS; as well as Mpped2, Slit2, Scn10a, and Tsc22d3 in the PF (Supplementary Fig. 23).

a (Top) Integrated heatmap/dotplot of CCS regulons, as inferred by the SCENIC algorithm. The heatmap color represents regular activities for each CCS component based on the area under the curve (AUC) scores. Dot size represents transcription factor expression in each CCS component. (Bottom) Motifs of representative transcription factors in the CCS regions. b Tbx3 and Irx3 show unique and overlapping expression patterns exclusively within the CCS. c SAN and cAVN show enrichment of Tbx3 with atrial genes (Myl4 and Myl7); LNB, HIS, and PBB show enrichment of both Tbx3 and Irx3 with ventricular genes (Myl2 and Myl3); and DBB and PF show enrichment of Irx3 with ventricular genes. d Schematic showing unique and overlapping expression patterns of Tbx3 and Irx3 throughout the CCS. Created with BioRender.com. e Genome browser views of chromatin accessibilities in the right atria, SA node, ventricle, and Tbx3-positive atrioventricular junction (Tbx3+ AVJ) as well as Tbx3 ChIP-seq, and the presence of Iroquois homeobox (Irx) transcription factor motifs, for the CCS component and regional marker genes. SAN sinoatrial node, cAVN compact atrioventricular node, LNB lower nodal bundle, HIS His bundle, PBB proximal bundle branch, DBB distal bundle branch, PF Purkinje fibers, Prolif. CCS proliferative CCS, CM contractile myocyte, VCS ventricular conduction system.
To deepen our understanding of transcriptional regulation of CCS heterogeneity, we focused on two TFs, Tbx3 and Irx3, known for their critical roles in CCS development and functions18,19,20,25,41 (Fig. 5b). Our scRNA-seq and ST analyses showed that Tbx3 and Irx3 exhibited distinct and overlapping expression patterns within the CCS: Tbx3 alone in the atrial components of the CCS (SAN and cAVN); both Irx3 and Tbx3 in the proximal VCS (LNB, HIS, PBB); and Irx3 alone in the distal VCS (DBB and PF) (Fig. 5c, d). Although Irx3 was not identified from SCENIC analysis, we observed strong VCS-enrichment of the Irx2 regulon, another member of the Irx TF family sharing the same Irx motif89,90. Given that Irx3, not Irx2, is well described to be essential for VCS development and function19,20,69, we hypothesized that individual and cooperative transcriptional regulation by Tbx3 and Irx3 contributed to the molecular heterogeneity of the CCS.
To test this possibility, we utilized publicly available datasets of the assay for transposase-accessible chromatin using sequencing (ATAC-seq) in different CCS regions: P0 mouse SAN91 and E13.5 Tbx3-positive atrioventricular junction (AVJ encompassing both AVN and HIS)25, along with their respective non-CCS counterparts: P0 right atria (RA) and mouse ventricle92. Additionally, we used Tbx3 ChIP-seq data performed in adult mouse cardiomyocytes93, and searched for two Irx TF binding motifs: ‘ACAnnTGT’89 and ‘ACATGT’90 (Fig. 5e). By intersecting these datasets, we examined chromatin accessibilities in CCS and non-CCS regions, Tbx3-binding, and the presence of Irx TF-binding motifs for the marker genes that we identified in this study. 45.9% (17/37) of the nodal markers showed SAN ATAC-seq peaks, which contrasts with 16.1% (5/31) of the proximal VCS genes and 11.4% (5/44) of the distal VCS genes, corroborating our findings. Similarly, 89.2% (33/37) of the nodal markers and 80.6% (25/31) of the proximal VCS markers showed chromatin accessibilities in AVJ, compared to 70.5% (31/44) of the distal VCS markers. Furthermore, 79.5% (89/112) of the CCS markers appeared to be potentially regulated by Tbx3 or Irx TFs. Specifically, Tbx3 ChIP-seq peaks were present in 70.3% (26/37) of the nodal genes, with a mildly lower incidence of 61.4% (27/44) in the distal VCS genes. In contrast, Irx motifs were found in 37.8% (14/37) of the nodal genes, while a greater prevalence of 61.4% (27/44) was noted in the distal VCS genes. These observations align with the expression patterns of the CCS markers and suggest the individual and cooperative regulatory role of Tbx3 and Irx TFs across the CCS.
Next, we closely examined the epigenomic landscape for several new and established CCS marker genes (Fig. 5e and Supplementary Fig. 24). SAN markers, Shox2 and Vsnl1, as well as a cAVN marker, Prdm6 displayed higher ATAC-seq peaks in the SAN and AVJ compared to the atria or ventricle, respectively. This suggests a spatial specificity in their genomic accessibility. Tbx3-binding peaks and Irx motif sequences were also found in Vsnl1 and Prdm6, indicating potential regulation by Tbx3 and Irx TFs. In accordance with its enriched expression in the proximal VCS, S100a6 exhibited higher chromatin accessibility peaks in Tbx3+ AVJ, compared to the ventricle. This aligned with the Tbx3 ChIP-seq peak, but not with Irx motifs, suggesting a possible direct transcriptional regulation by Tbx3, but not Irx TFs. Interestingly, genes enriched in the distal VCS where Tbx3 expression is absent, such as Nppa, Gja5, Mpped2, and Slit2, still displayed open chromatin regions in the Tbx3+ AVJ. They also showed Tbx3 ChIP-seq peaks and Irx motifs. On the other hand, another distal VCS gene, Scn10a, showed no apparent differences in ATAC-seq peaks between the Tbx3+ AVJ and the ventricle, although it did exhibit Tbx3 ChIP-seq peaks and Irx motifs. Lastly, consistent with their high expression in contractile myocytes, Gja1 and Ryr2 showed greater chromatin accessibility in the ventricle compared to AVJ, atria, and SAN, alongside the presence of Irx motifs and Tbx3 binding peaks. Collectively, these findings suggest a complex and region-specific potential transcriptional regulation of CCS regional marker genes by Tbx3 and/or Irx3.
Tbx3 and Irx3 contribute to CCS heterogeneity
To further investigate the individual and combined actions of Tbx3 and Irx3 on CCS gene regulation, we established an in vitro model that simulated various CCS regions by introducing Tbx3 and/or Irx3 in neonatal mouse atrial and ventricular cardiomyocytes (A-CMs and V-CMs, respectively) using adenovirus-mediated overexpression (Fig. 6a and Supplementary Fig. 25). Here, Tbx3 overexpression in A-CMs modeled the nodes (SAN and cAVN); Irx3 and Tbx3 overexpression in V-CMs, the proximal VCS (HIS and PBB); and Irx3 overexpression in V-CMs, the distal VCS (DBB and PF) (Fig. 6b). The regional properties of isolated A-CMs and V-CMs were validated by the significantly high atrial gene expression (e.g., Myl4 and Myl7) in A-CMs and ventricular gene expression (e.g., Myl2 and Myl3) in V-CMs (Fig. 6c). Subsequently, we assessed how Tbx3 and/or Irx3 influence the expression of CCS regional markers.

a Schematic of the experimental workflow to model the CCS regional heterogeneity in vitro. Neonatal mouse atrial cardiomyocytes (A-CMs) and ventricular cardiomyocytes (V-CMs) were infected with Ad-GFP, Ad-Irx3, Ad-Tbx3 or both Ad-Irx3 and Ad-Tbx3. Created with BioRender.com. b Schematic of the in vitro model of the CCS regions. Nodes were simulated by Tbx3 overexpression in A-CMs; the Proximal VCS by Tbx3 and Irx3 overexpression in V-CMs; and the Distal VCS by Irx3 overexpression in V-CMs. c Gene expression of atrial (Myl4 and Myl7) and ventricular (Myl2 and Myl3) genes in cultured A-CMs and V-CMs. n = 5 per group. Data are presented as the mean ± SEM. P-values were determined by unpaired two-tailed Student’s t-test. d–i Gene expression levels of the CCS component and regional markers in A-CMs and V-CMs upon Tbx3 and/or Irx3 overexpression. Checkered bar graphs represent A-CMs and solid bar graphs represent V-CMs. The schematic of the transcriptional regulatory model in each gene is displayed. Tested genes include (d) SA node marker, Vsnl1; (e) AV node marker, Prdm6; (f) His bundle marker, s100a6; (g) distal bundle branch marker, Nppa; (h) distal VCS marker, Mpped2; and (i) contractile myocyte marker, Gja1. n = 5 per group for A-CMs, and n = 5 (Prdm6, Mpped2) or 10 (Vsnl1, S100a6, Nppa, Gja1) for V-CMs. Data are presented as mean ± SEM. Statistical significance was determined using one-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file. VCS ventricular conduction system, A-CMs atrial cardiomyocytes, V-CMs ventricular cardiomyocytes.
We found that Vsnl1, exclusively expressed in the Tbx3+ SAN, was markedly increased by Tbx3 overexpression in A-CMs (Fig. 6d). Interestingly, the addition of Irx3 as well as Irx3 overexpression alone in A-CMs suppressed Vsnl1 expression, suggesting the antagonistic actions of Irx3 against Tbx3 outside nodal cells. In V-CMs, Vsnl1 was unaffected by Tbx3 or Irx3 overexpression, suggesting that Vsnl1 expression in the SAN is predominantly activated by Tbx3 in an atria-specific manner. On the other hand, another SAN marker, Shox2 was not affected by either Tbx3 or Irx3 overexpression in both A-CMs and V-CMs (Supplementary Fig. 26a), suggesting their insufficiency in regulating Shox2 expression in neonatal cardiomyocytes. This aligns with the previous data and supports the notion that Shox2 potentially acts earlier than Tbx3 in SAN development38. Similarly, cAVN genes, Prdm6 and Rspo3 were unaffected by Tbx3 and/or Irx3 in A-CMs (Fig. 6e and Supplementary Fig. 26b). However, in V-CMs, Prdm6 and Rspo3 were significantly repressed by Irx3 overexpression, supporting their absent expression in the Irx3+ VCS. This observation suggests that their expression in cAVN might be primarily driven by other transcription factors. Together, these data suggest that Tbx3 and Irx3 can play antithetical roles in gene expression: Tbx3 is an activator and Irx3 is a repressor of these tested nodal genes.
For genes enriched in the Tbx3+/Irx3+ proximal VCS, both Tbx3 and Irx3 appeared to function primarily as activators. For example, S100a6 was upregulated by Tbx3 and Irx3 together in the V-CMs (Fig. 6f). Interestingly, this upregulation was largely driven by Tbx3, in agreement with the enriched Tbx3-binding peaks, while Irx3 overexpression alone in V-CMs had no effect on its expression, consistent with the lack of Irx motifs in the S100a6 gene (Fig. 5d). Additionally, Pcp4 was also significantly increased upon overexpression of both Tbx3 and Irx3 in A-CMs and V-CMs (Supplementary Fig. 26c), supporting the cooperative activator properties of Irx3 and Tbx3 of the identified proximal VCS markers.
For the markers identified in the Tbx3−/Irx3+ distal VCS, we observed an inverse regulatory relationship between Tbx3 and Irx3 compared to nodal genes: Tbx3 as a repressor and Irx3 as an activator. For example, Irx3 overexpression significantly upregulated Nppa, Scn10a, and Gja5 in both A-CMs and V-CMs, supporting their enrichments in the distal VCS (Fig. 6g and Supplementary Fig. 26d, e). Importantly, when Irx3 and Tbx3 were overexpressed together, the activating role of Irx3 on Nppa, Scn10a, and Gja5 was largely abolished or reduced, demonstrating the dominant repressive role of Tbx3 on these genes. These findings corresponded with Tbx3-binding peaks in Nppa, Scn10a, and Gja5 (Fig. 5d), and explained their lower expressions in the His bundle. Notably, while Tbx3 overexpression alone significantly repressed Gja5 expression in A-CMs, it mildly increased Gja5 levels in the V-CMs, suggesting that Tbx3’s function on Gja5 depends on the chamber identity. Similarly, Tbx3 and Irx3 activated Mpped2 and Slit2 expression in the V-CMs, but had no impact on their expression in the A-CMs, suggesting chamber-specific actions of Tbx3 and Irx3 on these genes (Fig. 6h and Supplementary Fig. 26f).
Lastly, for genes enriched in the Tbx3−/Irx3− contractile myocytes, Tbx3 and Irx3 mainly play a repressive role. For instance, Gja1 and Ryr2 were significantly downregulated by Tbx3 and Irx3 overexpression together in V-CMs (Fig. 6i and Supplementary Fig. 26f), but not by Tbx3 or Irx3 alone. This demonstrates their synergistic repressive role in V-CMs and supports the absence of Gja1 in the His bundle, but not in the Purkinje fibers. On the contrary, in A-CMs, Tbx3 overexpression alone dominantly repressed the expression of both Gja1 and Ryr2, corroborating with the detection of Tbx3-binding peaks (Fig. 5d) and previous reports on reduced Gja1 and Ryr2 expression in the SAN94,95.
Together, our in vitro study using primary atrial and ventricular cardiomyocytes successfully recapitulated the unique expression patterns of several CCS regional marker genes in a dish. Furthermore, gene expression analysis integrated with ATAC-seq, ChIP-seq, and Irx motif data suggests that the transcriptional regulatory program mediated by Irx3 and Tbx3 is dependent on CCS regions as well as heart chamber identity, thereby contributing to the molecular heterogeneity within the CCS.
Discussion
In this study, we conducted a comprehensive molecular and spatial profiling of the transcriptional landscape of postnatal mouse CCS cells at a single-cell resolution. Our findings demonstrate how CCS components, from the SAN to PF, are molecularly continuous yet also distinct from each other. As noted in previous studies29, major limitations of investigating the CCS include the low number of CCS cells in the heart (~0.1–0.2% of the entire cardiomyocyte population) and difficulties in isolating viable postnatal CCS cells. For our scRNA-seq analysis, we utilized approximately 7000 CCS cells that were isolated from postnatal Cntn2Cre/+;Rosa26tdTomato/+ mouse hearts and purified using FACS. Among several Cre lines targeting the CCS, including Hcn4-CreERT296, Cx40-CreERT297, Gjd3-CreEGFP30 and Sema3a-CreERT266, the Cntn2Cre/+ model was chosen as it could mark all components of the early postnatal CCS in mice without tamoxifen induction, consistent with Cntn2-driven reporters and Cntn2-targeting antibody21,33,34. To our knowledge, this study has incorporated the highest number of purified entire mouse CCS cells to conduct a single-cell transcriptomic investigation to date.
Transcriptional profiling of the CCS using scRNA-seq was further facilitated by integration with spatial transcriptomics. Among various spatial transcriptomic technologies available, we chose NanoString GeoMx Digital Spatial Profiler (DSP) for its several advantages. First, given that all CCS components cannot be fully captured in one or two histological sections, the GeoMX DSP offered a larger slide scan area that accommodated over 10 sections of the P4 heart encompassing all CCS regions. Second, its segmentation tool enabled precise capture of tdTomato-positive CCS cells only, allowing transcriptomic assessments without the need for a cell-type deconvolution method. Lastly, it provided the whole transcriptome profiling. As a result, we were able to reliably acquire transcriptomic profiles of each CCS component. Noteworthy among our findings from ST is the molecular identification and comparison of CCS components within the atrioventricular junction: two sub-compartments of AVN26,27,28, the cAVN and LNB, as well as the HIS. Despite their anatomical proximity, our results indicate that the LNB shares more molecular similarities with the HIS than with the cAVN (Supplementary Fig. 6 and 7). This corroborates with the previous studies, demonstrating that LNB is developmentally originated from the ventricular myocardium, whereas cAVN is derived from Tbx2+ atrioventricular canal26,27,28,30. It is, therefore, important to highlight that the spatial information from ST significantly enhanced our ability to identify all CCS components in the scRNA-seq data. Furthermore, although GeoMX DSP presents technical limitations due to its probe-based transcript counting method, ST data compensated potential statistical biases in our scRNA-seq data that may arise from varying numbers of cells in CCS components. Collectively, this integrative approach, supported by histological validations, enabled the discovery of CCS marker genes with greater reliability.
Our study identified several marker genes of each CCS component, while also consolidating recently recognized CCS markers. In addition, zonation patterns of genes were identified within the CCS: specifically, nodes consisting of the SAN and cAVN, proximal VCS consisting of the LNB, HIS, and PBB, and distal VCS consisting of the DBB and PF. These marker genes include Gnao1 and Vsnl1 in SAN, Prdm6 in cAVN, Ppp1r17 in LNB, S100a6 and Igfbp7 in proximal VCS, and Nppa, Scn10a and Mpped2 in distal VCS. A recent study has shown that GNAO1 was found to be highly enriched in the human SAN, while PRDM6 was predicted as a potential TF in human nodal cells98. This suggests possible evolutionarily conserved roles of these genes in the CCS. While previous studies have demonstrated that Igfbp7 regulates pathological cardiac remodeling99 and that S100a6, a Ca2+ binding protein, modulates adverse ventricular remodeling100, it has not been tested whether they play physiological roles in the CCS. Together, it remains for future research to elucidate the functional implications of these CCS marker genes in both healthy and diseased hearts.
The completion of CCS development occurs postnatally. Since deficiencies in Nkx2-5, Tbx3, and Irx3 lead to CCS hypoplasia in the postnatal mouse (as early as P4), but not during embryonic development20,24,25, it was unexpected that no significant differences in gene expression between P1 and P4 were observed in our scRNA-seq results (Supplementary Fig. 9). This observation led us to consider a few possibilities: (a) the temporal duration between P1 and P4 may be too narrow to capture the transcriptional differences; (b) the major changes in postnatal maturation may occur at a later stage, such as P14, as shown previously101; and/or (c) postnatal CCS development may be primarily regulated and established by the embryonic transcriptional program. These possibilities were partially tested by directly comparing our postnatal CCS scRNA-seq data with embryonic CCS data29. We found high molecular similarities between E16.5 and postnatal stages within the same CCS regions, suggesting that cell specifications within the CCS, particularly for the SAN, cAVN and proximal VCS, may largely occur by E16.5. On the other hand, higher expression of early-development genes, such as Tagln, Cnn181, Bmp1083, and Corin83, was noted in the E16.5 CCS, compared to the postnatal CCS, indicating CCS maturation during pre-to-postnatal development. Corin is a notable gene that encodes a transmembrane cardiac serine protease, which activates Nppa-encoded ANP by cleaving pro-ANP102. Since ANP signaling is implicated in embryonic VCS development103, our findings raise a possibility that Corin-ANP signaling may be crucial for CCS development and maturation. Additionally, our observation that more than 3% of early postnatal CCS cells are still proliferating, highlights the ongoing progress of postnatal CCS maturation. Further studies are needed to examine the molecular profiles of fully mature adult CCS cells. As conducted in a series of postnatal cardiomyocytes (from P0-P28)101, it would also be informative to examine the trajectory of CCS postnatal maturation and identify a mediator in this developmental process. Meanwhile, the correlation analysis between E16.5 and our postnatal CCS data showed that the postnatal SAN cells were more strongly correlated with E16.5 SAN (tail) than with SAN (head). We also observed that Tbx18, which marks SAN (head)104 as well as Isl1105 were undetected in the postnatal SAN region of our scRNA-seq and ST data. Considering that Tbx18 and Isl1 are involved in early SAN development and their expression diminishes in the adult heart105, their absence in our data might indicate pre-to-postnatal maturation. However, it may also be attributed to the relatively lower number of nodal cells in our scRNA-seq data compared to VCS cells. Therefore, while the tdTomato signal in the postnatal CCS-tdT mouse heart overlapped with Shox2 and Vsnl1 expression in the SAN (Supplementary Fig. 13), further investigation to ascertain whether entire SAN regions are captured by Cntn2 could elucidate this discrepancy.
In the CCS, which is essentially a continuous structure, molecular heterogeneities govern distinct functional properties of each CCS component, ensuring harmonious contraction and relaxation of the heart. While the roles of individual transcription factors in the development and function of CCS components have been extensively studied15,16,17, our understanding of how these transcription factors cooperate across different regions of the CCS is comparatively limited. Our SCENIC analysis inferred gene regulatory networks within the CCS, controlled by established TFs such as Shox2, Gata685, Tbx324,25,41,86, Tbx522,41,87, and Etv121,88 as well as by less explored TFs, including Nr2f1, Atl5, and Bach1. It is also notable that CCS-specific regulons were largely clustered within the nodes only, nodes & proximal VCS, and the VCS, suggesting the zonation patterns of gene regulatory networks within the CCS. This was further tested functionally by examining the roles of two CCS-specific TFs, Tbx3, and Irx318,19,20,25,41. Our in vitro model using primary atrial and ventricular cardiomyocytes overexpressing Irx3 and Tbx3, along with ATAC-seq, ChIP-seq, and motif analyses, provides proof-of-principle evidence of their contributions to the establishment of molecular heterogeneities within the CCS. Specifically, our findings suggest that Tbx3 and Irx3 play individual, cooperative, or antagonistic actions in a chamber-specific, and CCS component-specific manner. The in vitro model we utilized has potential limitations due to adenovirus-mediated high TF expression, which exceeds physiological levels, and the relatively moderate effects of Tbx3 and Irx3 overexpression on gene regulation, which can be attributed to the maturity of neonatal mouse cardiomyocytes. Therefore, further investigation is warranted to elucidate the precise regulatory mechanisms by which the gene dosage balance between Tbx3 and Irx3 positively and negatively fine-tunes gene expression in a developmentally more potent system. We have previously demonstrated that Irx3 can promote transcription as a repressor (possibly through an unidentified repressor)19, suggesting a potential co-regulatory function of Irx3. Also, direct binding sites for Irx TFs remain to be identified. Thus, identifying the co-regulator(s) of Tbx3 and Irx3 in the context of chamber-specific or CCS region-specific gene regulation would significantly enhance our understanding of transcriptional regulatory networks in developing hearts. Indeed, our results are well aligned with a study demonstrating that the gene dosage balance between Tbx5 and Tbx3 orchestrates VCS-specific gene expression profiles and function87. Specifically, the VCS-specific removal of Tbx5 was sufficient to alter gene expression profiles of the VCS towards those of nodal cells by elevating Tbx3 expression. Since Irx3 physically interacts with Tbx520, this suggests a potential gene regulatory network among Tbx5, Tbx3, and Irx3 in establishing CCS heterogeneities.
In conclusion, our study generates a transcriptomic resource of the postnatal murine CCS and provides functional evidence of how molecular heterogeneities across the CCS can be transcriptionally established. These findings help advance our understanding of the molecular identities within the CCS beyond its anatomical structures, and therefore have implications for therapeutic approaches against conduction system defects.
Methods
Mice
All experimental protocols were approved by the Animal Care Committees of the University of Ottawa (HI-2950 and 3060), in accordance with the guidelines of the Canadian Council on Animal Care (CCAC). All mice had free access to standard chow (2019 Teklad Global Diet; Envigo) and water and were housed in a controlled environment with a 12 h light/12 h dark cycle, ambient temperature of 22-24 °C and humidity levels of 30–60%. Mice were humanely euthanized via CO2 inhalation and/or cervical dislocation based on CCAC guidelines on euthanasia of animals used in science.
For scRNA-seq, ST, and histological validation experiments, homozygous Cntn2tm1.1(cre/EGFP)Nmun (Cntn2Cre) and homozygous Rosa26tdTomato mice32, which were maintained on a CD-1 background, were crossed to generate CCS-fluorescent reporter mice (Cntn2Cre/+;Rosa26tdTomato/+, CCS-tdT). To achieve the maximum number of age-matched pups simultaneously, 8–15-weeks-old female mice were housed together without exposure to male mice for 2–3 weeks prior to mating to synchronize their estrous cycles. Following the mating setup, vaginal plugs were monitored daily. Neonatal mice on postnatal days 1, 2, and 4 were euthanized by cervical dislocation immediately before heart collection for cardiomyocyte isolation, ST or histological validation. A total of 103 CCS-tdT pups were used for scRNA-seq. For in vitro adenovirus-based overexpression studies, 2–3 P0 neonatal litters from CD-1 male and female breeders were used to isolate atrial and ventricular cardiomyocytes. Both males and females were included in all experiments, and sex differences were not considered.
Neonatal mouse cardiomyocyte isolation
The atria and ventricles of neonatal mice were collected separately in sterile ice-cold 1x phosphate-buffered saline (PBS). The ventricles were cut into 3-4 pieces for efficient digestion. Atrial and ventricular cardiomyocytes were separately placed into 50 mL centrifuge tubes containing filtered Hanks’ Balanced Salt Solution (1x HBSS, Thermofisher) with 0.0125% Trypsin-EDTA (Thermofisher) and 1% penicillin/streptomycin (P/S; Thermofisher). Cells were subjected to overnight trypsin digestion at 4 °C while gently rotating. Then, the trypsin solution was aspirated and discarded. 5 mL of filtered and warm (37 °C) L-15 + 0.075% Collagenase II/Dispase (Roche, 10269638001) + 1% P/S solution was added to the 50 mL centrifuge tube containing cardiomyocytes, and a fine magnetic stir bar was placed. The centrifuge tubes were placed in a tube rack, which was placed above the stir plate. After gentle stirring of the solution and cells for 5 min, the supernatant containing single cardiomyocytes was collected into 5 mL of cold fetal bovine serum (FBS) to prevent cells from over-digestion. An additional 5 mL of collagenase digestion solution was added to the cells, and this digestion-collection step was repeated until sufficient cells were collected. After the final collection, debris was removed using a 100 μm cell strainer (VWR International). Cells were then pelleted at 200 g for 5 min. Purified cells were resuspended and plated in a culture medium containing Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12, Thermofisher) with 10% FBS + 1% P/S. Pre-plating was done for 1 hr in the incubator to allow separation of fibroblasts from cardiomyocytes. Cardiomyocytes were then collected for either FACS or plated in new 12- or 24-well plates for adenovirus-mediated overexpression studies.
FACS, multiplexing of cells and single-cell RNA sequencing
Isolated P1, P2, and P4 neonatal cardiomyocytes suspended in culture medium (DMEM/F-12 with 10% FBS + 1% P/S) were filtered using a 100 μm cell strainer (VWR, 10199-658), then transferred to separate polypropylene tubes (Falcon). Cells were incubated with SytoxTM Blue dead cell stain (Thermofisher, S34857, 1:2000) prior to Fluorescence Activated Cell Sorting (FACS). Gating was done for size and granularity, then for Sytox-based viability, and finally for tdTomato-based identification of conduction cells. Approximately 0.1–0.2% of total viable cardiomyocytes were tdTomato-positive for all P1, P2, and P4 cells (Supplementary Fig. 2).
MULTI-seq reagents were generously provided by the developers, and multiplexing of P1, P2, and P4 cells was conducted following the developer’s protocols35. In brief, following separate FACS sorting of P1, P2, and P4 CCS cells into culture medium (DMEM/F-12 with 10% FBS + 1% P/S), cells were pelleted and washed using centrifugation (250 g, 5 min, 4 °C) and 1x PBS. Lipid-modified oligonucleotide anchor mix and unique barcodes were added to each sample at final concentrations of 200 nM. Cells were resuspended by gentle pipetting and were incubated at 4 °C for 10 min, ensuring to periodically mix by gently tapping. Then, a co-anchor lipid-modified oligonucleotide was added to each sample (final concentration of 200 nM), and cells were incubated at 4 °C for 5 min. Cells were then pelleted using centrifugation (250 g, 5 min, 4 °C) and resuspended in PBS containing 1% BSA to quench residual barcodes. P1, P2, and P4 cells were then pooled into a single tube and subjected to viability assessments and cell counting prior to scRNA-seq library generation using the 10x Chromium Single Cell 3’ v3 kit (10x Genomics). All gene expression libraries were sequenced with NextSeq500 (Illumina).
scRNA-seq data analysis
The generated fastq files were processed with CellRanger (version 7). Reads were aligned to the mm10-3.0.0 reference transcriptome annotation file. Sequences and annotations for tdTomato and Cre were manually added to the fasta and gtf files (Supplementary Data 5), respectively, using the function cellranger mkref. A total of 10,407 cells were sequenced, with a mean of 35,041 reads per cell and a median of 3055 genes detected per cell. Demultiplexing was performed using Python according to the unique MULTI-seq barcodes given to each P1, P2, and P4 sample prior to pooling.
Downstream bioinformatic analysis was performed on R (version 4.2.2) using the ‘Seurat’ R package (version 5.0.1)106. Briefly, pre-processing of the data was performed following quality assessment to include cells with >2000 and <5500 detected genes, and percentages of mitochondrial genes of >5% and <25% (Supplementary Fig. 3). Additional biological filtering was applied based on markers of common cell types known to exist in the heart. For instance, 1.03% of all cells (72 out of 6927 cells) that lacked expression of cardiomyocyte genes (Myh6, Myh7, Tnnt2, Tnni1, Ryr2) but expressed markers of fibroblasts (Postn, Col1a1, Col1a2, Col3a1) or smooth muscle cells (Cald1, Emilin1, Hexim1, Tagln, and Fabp4), were excluded from downstream analysis (Supplementary Fig. 4). Following pre-processing of the data, feature counts were normalized by log-transformation using the function NormalizeData. The top 5000 features were identified using “vst” as the selection method using FindVariableFeatures. Normalized counts for these variable genes were then scaled using ScaleData with regression of mitochondrial gene counts and the number of unique molecular identifiers (UMIs). Linear dimensional reduction was performed using principal component analysis (PCA) using RunPCA. For Uniform Manifold Approximation and Projection (UMAP) embedding using RunUMAP analysis using all cells, including non-cardiomyocytes (Supplementary Fig. 4), CCS cells (Supplementary Fig. 5b), ‘Nodes-HIS-BB’ (Supplementary Fig. 5c), ‘Nodes’ (Supplementary Fig. 5d), the first 25 principal dimensions were used. Optimal clusters were identified by FindNeighbors and FindClusters using resolutions of 0.25, 0.19, 0.4, and 0.97, respectively, determined by the examination of known marker expression patterns. For the ‘LNB-HIS-PBB’ (Supplementary Fig. 5e), the first 10 principal components were used with a clustering resolution of 0.6. For ‘Proliferative CCS’ (Supplementary Fig. 8), 20 principal components were used with a clustering resolution of 0.6. Heatmaps were generated using the ‘ComplexHeatmap’ R package (version 2.15.4).
E16.5 CCS data (GSE132658)29 was analyzed according to the author’s described methods as well as using markers demonstrated in the study. Integration of E16.5 and Postnatal CCS was performed using Seurat’s Integration pipeline. Briefly, anchors between the datasets were found using FindIntegrationAnchors followed by data integration using IntegrateData. For correlation between E16.5 and postnatal CCS components, averaged expressions of genes for each CCS component given by AverageExpression were used. Correlation analysis was then performed and visualized using the ‘corrplot’ R package (version 0.92). Direct comparison was performed on integrated data using FindMarkers.
Visualization of sequencing reads alignment to Scn10a was generated using the Integrative Genomics Viewer (IGV) (version 2.17.4), with the reference genome mm10 (Supplementary Fig. 12).
Gene regulatory network inference
Gene regulatory network inference was performed using ‘pySCENIC’ (version 0.12.1) following the SCENIC (single-cell regulatory network inference and clustering) pipeline84,107. The count matrix was provided as input after filtering out genes with zero expression. Repressive regulons were excluded, and putative regulons lacking expression of the TF in at least 40% of any region were filtered out, resulting in 97 retained regulons. Regulon activity was quantified in individual cells using AUCell and averaged across each region.
To construct regional gene regulatory networks, regulons were scored based on regional specificity108. The top 5 regulons specific to each region were retained after filtering for 40% expression of the TF in the respective region. Target genes differentially expressed in each region (adj. P < 0.05) were retained. The resulting regional networks with top 5 regulons were then visualized using the Fruchterman-Reingold layout (‘iGraph’, version 1.5.1), with node colors corresponding to the classification of the gene (transcription factors and CCS marker genes).
Spatial transcriptomics and data analysis
Spatial Transcriptomic (ST) analysis was performed in frozen tissue sections of P4 CCS-tdT mouse heart using the NanoString GeoMx Digital Spatial Profiler (DSP)109,110 in collaboration with the Advanced Genomics Core at the University of Michigan. The sections were subjected to in situ hybridization with the GeoMx Probe Mix for NGS readout (Mouse Whole Transcriptome Atlas RNA v1.0). CCS structure was visualized with anti-DsRed primary antibody (Takara, #632496, 1:200) and anti-rabbit AlexaFluorTM 594 secondary antibody (Thermofisher Scientific, A11012, 1:500). SYTO13 nuclear staining was performed for visualization of tissue morphology and cell counting. Regions of interest (ROI) were chosen using the fluorescent segmentation function on the DSP. Two ROIs were selected for each CCS region, including the SA node (SAN), lower nodal bundle (LNB), His bundle (HIS), proximal left and right bundle branch (PLBB/PRBB), distal left and right bundle branch (DLBB/DRBB), and left and right Purkinje fibers (LPF/RPF). One ROI was collected for the compact AV node (cAVN). Non-CCS regions adjacent to each CCS region were also selected, including right atrial appendage (RAA), left endomyocardium (LENDO), left epimyocardium (LEPI), basal interventricular septum (IVS-B), apical interventricular septum (IVS-A), and right ventricle (RV). Following ROI selection, DNA oligos were cleaved from RNAScope probes by directing UV light at each ROI using the digital micromirror device. Cleaved oligos were then collected into the wells of a 96-well collection plate, which were subjected to library preparation and next-generation sequencing. Sequencing depth was estimated by multiplying the total area of all the ROIs (µm2) and sequencing of the pooled library was performed on NovaSeq 6000 (Illumina).
Raw FASTQ files were processed using NanoString’s Automated Data Processing Pipeline, which includes adapter trimming and aligning stitched paired-end reads to the barcodes in the reference assay before the removal of PCR duplicates based on the UMI of each read. The resulting data collection container (DCC) files for each ROI were then processed with the ‘GeoMxTools’ package (version 3.0.1) using the corresponding NanoString DSP configuration file and the instrument-generated worksheet with ROI identifiers. Initial quality control filtering and third quartile (Q3) normalization were performed with minimum average negative control measurements for ROI retention and minimum counts above background for target retention. The resulting filtered and Q3 normalized data were analyzed for differential expression analysis using an unpaired t-test. A gene was considered CCS-specific as well as significantly differentially expressed (Fig. 2a, b) if it satisfied both of the following conditions: 1) |Fold change| > 1.5, P < 0.05 in the region of interest compared to all other CCS components, and; 2) |Fold change| > 1.5, in the region of interest compared to its respective non-CCS components. For SAN and cAVN, its non-CCS control was RAA; for LNB, HIS, PLBB, and PRBB, its control was IVS-B; for DLBB and DRBB, its control was IVS-A; for LPF its control was LENDO and; for RPF, its control was RV.
ATAC-seq and ChIP-Seq analyses
For chromatin accessibility and binding analyses, raw data was downloaded in fastq format using the fastq-dump function in the SRA toolkit. Fastq files were then uploaded onto the UseGalaxy server111. Quality control assessments were performed using FastQC (version 0.11.7)112. Reads were then aligned to the mm9 mouse reference genome using Bowtie2 (Galaxy Version 2.4.2+galaxy0) with the ‘--very-sensitive’ parameter applied for optimal mapping113. Mitochondrial genome reads and low mapping quality (mapQuality <30) were considered uninformative reads and filtered out using the Filter BAM datasets on a variety of attributes function (Galaxy Version 2.4.1)114. Duplicate reads were then removed using ‘Picard MarkDuplicates’ (Galaxy Version 2.18.2.2) (broadinstitute.github.io/picard). MACS2 callpeak function (Galaxy Version 2.1.1.20160309.6) was used to call peaks with the following parameters: ‘Single-end BED’, ‘--nomodel’, ‘--shift −100’, ‘--extend 200’, and FDR < 0.05115. The following datasets were analysed: E13.5 Tbx3+ AVJ ATAC-seq (GSE121464 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121464])25, adult ventricle ATAC-seq (GSE110209 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110209])92, P0 sinoatrial pacemaker ATAC-seq (GSE148515 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148515])91, P0 right atrial cardiomyocyte ATAC-seq (GSE148515 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148515]), and adult heart Tbx3 ChIP-seq (GSE35151 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE35151])93. Moreover, two Iroquois (Irx) motifs, ACATGT (Motif ID: UP00223_2)90 and ACAnnTGT89 were used for predicting genome-wide motif occurrences. Each motif sequence was uploaded onto FIMO (Find Individual Motif Occurrences), part of the MEME software package (tools.genouest.org/tools/meme/), and motifs were scanned in the UCSC mm9 mouse genome. Only motif occurrences with P values ≤ 1.0 × 10−4 were considered significant, which are automatically calculated by FIMO based on a log-likelihood ratio score conversion116.
Peaks were visualized in bigWig format by converting MACS2 Bedgraph output to bigWig via the Wig/BedGraph-to-bigWig function (Galaxy version 1.1.1) and uploading it onto the Integrative Genomics Viewer (IGV) genome browser (version 2.16.2)117. For datasets where peaks were readily available in bigWig format, peak files were directly downloaded from the NCBI Gene Expression Omnibus (GEO) and uploaded onto IGV. For datasets with multiple replicates, a mean bigWig file was generated using the deepTools2 ‘bigwigCompare’ function (Galaxy version 3.5.1.0.0)118.
Immunofluorescence staining
Freshly isolated neonatal hearts were fixed in a methanol-free 4% paraformaldehyde solution (Thermo ScientificTM PierceTM, #28906) in PBS overnight at 4 °C. Hearts were washed twice in PBS for 30 mins each, then were incubated in filtered 30% sucrose in PBS overnight at 4 °C. Fixed cryopreserved hearts were embedded in Tissue-Tek® O.C.T compound (Sakura, #4683) and immediately frozen on powdered dry ice. Embedded hearts were sectioned using a cryostat at 8 μm thickness and stored at −20 °C until use.
For immunofluorescence staining, heart sections were air-dried for 30 min at room temperature, then washed three times in PBS for 5 min each. The sections were permeabilized with 0.1% Triton X-100 in PBS for 15 min. Blocking was done using the appropriate host serum in 0.3% triton X-100 in PBS for 1 hr at room temperature. The sections were then incubated overnight at 4 °C with the following primary antibodies: chicken anti-RFP (Rockland Immunochemicals, 600-901-379, lot #42649, 1:100), goat anti-Cntn2 (R&D Systems, AF4439, lot #CDSO0219101, 1:50), rabbit anti-Gnao1 (Thermo Fisher Scientific, PA5-30044, lot #YH4027897A, 1:100), rabbit anti-Scn10a (Alomone Labs, ASC-016, lot #, ASC016AN2550, 1:50), rabbit anti-Igfbp7 (Abcam, ab74169, lot #GR242021-1, 1:50), rabbit anti-S100a6 (Abcam, ab181975, lot #GR3438351-1, 1:100), rabbit anti-Nppa (EMD Millipore, ab5490, lot #Q3015715, 1:250), mouse anti-Shox2 IgG2a (Santa Cruz Biotechnology, sc-81955, lot #B2822, 1:50) or rabbit anti-Vsnl1 (GeneTex, GTX115039, lot #40247, 1:100), rabbit anti-Ppp1r17 (Thermo Fisher Scientific, PA5-61599, lot #YH4028316B, 1:200). The following day, sections were washed three times with PBS for 5 min each, and then incubated with appropriate secondary antibodies: goat anti-chicken IgY Alexa FluorTM 568 (Thermo Fisher Scientific, A11041, lot #2482953, 1:200), donkey anti-chicken IgY Alexa FluorTM 568 (Thermo Fisher Scientific, A78950, lot # 2622381, 1:200), goat anti-rabbit Alexa FluorTM 488 (Thermo Fisher Scientific, A-11008, lot #2420730, 1:500), goat anti-mouse IgG2a Alexa FluorTM 488 (Thermo Fisher Scientific, A21131, lot #2420706, 1:500), donkey anti-rabbit Alexa Fluor® 647 (Jackson ImmunoResearch Laboratories Inc., 711-605-152, 1:200), goat anti-mouse IgG2b Alexa FluorTM 488 (Thermo Fisher Scientific, A21141, lot #2432059, 1:500) for 1 hr at room temperature in the dark. Finally, the sections were subjected to the Vector® TrueVIEW Autofluorescence quenching kit (Vector laboratories, SP-8400) to minimize autofluorescence of the heart. A mounting medium with DAPI (Abcam, ab104139) was used for nuclei staining. Imaging was performed using LSM 880 confocal microscope with Airyscan (Zeiss) or AxioObserver 7 microscope (Zeiss), and processing was performed using ZEN microscopy software with Airyscan processing and orthogonal projection. Whole tissue fluorescence imaging to examine Cntn2Cre-mediated tdTomato expression of the CCS was performed using the Zeiss SteREO Discovery V8 stereomicroscope. Fluorescence slide scanning was performed using Aperio VERSA 8 Scanner (Leica Biosystems).
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) was performed using the RNAscope Multiplex Fluorescent Reagent Kits v2 (Advanced Cell Diagnostics, ACD) as per the manufacturer’s protocol. In brief, frozen tissue sections (8 μm thickness) of P4 CCS-tdT mouse heart were baked at 60 °C for 30 min, followed by post-fixation in 4% PFA for 30 min at 4 °C. After target retrieval for 5 min and Protease Plus incubation for 15 min, the sections were incubated for 2 hrs at 40 °C with the following probes: Prdm6 (mouse, Cat #456891), Mpped2 (mouse, ACD, #848371), Pcp4 (mouse, ACD, #402311) and tdTomato (mouse, ACD, #317041-C2). Opal 690 Reagent Pack (Akoya Biosciences, #FP1497001KT, 1:200) and Opal 570 Reagent Pack (SKU FP1488001KT, 1:200) were used to visualize the FISH signal. Fluorescence microscopy was performed using LSM 880 confocal microscope (Zeiss) or AxioObserver 7 microscope (Zeiss), followed by image processing with ZEN microscopy software.
Trichrome staining
To examine the cellular morphology and fibrous insulation in the atrioventricular region of the cardiac conduction system (i.e., cAVN, LNB and HIS), P4 mouse hearts were fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned at 5 μm thickness. Fibrosis was visualized by Masson’s trichrome staining, and images were captured using the Aperio VERSA 8 Scanner (Leica Biosystems).
Adenovirus-mediated overexpression
24 hrs following the isolation and plating of atrial and ventricular cardiomyocytes, debris and dead cells were washed with pre-warmed PBS. After washing, pre-warmed fresh culture medium (DMEM/F-12 + 10% FBS + 1% P/S) containing adenoviruses expressing GFP, Irx3, Tbx3, or both Irx3 and Tbx3 was added to the cells at a multiplicity of infection of 10 virus particles per cell. Adenovirus was washed off after 7-12 hrs, and the cells were cultured for an additional 48 hrs before being collected for RNA extraction.
Quantitative PCR
Cultured cardiomyocytes were lysed in TrizolTM Reagent (InvitrogenTM) through vigorous pipetting. For every 1 mL of TrizolTM, 0.2 mL of chloroform (C607-4, Fisher Chemical) was added to the cell lysate, followed by vigorous shaking for 15 sec and incubation at room temperature for 5 min. Centrifugation was done at 12,000 g for 15 min at 4 °C to allow separation of the aqueous layer collection. Using the aqueous layer, RNA was purified with PureLinkTM RNA Mini Kit (InvitrogenTM) according to the manufacturer’s protocol. Nanodrop2000 spectrophotometer (Thermo Fisher Scientific) was used for spectrophotometric quality and quantity assessments. cDNA was synthesized from purified RNA through reverse transcription using the High-Capacity cDNA Reverse Transcription Kit (Applied BiosystemsTM) following the manufacturer’s instructions, and to inhibit RNA degradation, RNaseOUTTM Recombinant Ribonuclease Inhibitor (InvitrogenTM) was used. Quantitative PCR (qPCR) was performed using PowerUpTM SYBRTM Green Master Mix (Applied BiosystemsTM) with QuantStudioTM 5 Real-Time PCR System (Applied Biosystems). Relative fold change and normalization of genes of interest were performed using the 2−ΔΔCT method, with Tbp serving as the housekeeping gene after ensuring its stable expression across all conditions. Primer sequences used are listed in the supplemental data (Supplementary Data 6).
Statistical analysis and reproducibility
Fluorescence stereomicroscope imaging of the CCS-tdT heart (Fig. 1a) was performed on more than five hearts. All immunofluorescence experiments (Fig. 3a, c–f and Supplementary Figs. 12, 13, 15–17, 19, and 20) were independently performed at least two times with varying antibody concentrations. All fluorescence in situ hybridization (FISH) experiments (Supplementary Fig. 14,18, 21) were conducted at least twice. Comparison between the embryonic (E14.5) and postnatal CCS data (Fig. 4d) was conducted using the Wilcoxon Rank Sum Test. Data are provided in Supplementary Data 3. Statistical analyses of qPCR experiments (Fig. 6 and Supplementary Figs. 24 and 25) were performed using GraphPad Prism 10 (GraphPad Software). Results are reported as mean ± standard error of the mean (SEM). Statistical differences between the means of two or more groups were tested using unpaired two-tailed Student’s t-test or one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test. P-values less than 0.05 were considered statistically significant. The number of samples (n) is stated in figure legends. No statistical method was used to predetermine the sample size.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The scRNA-seq data and spatial transcriptomics data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession code, GSE231547. Source data for Fig. 6 and Supplementary Fig. 25 and 26 are provided with this paper. Source data are provided with this paper.
Code availability
All codes used to generate the results and figures in this study are available in Zenodo (https://doi.org/10.5281/zenodo.12520577).
References
Haissaguerre, M. et al. Role of Purkinje conducting system in triggering of idiopathic ventricular fibrillation. Lancet 359, 677–678 (2002).
Scheinman, M. M. Role of the His-Purkinje system in the genesis of cardiac arrhythmia. Heart Rhythm 6, 1050–1058 (2009).
Boyden, P. A., Dun, W. & Robinson, R. B. Cardiac Purkinje fibers and arrhythmias; the GK Moe Award Lecture 2015. Heart Rhythm 13, 1172–1181 (2016).
Goodyer, W. R. & Wu, S. M. Fates aligned: origins and mechanisms of ventricular conduction system and ventricular wall development. Pediatr. Cardiol. 39, 1090–1098 (2018).
Itoh, T. & Yamada, T. Multifocal ventricular arrhythmias originating from the His-Purkinje system. JACC Clin. Electrophysiol. 4, 1248–1260 (2018).
Haissaguerre, M., Vigmond, E., Stuyvers, B., Hocini, M. & Bernus, O. Ventricular arrhythmias and the His-Purkinje system. Nat. Rev. Cardiol. 13, 155–166 (2016).
Keith, A. & Flack, M. The form and nature of the muscular connections between the primary divisions of the vertebrate heart. J. Anat. Physiol. 41, 172–189 (1907).
Roguin, A. Wilhelm His Jr. (1863-1934)–the man behind the bundle. Heart Rhythm 3, 480–483 (2006).
Suma, K. Sunao Tawara: a father of modern cardiology. Pacing Clin. Electrophysiol. 24, 88–96 (2001).
Steiner, I. History of the Purkyne fibres in the heart. On the occasion of bicentenary of Jan Evangelista Purkyne’s birth (December 17, 1787). Sb. Ved. Pr. Lek. Fak. Karlov. Univerzity Hradci Kralove 30, 7–20 (1987).
Mangoni, M. E. & Nargeot, J. Genesis and regulation of the heart automaticity. Physiol. Rev. 88, 919–982 (2008).
Kupersmith, J., Krongrad, E. & Waldo, A. L. Conduction intervals and conduction velocity in the human cardiac conduction system. Studies during open-heart surgery. Circulation 47, 776–785 (1973).
Verheijck, E. E. et al. Pacemaker synchronization of electrically coupled rabbit sinoatrial node cells. J. Gen. Physiol. 111, 95–112 (1998).
Kreuzberg, M. M. et al. Functional properties of mouse connexin30.2 expressed in the conduction system of the heart. Circ. Res. 96, 1169–1177 (2005).
Bhattacharyya, S. & Munshi, N. V. Development of the cardiac conduction system. Cold Spring Harb. Perspect. Biol. 12, a037408 (2020).
van Eif, V. W. W., Devalla, H. D., Boink, G. J. J. & Christoffels, V. M. Transcriptional regulation of the cardiac conduction system. Nat. Rev. Cardiol. 15, 617–630 (2018).
Choquet, C., Boulgakoff, L., Kelly, R. G. & Miquerol, L. New insights into the development and morphogenesis of the cardiac Purkinje fiber network: linking architecture and function. J. Cardiovasc. Dev. Dis. 8, 95 (2021).
Hoogaars, W. M. et al. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc. Res. 62, 489–499 (2004).
Zhang, S. S. et al. Iroquois homeobox gene 3 establishes fast conduction in the cardiac His-Purkinje network. Proc. Natl Acad. Sci. USA 108, 13576–13581 (2011).
Kim, K. H. et al. Irx3 is required for postnatal maturation of the mouse ventricular conduction system. Sci. Rep. 6, 19197 (2016).
Shekhar, A. et al. Transcription factor ETV1 is essential for rapid conduction in the heart. J. Clin. Invest. 126, 4444–4459 (2016).
Moskowitz, I. P. et al. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell 129, 1365–1376 (2007).
Jongbloed, M. R. et al. Expression of Id2 in the second heart field and cardiac defects in Id2 knock-out mice. Dev. Dyn. 240, 2561–2577 (2011).
Frank, D. U. et al. Lethal arrhythmias in Tbx3-deficient mice reveal extreme dosage sensitivity of cardiac conduction system function and homeostasis. Proc. Natl Acad. Sci. USA 109, E154–E163 (2012).
Mohan, R. A. et al. T-box transcription factor 3 governs a transcriptional program for the function of the mouse atrioventricular conduction system. Proc. Natl Acad. Sci. USA 117, 18617–18626 (2020).
Aanhaanen, W. T. et al. Developmental origin, growth, and three-dimensional architecture of the atrioventricular conduction axis of the mouse heart. Circ. Res. 107, 728–736 (2010).
George, S. A. et al. At the atrioventricular crossroads: dual pathway electrophysiology in the atrioventricular node and its underlying heterogeneities. Arrhythm. Electrophysiol. Rev. 6, 179–185 (2017).
Temple, I. P., Inada, S., Dobrzynski, H. & Boyett, M. R. Connexins and the atrioventricular node. Heart Rhythm 10, 297–304 (2013).
Goodyer, W. R. et al. Transcriptomic profiling of the developing cardiac conduction system at single-cell resolution. Circ. Res. 125, 379–397 (2019).
Bhattacharyya, S. et al. Using Gjd3-CreEGFP mice to examine atrioventricular node morphology and composition. Sci. Rep. 9, 2106 (2019).
Liang, D. et al. Cellular and molecular landscape of mammalian sinoatrial node revealed by single-cell RNA sequencing. Nat. Commun. 12, 287 (2021).
Bhattacharyya, S., Bhakta, M. & Munshi, N. V. Phenotypically silent Cre recombination within the postnatal ventricular conduction system. PLoS ONE 12, e0174517 (2017).
Goodyer, W. R. et al. In vivo visualization and molecular targeting of the cardiac conduction system. J. Clin. Invest. 132, e156955 (2022).
Pallante, B. A. et al. Contactin-2 expression in the cardiac Purkinje fiber network. Circ. Arrhythm. Electrophysiol. 3, 186–194 (2010).
McGinnis, C. S. et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods 16, 619–626 (2019).
Liu, F. et al. GATA-binding factor 6 contributes to atrioventricular node development and function. Circ. Cardiovasc. Genet. 8, 284–293 (2015).
van Eif, V. W. W., Stefanovic, S., Mohan, R. A. & Christoffels, V. M. Gradual differentiation and confinement of the cardiac conduction system as indicated by marker gene expression. Biochim. Biophys. Acta Mol. Cell Res. 1867, 118509 (2020).
Espinoza-Lewis, R. A. et al. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev. Biol. 327, 376–385 (2009).
Ye, W. et al. A common Shox2-Nkx2-5 antagonistic mechanism primes the pacemaker cell fate in the pulmonary vein myocardium and sinoatrial node. Development 142, 2521–2532 (2015).
Hoogaars, W. M. et al. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 21, 1098–1112 (2007).
Bakker, M. L. et al. Transcription factor Tbx3 is required for the specification of the atrioventricular conduction system. Circ. Res. 102, 1340–1349 (2008).
Zhao, J. & Mommersteeg, M. T. M. Slit-Robo signalling in heart development. Cardiovasc Res. 114, 794–804 (2018).
Mommersteeg, M. T., Yeh, M. L., Parnavelas, J. G. & Andrews, W. D. Disrupted Slit-Robo signalling results in membranous ventricular septum defects and bicuspid aortic valves. Cardiovasc. Res. 106, 55–66 (2015).
Franco, D. & Icardo, J. M. Molecular characterization of the ventricular conduction system in the developing mouse heart: topographical correlation in normal and congenitally malformed hearts. Cardiovasc. Res. 49, 417–429 (2001).
Spielmann, N. et al. Extensive identification of genes involved in congenital and structural heart disorders and cardiomyopathy. Nat. Cardiovasc. Res. 1, 157–173 (2022).
Ma, L., Lu, M. F., Schwartz, R. J. & Martin, J. F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 132, 5601–5611 (2005).
Li, G. et al. Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev. Cell 39, 491–507 (2016).
Zhang, Q. et al. Expression and roles of Cav1.3 (alpha1D) L-type Ca(2)+ channel in atrioventricular node automaticity. J. Mol. Cell Cardiol. 50, 194–202 (2011).
Gewies, A. et al. Prdm6 is essential for cardiovascular development in vivo. PLoS ONE 8, e81833 (2013).
Hong, L. et al. Prdm6 controls heart development by regulating neural crest cell differentiation and migration. JCI Insight 7, e156046 (2022).
Zou, M. et al. Prdm6 drives ductus arteriosus closure by promoting ductus arteriosus smooth muscle cell identity and contractility. JCI Insight 8, e163454 (2023).
Kinameri, E. et al. Prdm proto-oncogene transcription factor family expression and interaction with the Notch-Hes pathway in mouse neurogenesis. PLoS ONE 3, e3859 (2008).
Verweij, N., van de Vegte, Y. J. & van der Harst, P. Genetic study links components of the autonomous nervous system to heart-rate profile during exercise. Nat. Commun. 9, 898 (2018).
Galdos, F. X. et al. devCellPy is a machine learning-enabled pipeline for automated annotation of complex multilayered single-cell transcriptomic data. Nat. Commun. 13, 5271 (2022).
Caglar, C. & Friedman, J. Restriction of food intake by PPP1R17-expressing neurons in the DMH. Proc. Natl Acad. Sci. USA 118, e2100194118 (2021).
Kim, E. E. et al. PCP4 regulates Purkinje cell excitability and cardiac rhythmicity. J. Clin. Invest. 124, 5027–5036 (2014).
Delgado, C. et al. Neural cell adhesion molecule is required for ventricular conduction system development. Development 148, dev199431 (2021).
Christoffels, V. M. et al. Chamber formation and morphogenesis in the developing mammalian heart. Dev. Biol. 223, 266–278 (2000).
Zeller, R., Bloch, K. D., Williams, B. S., Arceci, R. J. & Seidman, C. E. Localized expression of the atrial natriuretic factor gene during cardiac embryogenesis. Genes Dev. 1, 693–698 (1987).
Tian, X. et al. Identification of a hybrid myocardial zone in the mammalian heart after birth. Nat. Commun. 8, 87 (2017).
Mishra, A., Tavasoli, M., Sokolenko, S., McMaster, C. R. & Pasumarthi, K. B. S. Atrial natriuretic peptide signaling co-regulates lipid metabolism and ventricular conduction system gene expression in the embryonic heart. iScience 27, 108748 (2024).
Vedantham, V., Galang, G., Evangelista, M., Deo, R. C. & Srivastava, D. RNA sequencing of mouse sinoatrial node reveals an upstream regulatory role for Islet-1 in cardiac pacemaker cells. Circ. Res. 116, 797–803 (2015).
Mukharji, A., Drucker, D. J., Charron, M. J. & Swoap, S. J. Oxyntomodulin increases intrinsic heart rate through the glucagon receptor. Physiol. Rep. 1, e00112 (2013).
Luo, X. et al. MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J. Clin. Invest. 123, 1939–1951 (2013).
Mofid, A. et al. Cardiac overexpression of S100A6 attenuates cardiomyocyte apoptosis and reduces infarct size after myocardial ischemia-reperfusion. J. Am. Heart Assoc. 6, e004738 (2017).
Li, Y. et al. Genetic targeting of Purkinje fibres by Sema3a-CreERT2. Sci. Rep. 8, 2382 (2018).
Choquet, C., Kelly, R. G. & Miquerol, L. Nkx2-5 defines distinct scaffold and recruitment phases during formation of the murine cardiac Purkinje fiber network. Nat. Commun. 11, 5300 (2020).
Pernot, M. et al. Decrease of Pdzrn3 is required for heart maturation and protects against heart failure. Sci. Rep. 12, 8 (2022).
Lebel, M. et al. The Iroquois homeobox gene Irx2 is not essential for normal development of the heart and midbrain-hindbrain boundary in mice. Mol. Cell. Biol. 23, 8216–8225 (2003).
Malhotra, R. et al. Matrix gla protein levels are associated with arterial stiffness and incident heart failure with preserved ejection fraction. Arterioscler. Thromb. Vasc. Biol. 42, e61–e73 (2022).
Peng, W. et al. Dysfunction of myosin light-chain 4 (MYL4) leads to heritable atrial cardiomyopathy with electrical, contractile, and structural components: evidence from genetically-engineered rats. J. Am. Heart Assoc. 6, e007030 (2017).
Yamada, N. et al. Mutant KCNJ3 and KCNJ5 potassium channels as novel molecular targets in bradyarrhythmias and atrial fibrillation. Circulation 139, 2157–2169 (2019).
Mangoni, M. E. et al. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/alpha1G T-type calcium channels. Circ. Res. 98, 1422–1430 (2006).
Graham, V., Zhang, H., Willis, S. & Creazzo, T. L. Expression of a two-pore domain K+ channel (TASK-1) in developing avian and mouse ventricular conduction systems. Dev. Dyn. 235, 143–151 (2006).
Makara, M. A. et al. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ. Res. 115, 929–938 (2014).
Man, J. C. K. et al. Variant intronic enhancer controls SCN10A-short expression and heart conduction. Circulation 144, 229–242 (2021).
Yamashita, N., Ilg, E. C., Schafer, B. W., Heizmann, C. W. & Kosaka, T. Distribution of a specific calcium-binding protein of the S100 protein family, S100A6 (calcyclin), in subpopulations of neurons and glial cells of the adult rat nervous system. J. Comp. Neurol. 404, 235–257 (1999).
Kuznicki, J., Kordowska, J., Puzianowska, M. & Wozniewicz, B. M. Calcyclin as a marker of human epithelial cells and fibroblasts. Exp. Cell Res. 200, 425–430 (1992).
Akazawa, H. & Komuro, I. Roles of cardiac transcription factors in cardiac hypertrophy. Circ. Res 92, 1079–1088 (2003). [pii].
Owens, G. K., Kumar, M. S. & Wamhoff, B. R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 (2004).
Miano, J. M. & Olson, E. N. Expression of the smooth muscle cell calponin gene marks the early cardiac and smooth muscle cell lineages during mouse embryogenesis. J. Biol. Chem. 271, 7095–7103 (1996).
Chen, H. et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 131, 2219–2231 (2004).
Yan, W., Sheng, N., Seto, M., Morser, J. & Wu, Q. Corin, a mosaic transmembrane serine protease encoded by a novel cDNA from human heart. J. Biol. Chem. 274, 14926–14935 (1999).
Aibar, S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Gharibeh, L. et al. GATA6 is a regulator of sinus node development and heart rhythm. Proc. Natl Acad. Sci. USA 118, e2007322118 (2021).
van Eif, V. W. W. et al. Transcriptome analysis of mouse and human sinoatrial node cells reveals a conserved genetic program. Development 146, dev173161 (2019).
Burnicka-Turek, O. et al. Transcriptional patterning of the ventricular cardiac conduction system. Circ. Res. 127, e94–e106 (2020).
Bhattacharyya, S. et al. Global chromatin landscapes identify candidate noncoding modifiers of cardiac rhythm. J. Clin. Invest. 133, e153635 (2023).
Bilioni, A., Craig, G., Hill, C. & McNeill, H. Iroquois transcription factors recognize a unique motif to mediate transcriptional repression in vivo. Proc. Natl Acad. Sci. USA 102, 14671–14676 (2005).
Berger, M. F. et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133, 1266–1276 (2008).
Galang, G. et al. ATAC-Seq reveals an Isl1 enhancer that regulates sinoatrial node development and function. Circ. Res. 127, 1502–1518 (2020).
van Duijvenboden, K. et al. Conserved NPPB+ border zone switches from MEF2- to AP-1-driven gene program. Circulation 140, 864–879 (2019).
van den Boogaard, M. et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J. Clin. Invest. 122, 2519–2530 (2012).
Boyett, M. R. et al. Connexins in the sinoatrial and atrioventricular nodes. Adv. Cardiol. 42, 175–197 (2006).
Marionneau, C. et al. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J. Physiol. 562, 223–234 (2005).
Liang, X. et al. HCN4 dynamically marks the first heart field and conduction system precursors. Circ. Res. 113, 399–407 (2013).
Beyer, S., Kelly, R. G. & Miquerol, L. Inducible Cx40-Cre expression in the cardiac conduction system and arterial endothelial cells. Genesis 49, 83–91 (2011).
Kanemaru, K. et al. Spatially resolved multiomics of human cardiac niches. Nature 619, 801–810 (2023).
Zhang, L. et al. Insulin-like growth factor-binding protein-7 (IGFBP7) links senescence to heart failure. Nat. Cardiovasc. Res. 1, 1195–1214 (2022).
Tsoporis, J. T., Izhar, S., Desjardins, J. F., Leong-Poi, H. & Parker, T. G. Conditional cardiac overexpression of S100A6 attenuates myocyte hypertrophy and apoptosis following myocardial infarction. Curr. Pharm. Des. 20, 1941–1949 (2014).
Murphy, S. A. et al. PGC1/PPAR drive cardiomyocyte maturation at single cell level via YAP1 and SF3B2. Nat. Commun. 12, 1648 (2021).
Yan, W., Wu, F., Morser, J. & Wu, Q. Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proc. Natl Acad. Sci. USA 97, 8525–8529 (2000).
Govindapillai, A. et al. Characterizing the role of atrial natriuretic peptide signaling in the development of embryonic ventricular conduction system. Sci. Rep. 8, 6939 (2018).
Wiese, C. et al. Formation of the sinus node head and differentiation of sinus node myocardium are independently regulated by Tbx18 and Tbx3. Circ. Res. 104, 388–397 (2009).
Liang, X. et al. Transcription factor ISL1 is essential for pacemaker development and function. J. Clin. Invest. 125, 3256–3268 (2015).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
Van de Sande, B. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247–2276 (2020).
Suo, S. et al. Revealing the critical regulators of cell identity in the mouse cell atlas. Cell Rep. 25, 1436–1445 e1433 (2018).
He, S. et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nat. Biotechnol. 40, 1794–1806 (2022).
Williams, C. G., Lee, H. J., Asatsuma, T., Vento-Tormo, R. & Haque, A. An introduction to spatial transcriptomics for biomedical research. Genome Med. 14, 1–68 (2022).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544 (2018).
Wingett, S. W. & Andrews, S. FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res 7, 1338 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Stromberg, M. P. & Marth, G. T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692 (2011).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Batut, B. et al. Community-driven data analysis training for biology. Cell Syst. 6, 752–758.e751 (2018).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Acknowledgements
This study was supported primarily by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant (RGPIN-2018-06870) and partly the Canadian Institutes of Health Research (CIHR) Project Grant (PJT 173281) to Dr. Kyoung-Han Kim. He is also a recipient of the National New Investigator Award from the Heart and Stroke Foundation of Canada (HSFC) and the Early Researcher Award from the Government of Ontario, Canada. The infrastructure was supported by the Canadian Foundation for Innovation, John R. Evans Leaders Fund (CFI-JELF; 37735) to Dr. Kyoung-Han Kim. Yena Oh was supported by the Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award (CGS-D), UOHI Endowed Graduate Scholarship, and the Queen Elizabeth II Graduate Scholarship in Science and Technology. Rimshah Abid was supported by the Canada Graduate Scholarships – Master’s program. Termeh Aslani was supported by the UOHI Endowed Graduate Scholarship. Marwan Bakr was supported by the NSERC Undergraduate Student Research Award (USRA).
We thank Fernando Ortiz, Kathy Sheikheleslamy and Pearl Campbell in the StemCore Laboratories, the Ottawa Hospital Research Institute, for the FACS and 10x Genomics scRNA-seq experiments; Dana King and Margaret Hogan in the University of Michigan Advanced Genomics Core for the NanoString GeoMX DSP experiment; and XiaoLing Zhao in the Cell Imaging & Histopathology Core Facility at the University of Ottawa Heart Institute for histological assessments. We also thank Dr. Paul Delgado-Olguin at the Hospital for Sick Children and Dr. Joe Eun Son at the Kyungpook National University for their feedback and discussion.
Author information
Authors and Affiliations
Contributions
K-H.K. and Y.O. conceived the project and designed experiments. Y.O., R.A., T.A., and K-H.K. performed experiments. Y.O., S.D., M.B., and K-H.K. conducted bioinformatic analysis. Y.O., R.A., M.B., and K-H.K. prepared the figures. D.P.C., B.V., N.V.M., and C.-C.H. provided reagents. D.P.C. and J.G.P provided technical support. K-H.K. directed the project. Y.O. and K-H.K. drafted, edited, and revised the manuscript with input from all authors. All authors, Y.O., R.A., S.D., M.B., T.A., D.P.C., B.V., J.G.P., N.V.M., C-C.H., and K-H.K. read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Vincent Christoffels, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Oh, Y., Abid, R., Dababneh, S. et al. Transcriptional regulation of the postnatal cardiac conduction system heterogeneity. Nat Commun 15, 6550 (2024). https://doi.org/10.1038/s41467-024-50849-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50849-1
- Springer Nature Limited