

Abstract
Immunochemotherapy has been the mainstay of treatment for newly diagnosed diffuse large B-cell lymphoma (ndDLBCL) yet is inadequate for many patients. In this work, we perform unsupervised clustering on transcriptomic features from a large cohort of ndDLBCL patients and identify seven clusters, one called A7 with poor prognosis, and develop a classifier to identify these clusters in independent ndDLBCL cohorts. This high-risk cluster is enriched for activated B-cell cell-of-origin, low immune infiltration, high MYC expression, and copy number aberrations. We compare and contrast our methodology with recent DLBCL classifiers to contextualize our clusters and show improved prognostic utility. Finally, using pre-clinical models, we demonstrate a mechanistic rationale for IKZF1/3 degraders such as lenalidomide to overcome the low immune infiltration phenotype of A7 by inducing T-cell trafficking into tumors and upregulating MHC I and II on tumor cells, and demonstrate that TCF4 is an important regulator of MYC-related biology in A7.
Similar content being viewed by others

Introduction
Diffuse Large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin Lymphoma with a 5-year relative survival rate of 60%1. While most patients with newly diagnosed (nd) DLBCL are treated and cured with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), about 40% of patients are either refractory or experience relapse, highlighting the need for novel targeted agents2. Current approaches for high-risk patient identification in clinical practice include International Prognostic Index Score (IPI), MYC, BCL2 and/or BCL6 translocations by FISH, and activated B-cell (ABC)/non-germinal center B-cell (non-GCB) cell-of-origin (COO) subtyping by gene expression or immunohistochemistry3,4. COO has been used prospectively for patient selection in two randomized control trials ROBUST (NCT02285062) and PHOENIX (NCT01855750); however, neither reached their primary endpoint5,6. Over the last several years, multiple genetic classifications have been published and each reported predominantly ABC subtypes, such as C57, MCD8,9, and MYD8810 all with poor survival with R-CHOP. Gene expression signatures such as double hit/dark zone signature (DHITsig/DZsig)11,12 and molecular high grade13 identified mostly high-risk GCB patients; while TME26 categorized DLBCL tumors into high and low infiltration of T cells, macrophage, and dendritic cells14 Only recently with seminal work on Lymphoma Microenvironment ((LME15) and EcoTyper16 has the crosstalk between tumor cells and their microenvironment come into focus. These molecular classifications may eventually take DLBCL into the era of tailored targeted therapies, exemplified by a recent report in an R-CHOP + X trial, X being an agent that could specifically target the biology of a predefined patient segment17.
In this work, we sought to identify high-risk patients using a combination of both tumor-intrinsic and tumor microenvironment features for two purposes: (1) to develop a biomarker to be used in the clinic for identifying high-risk ndDLBCL patients for selection, stratification, or enrichment in clinical trials; and (2) to identify biological targets for these high-risk patients for future precision medicine approaches.
We performed unsupervised clustering on transcriptomic features derived from RNAseq data generated from routine diagnostic formalin-fixed paraffin-embedded (FFPE) samples of 1208 excisional lymph node biopsies of ndDLBCL patients. The results showed seven unique biological clusters for which we subsequently developed a gene expression classifier called SubLymE that allows the classification of ndDLBCL patients into one of the seven clusters. We found cluster A7 to have poor outcomes on R-CHOP treatment, and key biological features included enrichment for ABC COO, upregulation of MYC and MYC downstream pathways without MYC amplifications or translocations, and low immune cell infiltration. Significant genomic features of A7 included enrichment of chromosome 3 and 18 gains and 9p21 loss. Using pre-clinical models, we further propose TCF4 as a potential therapeutic target for A7 biology and patients. Retrospective analysis of the lenalidomide plus R-CHOP (R2-CHOP) experimental arm in the ROBUST trial indicated R2-CHOP has significant clinical benefit in A7 patients, for which we describe a mechanism of action of how lenalidomide overcomes high-risk features in A7 patients via enhanced immune infiltration.
Results
Unsupervised clustering analysis identifies seven molecular subpopulations of ndDLBCL
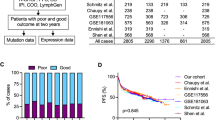
Unsupervised clustering using iClusterPlus18 was performed on a Discovery cohort of 1208 ndDLBCL patients including both ABC and GCB subtypes (see Methods and Table 1). While there are currently many publicly datasets available, this discovery cohort was selected based upon its large size, consistent profiling methodology, and availability of genomic and clinical data at the inception of this research. In addition to normalized RNAseq gene expression data, three sets of gene-expression-derived features were included as input, including Gene Set Variation Analysis (GSVA) scores from the MSigDB Hallmark pathways (H), chromosomal copy number alterations (C1)19, and inferred immune cell type abundance scores (see Methods). No clinical features or survival data were included as input features (Fig. 1A).

Unsupervised clustering was applied to a large cohort of patient-derived RNAseq data to identify biologically homogeneous subtypes of DLBCL. A Schematic of data transformation, unsupervised clustering, and classifier training methodology. Steps in black represent data objects, while steps in blue represent algorithmic processes. B Co-clustering frequency heatmap identifies sample clusters that consistently group together over repeated subsampling runs. C Cluster prevalence and breakdown by COO and TME26 classification. Bar heights represent the observed proportion in each cohort, and error bars represent the 95% confidence interval. D Top 50 up- and down-regulated genes per cluster from the Discovery dataset, replicated in each cohort. Source data are provided as a Source Data file.
Unsupervised clustering identified eight clusters (A1–A8 for aggressive lymphoma) in the Discovery cohort (Fig. 1B). COO and TME26 classification are shown for each cluster as measurements of cluster biological properties. For determination of COO we applied the Reddy method20 due to its wide applicability to gene expression data, and limited availability of Nanostring LST calls21. A comparison of Reddy versus Nanostring COO scores in samples where both data were available showed high concordance (r = 0.923) (Supplementary Fig. 1).
Certain clusters are significantly enriched for ABC (A7: 86% ABC, p < 2.2e−16) or GCB patients (A2: 90% GCB, p < 2.2e−16), TME26-positive (A6: 98% TME26 + , p < 2.2e−16) or TME26-negative (A1: 62% TME26-, p = 0.009; A2: 62% TME26-, p = 0.001; A7: 70% TME26-, p < 2.2e−16). Mean tumor purity (74% overall) varied by cluster, with A1 and A7 showing higher (80% and 83%) and A4 and A6 showing lower purity (70% and 68%). The smallest cluster A8 (n = 25, 2% of the discovery cohort) was found to be highly distinct from the rest, with A8 cases almost never clustering among other subtypes and having virtually no effect on the clustering in the rest of the data. Further analysis showed that despite initial quality control filters, A8 contained samples of borderline technical quality, evidenced by aberrant alignment metrics including a high number of unaligned, intergenic, and ribosomal reads, and low number of reads assigned to coding regions (Supplementary Fig. 2A). As we sought to classify groups defined by lymphoma biology, A8 was omitted from classifier training. Using the A1-A7 cluster labels, we then trained a multinomial generalized linear model (named as SubLymE) on the Discovery cohort to generate a classifier for identifying these clusters in independent ndDLBCL cohorts.
Using the SubLymE classifier, the seven clusters were reproduced in four replication cohorts: Molecular Epidemiology Resource (MER) of the University of Iowa/Mayo Clinic Lymphoma (n = 343)22, REMoDL-B (n = 928)23, GOYA (R-CHOP arm only, n = 271)24, and Reddy (n = 442)20. All seven clusters have consistent molecular characteristics as measured by COO or TME26 status across discovery and replication cohorts (Fig. 1C, Table 1), although cluster prevalence varies somewhat from cohort to cohort, with A6 and A7 being more stable than others possibly attributable to variations across patient populations (Fig. 1C). We further demonstrated biological consistency of the clusters by showing the top 50 up- and down-regulated differentially expressed genes for each cluster which exhibited similar expression patterns in the replication datasets (Fig. 1D). Furthermore, the directionality and magnitude of cluster-specific dysregulation of Hallmark pathways as measured by normalized enrichment score (NES) were consistent between the discovery and MER cohorts for all clusters with the exception of A4 (Supplementary Fig. 2B).
Cluster A7 is a reproducible high-risk patient subpopulation in newly diagnosed DLBCL treated with R-CHOP
We next examined EFS (in MER, REMoDL-B, GOYA) and OS (in Reddy) of the seven clusters under R-CHOP or R-CHOP-like treatment regimens. In the MER cohort, cluster A7 displayed the shortest median event-free survival (EFS) of 38.2 months compared to 165.6 months in non-A7 patients (Fig. 2A), and a binarized model showed a statistically significant difference in survival between A7 and non-A7 patients (p = 0.0052, Fig. 2B). A7 identifies 18% of the patients who will fail to achieve 24 months of event-free survival (EFS24)25, marginally enriching for this clinically relevant pattern of early treatment failure when compared to non-A7 patients (p = 0.058). These patterns are replicated in R-CHOP-treated patients in the REMoDL-B, GOYA, and Reddy cohorts (Fig. 2A, B), with significant differences in EFS/OS between A7 and non-A7 patients (p = 0.0019, p = 0.012, p = 0.0029, respectively), and significant enrichment of EFS24 (but not OS24) failures (p = 0.004, p = 0.03, p = 0.10, respectively). No other cluster consistently showed a significantly different hazard rate when compared to all other clusters combined (Supplementary Fig. 3).

Kaplan Meier curves showing event-free (MER, REMoDL-B, GOYA) or overall (Reddy) survival among RCHOP-treated patients in the replication cohorts, with two-sided, unadjusted log-rank p-values shown. Survival is shown stratified (A) by all seven of the SubLymE clusters; (B) by a binarized model comparing A7 to non-A7, and (C) by a binarized model comparing A7 to non-A7 in the ABC subpopulation only. Source data are provided as a Source Data file.
Given the high prevalence of ABC patients within A7, we constrained the survival analysis to the ABC subtype and found the high-risk nature of A7 was not solely due to its enrichment of the ABC subtype, with the A7 vs. non-A7 hazard ratio (HR) among ABCs being in MER 1.71 (95% confidence interval: 0.98–3.00; p = 0.071), in REMoDL-B 1.50 (0.89–2.53; p = 0.13), in ROBUST 1.64 (1.08–2.51, p = 0.01), in GOYA 1.67 (0.91–3.05; p = 0.093), and in Reddy 1.61 (1.0–2.58; p = 0.048) (Fig. 2C). Even though the ABC-only subpopulation comparisons in MER, REMoDL-B, and GOYA are non-significant, the subpopulations are too small to reliably call effects of this magnitude significant (power < 50%), and the effect sizes are consistent with the appropriately powered and significant estimates from ROBUST and Reddy.
A7 status is not strongly associated with known clinical prognostic factors such as IPI, age, extranodal involvement, or disease stage. There is a trend of high-risk clinical factors being associated with the high-risk A7 class, and there is a nominal association of some features in certain cohorts (Supplementary Fig. 4). A Cox proportional hazards model showed that A7 status is significantly associated with EFS in the MER cohort when considered alone in a univariate model (HR = 2.00 (1.24–3.26), p = 0.006). Furthermore, multivariate Cox models showed that A7’s risk association was not accounted for by other risk factors, as A7 status remains a significant risk predictor even when controlling for other clinical or molecular risk factors such as age, COO, and IPI (Table 2).
Comparison among lymphoma subtypes identifies cluster-specific biological themes
The biology of the SubLymE clusters can be explored in contrast to one another to identify points of differentiation. Figure 3A shows a heatmap of GSVA signature scores sorted by cluster in ROBUST, arranged into six categories of lymphoma-related processes. For example, Clusters A1, A2, and A7 show strong downregulation of tumor microenvironment signatures, in contrast to cluster A6 which shows strong upregulation of these signatures. Clusters A2, A5, and A7 also show upregulation of malignant processes and metabolism signatures including MYC signaling pathways, while clusters A3 and A6 showed downregulation of these signatures. Differential B-cell transcription factors were related to the COO makeup of the clusters, with A2 showing upregulation of GCB-related signatures (IRF4Dn-1) and A7 showing strong upregulation of ABC-related signatures (IRF4Up-7, OCT2Up-1). Overall, cluster A7 is unique in its combination of high ABC signals, upregulated metabolic and malignant processes, upregulation of B-cell differentiation pathways, and low TME expression. Clusters similar to A7 include A2, which differentiates from A7 primarily along the COO axis, as well as A5, which shares many tumor-intrinsic features with A7 but has a more active immune microenvironment. Expression patterns of these pathways are replicated in the other cohorts, with similarities in the directionality and significance of cluster-associated pathways (Supplementary Figs. 5, 6).

A Heatmap of biologically relevant pathways across SubLymE classes in the ROBUST dataset. Heatmap colors represent signature-level normalized enrichment scores. B Association of class calls by SubLymE and other classification methods. The heatmap shows Cohen’s Kappa values measuring strength of association between specific pairs of classes, tested by binarizing each classifier with respect to the classes being compared. Positive values indicate classes that are commonly called together, while negative values indicate classes that are rarely called together. Kappa values not significantly different from 0 (p < 0.05) were set to 0 to highlight statistically significant associations. Associations are shown for MER, with the exception of the MHG comparisons, which were tested in REMoDL-B. Source data are provided as a Source Data file.
To characterize the SubLymE clusters in the context of recently described DLBCL subtyping methods, we applied eleven other molecular classification approaches to the MER dataset, which was chosen for its availability of transcriptomic, genomic and clinical data. These methods included B-cell states and Lymphoma Ecotypes (LEs)16, Lymphoma Microenvironment (LME)15, LymphGen9, HMRN10, Dark Zone Signature (DZsig)11, TME2614, Reddy COO20, Molecular High Grade (MHG, applied to REMoDL-B instead of MER)13, LymphProg26, and HRsig-Mayo classifiers27,28. The association between SubLymE and the other subtypes showed varying effect sizes in the range of moderate-to-large (0.22 < Cramer’s V < 0.55, Supplementary Fig. 7), consistent with the levels of association between other pairs of methods. Although none of the classifiers tested were statistically independent of all others, moderate association metrics suggest that while certain methods may leverage common biological themes, the patient-level classifications between methods are heterogeneous.
A pairwise comparison of specific class labels provides insight into similarities between patient groups identified under different methods (Fig. 3B and Supplementary Fig. 7). Cluster A7 was associated with groups from several different classification methods, including the S5 high-risk B-cell state, the Unclassified Ecotype, the High-Risk HRsig-Mayo class, the MYD88 HMRN group, the Depleted LME, ABC Reddy and MHG COO, LymphProg-High, and TME26 negativity. In contrast, cluster A6 showed association with multiple clusters hallmarked by their relative abundance of immune activity, including the S2 B-cell State, Ecotypes LE4 and LE7, the Low Risk HRSig-Mayo group, the Infiltrated LME, LymphProg-Low, and TME26 positivity.
Cluster A2 was strongly associated with clusters hallmarked by GCB biology, including the S1 B-cell state, DZSig-Pos, the LE8 Ecotype, the EZB LymphGen cluster, and GCB Reddy COO, and both GCB and MHG under MHG-COO. The cluster was not solely defined by its GCB biology but also its lack of immune infiltration, as observed by its associations with both the Depleted LME class and the TME26 negative class. Cluster A3 was another cluster showing significant overlap with GCB-related classes like GCB COO, LymphGen EZB, and LME-GC. In contrast to the low-TME A2 GCB cluster, however, A3 exhibited overlap with TME26 positivity and Treg-enriched LE5. Sankey plots illustrating cluster co-incidence may be found in Supplementary Figs. 8–11. Although some SubLymE classes significantly enrich for particular subtypes called by other classifiers, there exists no clear one-to-one correspondence between SubLymE classes and other methods.
A7 DLBCL tumors are associated with copy number alterations
Focusing on A7 as the high-risk cluster, we assessed genomic features associated with A7 and discovered no significantly enriched non-synonymous somatic mutations after multiple hypothesis corrections in comparison to other clusters using the ROBUST data. However, several mutations associated with ABC-DLBCL and particularly MCD genetic subtype such as ETV6 and PIM1 were nominally enriched in A7, reflecting the mostly ABC-DLBCL nature of A7 tumor biology (Supplementary Fig. 12A). Certain links may be drawn between nominally enriched mutations in SubLymE and cluster-defining mutations from other classification methodologies. A2 exhibits enrichment of EZH2 mutations, a feature of the LymphGen EZB cluster; A5 is enriched for MYD88, similar to the MYD88 HMRN cluster.
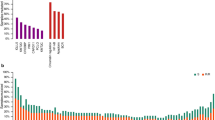
On the other hand, copy number alteration analysis demonstrated a number of significantly enriched CNAs in A7 (FDR < 0.05), suggesting copy number changes involving multiple genes may be genetic drivers of A7 biology rather than point mutations. Approximately 60% of A7 DLBCL patients exhibit arm-level copy number gains on chromosomes 3p, 3q, and 18q, and 44% exhibit focal deletions on chromosome 9p (Fig. 4, Supplementary Fig. 12B). Several important genes related to DLBCL etiology have been identified at these loci, including FOXP1 (3p), BCL6 (3q), CDKN2A and MTAP (9p21), BCL2, MALT1, and TCF4 (18q).

Prevalence of copy number gains and losses in A7 and non-A7 subpopulations (ROBUST), with FDR-corrected significant differences highlighted with asterisks. Source data are provided as a Source Data file.
A7 tumors exhibit TCF4-linked upregulation of MYC and MYC signatures
Differential expression and pathways analysis of A7 vs. non-A7 cases showed that A7 exhibited elevated expression of pathways defined by MYC targets, E2F targets, and G2M checkpoint signatures (Fig. 5A), as well as upregulation of MYC itself (Fig. 5B), indicative of a highly proliferative malignant DLBCL clone. As MYC target pathways were upregulated in A7, we investigated MYC gene and protein expression through gene expression and IHC analyses, respectively. Pathways dysregulated in other clusters may be found in Supplementary Fig. 13.

A Results of GSEA Hallmark pathway analysis presented as positive or negative association with A7 cluster membership. B c-myc gene expression by A7 status across clinical cohorts. In each cohort, MYC is expressesd at significantly higher levels in A7 compared to non-A7 (two-sided, unadjusted Wilcoxon p-values: ROBUST, <2.2e16; MER, 2.9e-7; REMoDL-B, 2.2e-9; GOYA, 5.5e-8; Reddy, 1.8e−9). C Representative c-myc IHC staining in multiple A7 cases and non-A7 cases; samples were selected for similar tumor cellularity. D Gene expression of TCF4 in patients plotted with copy number alteration status of TCF4 (ROBUST, N = 299), value in parentheses, copy number of TCF4;. Box plots represent the median and upper/lower quartiles, with whiskers extending to the most extreme values. **** indicates p < 0.0001 (TCF4 Diploid vs. Gain, p = 1.8e−5; Diploid vs. Amplification, p = 4.4e−14), N.S., not significant (two-sided, unadjusted Wilcoxon test). E TCF4 mRNA expression in DLBCL cell lines, value in parentheses is the copy number of TCF4 in each model. F Western blot analyzing expression of TCF4 in ABC-DLBCL cell lines. Representative blots from 3 independent experiments. G, H Western blot analysis and quantitation of expression of MYC and TCF4 in TCF4 knockdown ABC-DLBCL cell lines. GAPDH, loading control. Represetative blots from 2 independent experiments. I Cell proliferation assay of control (shNT) and TCF4 knockdown (shTCF4) in ABC-DLBCL cell lines. Each point represents the mean of technical triplicates with standard error of the mean too small to visualize. Source data are provided as a Source Data file.
Although A7 patients have somewhat elevated tumor purity (median 90% in A7, median 80% in non-A7, p = 2.7e−10, ROBUST), MYC expression is not correlated with tumor cellularity, indicating that high MYC expression in A7 is a true biological feature of A7 tumors not simply driven by high tumor cellularity (Supplementary Fig. 14A). Myc protein expression was elevated in A7 tumors compared to non-A7 tumors as indicated by a higher percentage of c-myc positive tumors as defined by a cutoff of ≥40% positive cells in the discovery cohort and REMoDL-B, (Fig. 5C; Supplementary Fig. 14B). Known genetic drivers of increased MYC expression and signaling such as MYC translocations, amplifications and mutations are not enriched in A7 DLBCL tumors, implicating other mechanisms of MYC upregulation.
As shown in Fig. 4, chromosome 18q is amplified at an arm level in A7 DLBCL tumors. TCF4, a basic helix-loop-helix (bHLH) transcription factor residing at 18q21.2 was previously reported to drive MYC gene expression in ABC-DLBCL through binding to MYC enhancer29. Interrogation of DLBCL patient tumor gene expression and DNA copy number alterations demonstrated the association between TCF4 copy number and elevated expression of both TCF4 and MYC in ROBUST (Fig. 5D). We identified two cell line models through genomic and COO profiling (RIVA and U2932), which showed them to be ABC cell lines that uniquely exhibited both of the largest and most significant A7-specific genomic features, arm-level gains in chr3 and chr18 (Supplementary Fig. 15). Examination of these cell lines demonstrated a strong correlation between TCF4 gene expression and TCF4 copy number leading to elevated TCF4 protein levels compared to non-A7-like cell lines (Fig. 5E, F).
To characterize TCF4 function in an A7-like DLBCL setting, we performed a TCF4 knockdown in DLBCL cell line models (Fig. 5G, H, Supplementary Fig. 16A, B) which led to a reduction of c-myc protein expression in the TCF4 amplified cell lines (RIVA and U2932), but not in those without TCF4 amplification (SU-DHL-2 and TMD8), suggesting TCF4 amplification contributes to MYC over-expression in 18q gain DLBCL models. Additionally, qPCR analysis further confirmed the reduction of myc expression led to decreased myc transcriptional activity of known myc target genes (Supplementary Fig. 16C) In line with these observations, knockdown of TCF4 strongly and significantly inhibited cell proliferation and increased apoptosis in TCF4-amplified cell lines, whereas induction of the same shRNAs had only about half the effect size in cell lines without TCF4 amplification (Fig. 5I, Supplementary Fig. 16D, E). Further studies utilizing shRNA targeting MYC confirmed the broad role myc has in regulating proliferative capacity of all four DLBCL cell lines, independent of TCF4 amplification status (Supplementary Fig. 16F, G). Taken together, these data suggest TCF4 as an important regulator of myc-related biology within the A7 DLBCL population.
A7 DLBCL tumors exhibit decreased MHC expression and a less immune infiltrated tumor microenvironment
Utilizing computational immune deconvolution algorithms (see Methods, Cell Type Signatures) to interrogate bulk RNAseq data generated from DLBCL tumor biopsies, A7 tumors were revealed to have an enrichment of B-cell populations and a diminished infiltration of multiple T-cell populations, dendritic cells, and macrophages (Fig. 6A). Utilizing COO and TME26 as continuous variables, it becomes apparent that A7 exhibits a combination of highest ABC scores and lowest TME26 scores, in contrast to the other clusters (Fig. 6B). However, neither COO and TME26 alone or combined are sufficient to uniquely identify A7. Using the COO or TME26 scores as univariate predictors of A7 membership yields prediction AUCs between 0.82 and 0.86 in both ROBUST and MER datasets, with optimized classifiers achieving roughly 80% sensitivity. However, the specificity in classifying A7 with these parameters is only 70% as over half of ABC-DLBCL patients with a low tumor microenvironment infiltration are non-A7.

A Deconvolution plot of inferred immune cell populations as enriched or depleted in A7 relative to non-A7 patients. B Scatter plot of the Discovery cohort in the space of Reddy COO score vs. TME26 score. C Single channel MIBI images for indicated markers in representative A7 (red) and non-A7 DLBCL cases (gray). D Multiplexed MIBI images for indicated markers in representative A7 (red) and non-A7 DLBCL cases (gray). Source data are provided as a Source Data file.
To confirm the computational inferences of low immune infiltration in A7 DLBCL tumors, we generated an antibody panel appropriate for multiplexed ion beam imaging (MIBI) which could interrogate tumor and immune features of the DLBCL tumor microenvironment. Antibodies were validated on four different tissue types (tonsil, thymus, liver and placenta). As shown in Supplementary Figs. 17–19, staining of tonsil shows expected histological distribution of cells and immune markers such as T cells with membranous staining of CD3 in the intrafollicular area and B cells expressing CD20 and MHC class II in the germinal center. Additionally, transcription factors such as Ki67 and FoxP3 were located in the nucleus and demonstrated known colocalization patterns in appropriate cell types (e.g., FoxP3 staining primarily in CD4+ T cells) (Supplementary Figs. 17–19). Initial staining of DLBCL tumor samples demonstrated significant interpatient heterogeneity in staining patterns with A7 DLBCL tumors exhibiting high levels of CD20 cells with low overall antigen presentation marker expression and a lack of T-cell infiltration (Fig. 6C, Supplementary Figs. 20, 21). Overlaying multiple channels to visualize markers of tumor and immune populations demonstrated that A7 tumors were indeed enriched for proliferative (Ki67 + ) CD20 + DLBCL cells and absent of CD3 + T-cell infiltration compared to non-A7 tumors (Fig. 6D, top panel, Supplementary Figs. 17–19). Reports in the literature30 have linked T-regulatory cells to a highly proliferative tumor through secretion of inhibitory cytokines such a TGF-β and expression of high avidity receptors for essential cytokines such as IL-2 leading to inhibition of T-cell activation and lack of recruitment to the TME, however this was not differentially observed between tumors from A7 and non-A7 patients in our cohort (Fig. 6D, middle panel). Interestingly, we were able to confirm that A7 DLBCL tumor cells do not exhibit prominent surface expression of MHC class I or II molecules, in line with reported data for solid and hematologic tumors with dysregulated c-myc expression31 (Fig. 6D, bottom panel, Supplementary Figs. 17–19). We also hypothesize that chromosome 9p21 loss, which is known to be associated with cold tumors including B-cell neoplasms32, may be one of the genetic drivers for low TME in A7.
Lenalidomide treatment increased T-cell infiltration in vivo and overcomes poor prognosis of A7 when combined with R-CHOP in DLBCL patients
Lenalidomide is a known T cell activator33,34,35 and based on this rationale, was hypothesized to overcome the low T cell phenotype of A7 tumors and perhaps overcome its poor prognostic behavior. In addition, another Aiolos/Ikaros degrader avadomide has been shown to enhance T cell trafficking into DLBCL tumor tissue in a clinical trial36. Given this finding, we sought to demonstrate a mechanism by which lenalidomide may overcome high-risk aspects of A7 biology.
To this end, we performed ChIP-seq for Aiolos and Ikaros in DLBCL cell lines. We found binding of these two transcriptional repressors to the promoter region of multiple members of MHC class I/II such as HLA-A, HLA-C,and CD58, respectively as well as other costimulatory genes including β2M and CD86 (Fig. 7A)37. Subsequent flow cytometry experiments demonstrated that degradation of Aiolos and Ikaros by lenalidomide treatment in HLA-A2 positive DLBCL cell line RL resulted in upregulation of both MHC class I and II expression on the cell surface (Fig. 7B). This lenalidomide mediated upregulation of MHC expression resulted in increased CD8+ T-cell specific cytotoxicity as measured by decreased target cell viability and increased secretion of granzyme B detected in the supernatant of a co-culture model of DLBCL cells loaded with an HLA-A2 specific EBV peptide (Fig. 7C). Degradation of Aiolos and Ikaros within T cells is known to induce immunomodulatory activities such as activation, increased proliferation, cytokine expression and phenotypic shifts towards more effector populations33,36,38. To evaluate the effect of lenalidomide on immune-mediated activities in vivo we performed a cross between a humanized CRBN mouse, where exons 2-12 of the human CRBN sequence were inserted into the genomic locus of the mouse CRBN and the insertion of a stop codon to prevent translation of the endogenous mouse gene, with an eµ-myc DLBCL mouse. Diseased spleens from the resulting progeny were harvested and utilized in an adoptive transfer into recipient hCRBN mice to generate a cohort of homogenous animals where drug-mediated effects could be measured. As shown in Supplementary Fig. 22A, transplant of diseased splenocytes results in effacement of the normal germinal center architecture and the presence of large B cells (red arrow) in a diffuse pattern within the spleen compared to non-transplanted mice. Four days post-transplantation of hCRBN/eµ-myc DLBCL cells, mice were randomized to either vehicle control or lenalidomide treatment groups (n = 8 mice/cohort). Mice were then treated once daily for four more days prior to euthanasia and collection of the spleen. Multiplex immunofluorescence for markers to quantify the number of tumor and immune cells within the spleen was performed, leading to the observation that lenalidomide treatment significantly increased the infiltration of CD8+ T cells but not CD4+ T cells within the spleen compared to vehicle controls while also decreasing B cells including proliferating B cells (Fig. 5D, E; Supplementary Fig. 22B). Additionally, we observed increased HLA-DR expression on malignant DLBCL cells within the spleen of mice treated with lenalidomide and this correlated with increased DLBCL cells staining positive for cleaved caspase 3, a marker of apoptosis (Supplementary Fig. 22C). Overall, lenalidomide can improve immune visibility of DLBCL cells through increased MHC I/II expression and increase trafficking of CD8+ T cells to the DLBCL tumor.

A ChIP-seq peaks demonstrating direct binding of Aiolos and Ikaros at representative promoters of MHC class I and II genes in U2932 DLBCL cells. B Surface mean fluorescent intensity of MHC class I (black bars) and MHC class II (gray bars) in DLBCL cells treated with vehicle or lenalidomide for 24 h as measured by flow cytometry. Error bars represent standard error of the mean. C Vehicle and lenalidomide treated target DLBCL cell viability following co-culture with EBV reactive CD8+ T cells overnight. % live target cells are marked by inverted triangles. ELISA measurements for granzyme B in resulting co-culture supernatants are marked by round dots. Error bars represent standard error of the mean. D Cellular composition for indicated population in hCRBN/eu-myc transplanted mice treated with vehicle or lenalidomide (30 mg/kg) for four days). Scale bars = 300 µm. N = 5. E Intratumoral cell counts in transplanted mice and transplanted mice treated with vehicle or lenalidomide by multiplex immunofluorescence for indicated cell population (**p < 0.01, ****p < 0.0001; B cells No transplant vs. Vehicle p = 5.7e−10, Vehicle vs. Len p = 1.4e−7; CD8+ T cells No transplant vs Vehicle, p = 1.8e−12, Vehicle vs. Len p = 4.4e−6). F Kaplan Meier EFS curves for A7 and non-A7 ABC-DLBCL populations treated with either R-CHOP or lenalidomide plus R-CHOP (R2-CHOP) in the ROBUST trial, with accompanying table of group-pairwise log-rank p-values. Source data are provided as a Source Data file.
We next examined the effect of lenalidomide in combination with R-CHOP (R2-CHOP) compared to R-CHOP in the ROBUST trial, and found an interaction between A7 status, treatment arm, and outcome. While there was no significant different in event-free survival rates between treatment arms in the overall ITT population5, a retrospective analysis restricted to A7 patients showed that R2-CHOP is significantly associated with a superior EFS compared to R-CHOP (Fig. 7F; A7 R2-CHOP vs. A7 R-CHOP HR = 0.50 (0.30-0.83)). Interestingly, the clinical performance of R2-CHOP in non-A7 ABC-DLBCL patients is not significantly different from R-CHOP (HR = 1.05; (0.69–1.59).
Discussion
While many patients with newly diagnosed DLBCL are cured, about 40% have progressive disease and are in need of novel therapies. Given the clonal heterogeneity of DLBCL and the involvement of both tumor intrinsic and extrinsic factors, prospectively identifying these patients and developing targeted agents remains a challenge in the field. Newer classification systems based on genetics and gene expression have identified subtypes with poor clinical outcome but have proven challenging to implement in the clinic. Here we apply an unsupervised clustering approach called iClusterPlus, in order to identify patient clusters among transcriptomic features representing both the tumor and the microenvironment. We identify a high-risk patient population called A7 in ndDLBCL whose poor outcome is not explained by clinical prognostic indicators such as IPI, and therefore provides a biologically meaningful way to identify high-risk patients that clinical factors do not.
The high-risk A7 patient population is constituted of approximately 80% ABC-DLBCL and accounts for approximately 40% of all ABC patients. One hallmark of A7 is elevated expression of MYC and its downstream pathways, however this is likely driven by upstream regulators of MYC as the MYC gene is not amplified or the recipient of an Ig translocation in A7 patients. Elevated levels of c-myc expression and downstream affected pathways are associated with a reorganization of the tumor microenvironment leading to lack of infiltration of immune cells such as T and NK-cells39. Others have shown that elevated MYC mRNA and protein expression is associated with poor survival in ABC patients where MYC translocations are rare40; and MYC over-expression is not associated with translocation events or copy number gain41. While somatic mutations are not significantly associated with A7, the group is enriched in CNAs including amplifications on chromosomes 3 and 18. Copy number gains on chromosome 3 could contribute to immune escape (FOXP1, 3p14), NF-κB pathway activation (NFKBIZ, 3q12) and B cell differentiation arrest (BCL6, 3q27)42. This finding is not surprising as the discovery of A7 was based on gene expression which may be more a function of copy number alterations than single nucleotide variants.
Limitations of our analysis include the bulk nature of the RNAseq data, which hampers analysis of cell-type specific signatures found only in tumor or immune cells, for example. Further analysis may be conduced in single cell RNAseq data to better understand specificity of our signatures with regard to certain cell types, as well as the cellular composition of different SubLymE clusters.
Comparisons to other subtyping methodologies showed that our classifier shares points of commonality and differentiation with other methods, respecting the combination of both tumor-intrinsic and microenvironment features of DLBCL. A7 showed strong association with ABC-associated classes including ABC COO, the HMRN MYD88 group, and DZsig-Neg. In addition, A7 is characterized by an immune-low microenvironment and has strong associations with LME-DE and TME26-negative signatures. A7 was associated with the Unclassified Ecotype and showed no enrichment in any LymphGen cluster. From an outcome perspective, A7 enriches for EFS24 failure and is associated with the HRSig-Mayo High-risk/ARID1A signature, as well as the LymphProg-High risk class. Finally, A7 is associated with B-cell state S5 which describes ABC/pre-plasmablast biology.
Pairwise associations between classification methods showed that no method was statistically independent from all other methods, and a hierarchical clustering of the association metrics (Supplementary Fig. 7) showed that the methodologies associate in three main groups. These include a group of genomic/tumor-intrinsic classifiers (LymphGen, HMRN, DZsig), a group dominated by microenvironment-based classifiers (TME26, Ecotyper, LME, HRSig-Mayo), and a group dependent on relationships between tumor-intrinsic and microenvironment signal (SubLymE, B-cell State, COO by Reddy and LymphProg). Although Reddy does classify tumor-intrinsic COO signal, it also bears a nonlinear relationship to microenvironment infiltration (Fig. 6B).
Survival analysis of the different clustering methods (Supplementary Figs. 8–11) showed that four of the tested classifiers resulted in significant stratification of EFS in MER (or REMoDL-B, for MHG), which were the SubLymE classifier, the HRsig-Mayo classifier, DZsig, MHG, and LymphProg. The SubLymE classification method provides a complementary approach to other methods, identifying unique high-risk patient subsets not identifiable by other methods.
TCF4 is a transcription factor known to regulate MYC expression29 and our in vitro data demonstrated this linkage supporting TCF4 as one putative therapeutic target for A7 patients. It will be important in future work to characterize TCF4 protein expression in patient biopsies as a potential biomarker and therapeutic target. Additional pathway changes enriched in A7 include upregulation of G2M checkpoint and mitotic spindle checkpoint indicative of cell cycle dysregulation. Upregulation of DNA repair pathways and down-regulation of the p53 pathway were also observed but in the absence of enrichment of TP53 mutations. Another feature of A7 is upregulation of the oxidative phosphorylation pathway which was previously reported in in a subset of DLBCL43,44.
An important hallmark of A7 is the low abundance of immune infiltrating cells including T cells and macrophages as well as low MHC I and II expression on the malignant B cells. Interestingly, we show retrospectively in a large Phase 3 trial that lenalidomide, a cereblon regulator and IKZF1/3 degrader, in combination with R-CHOP can overcome the poor prognosis of R-CHOP alone in A7 patients. These findings are supported by our prior study that identified a cohort of DLBCL with early clinical failure that were enriched for ABC, had enhanced activation of inflammatory pathways, and were sensitive to R2CHOP45. Upon binding of lenalidomide to CRBN, CRBN converts to a closed active form and binds substrates IKZF1 and 3 (Ikaros and Aiolos) thereby recruiting them for ubiquitination by the CRL4-CRBN complex and subsequent degradation via the proteosome33,35,46,47. Ikaros and Aiolos directly repress the transcription of multiple families of genes including interferon stimulated genes whose function results in decreased proliferation of DLBCL cells48. Mechanistically, lenalidomide has been shown to induce a proliferative block and increased apoptosis in ABC-DLBCL cells48,49,50. Additional data presented herein demonstrate that lenalidomide may also stimulate immune cell trafficking into the DLBCL tumor as well as increased MHC expression leading to immune recognition of tumor cells by CD8+ T cells. Although these data do not support use of lenalidomide as part of front-line therapy for DLBCL, they provide a mechanistic rationale for the use of more active IKZF1/3 degraders such as CC-99282/golcadomide in both standard risk and high-risk A7 ndDLBCL patients, which requires testing in the clinic51. It also opens the potential for epigenetic agents that may enhance MHC expression. Application of SubLymE to the bortezomib arm of REMoDL-B showed that A7’s poor prognosis was mitigated somewhat (p = 0.16), although by an unknown mechanism of action and to a lesser extent than lenalidomide in ROBUST. Newer therapies such as Polatuzumab in combination with R-CHP and T cell engagers are emerging52, although mechanisms of resistance such as lower baseline CD8 T-cell levels and Myc overexpression53 may pose challenges. The clinical benefit of these promising agents as well as others like BTK inhibitors in the A7 population remains an open question, but application of SubLymE to these patient cohorts would provide insights. Future prospective application of SubLymE to cohorts treated with drugs of different mechanisms of action beyond immunochemotherapy may provide further insight into the interplay between the biology defining the SubLymE classes and the biological drivers of disease progression, as well as establishing A7 as a distinct subgroup amenable for clinical intervention.In summary, this work identified a biologically homogeneous high-risk DLBCL patient population that needs prospective validation but holds promise for future targeted drug development in DLBCL.
Methods
Ethics Statement
Our research methodology complies with all relevant ethical regulations, including FDA approval for clinical trial protocols and patient consent, and Mayo Clinic IRB approval for the MER observational study. Animal studies were approved by the internal BMS Institutional Animal Care and Use Committee. Tumor size/burden was not used as an inclusion/exclusion criterion for the cohorts analyzed in this work.
Datasets
The Discovery cohort of 1208 patients was a combination of the ROBUST clinical trial screening population (NCT02285062, n = 1016)5 which included all DLBCLs irrespective of treatment or COO, and a set of commercially sourced newly diagnosed patient samples (n = 192). The replication cohorts included MER (Molecular Epidemiology Resource), an observational epidemiology cohort study of prospectively enrolled newly diagnosed DLBCL patients at Mayo Clinic (Rochester, MN) and the University of Iowa (Iowa City, IA) (n = 343)22 as part of Lymphoma Genome Project (LGP). Patient samples in these studies were collected with informed consent for research use and were approved by the Institutional Review Board at Mayo Clinic (for MER) and each study site’s Institutional Review Board (for ROBUST) in accordance with the Declaration of Helsinki. Other replication cohorts were obtained from public sources include REMoDL-B; the REMoDL-B clinical trial cohort (n = 928, n = 469 on R-CHOP arm;23 the GOYA clinical trial (n = 271, R-CHOP); and the Reddy cohort (n = 442, R-CHOP). All cohorts were unblinded with respect to treatment during analysis in order to assess differential effects by treatment arm.
SubLymE was trained entirely on DLBCL NOS patients without HGBL. Samples from the MER cohort generally had high tumor cellularity by pathology review, with 83% of samples having >50% purity, and 57% having >70% purity. Unless otherwise specified, analysis of clinical outcome was conducted only in R-CHOP-treated patients, or in the MER dataset, R-CHOP-like-treated patients, which included a minority of patients treated with MR-CHOP (9% of the RCHOP-like treated cohort), R-EPOCH (7%), Other immunochemotherapy (3%), ER-CHOP (1%), RAD-RCHOP (1%), and RCHOP/Zevalin (1%). For ROBUST, clinical outcome was available only for ABC patients. Demographics and clinical characteristics of the discovery and replication cohorts are summarized in Table 1.
RNAseq and DNA sequencing
ROBUST, MER, and the Commercial samples were sequenced at Expression Analysis, Inc (Durham, HC, USA) according to standard protocols. The Allprep DNA/RNA FFPE kit was used to simultaneously purify genomic DNA and total RNA from formalin-fixed, paraffin embedded (FFPE) tissue sections. RNAseq libraries (75PE, 50M) were constructed using Illumina TruSeq RNA Access method.
Whole exome sequencing (WES) libraries (200x for tumor, 100x for germline control) were created for MER and Commercial cohorts using the Agilent SureSelect Human All Exon v6. Whole genome (WGS) libraries (60X for tumor, 30X for germline) were prepared for ROBUST samples using the Swift Accel-NGS 2S Plus DNA library kit (#21024 or 21096, Swift) with modifications to the Ampure Bead cleanup steps in the procedure.
Sequencing data QC and processing
RNAseq samples met minimum quality thresholds in terms of library size, duplication rate, and alignment. Cohort-level QC was performed by removing samples failing multiple outlier detection metrics. After an initial round of unsupervised clustering, a small, highly distinct cluster of borderline quality samples were removed for further analysis.
Sequencing data were processed through an internal cloud-based platform which runs the Sentieon implementation of the GATK best practices, using BWA-mem for alignment and the Sentieon implementation of Mutect2 (tnhaplotyper). Variants were annotated with SnpEff using the dbnsfp database. For WGS, data copy number aberrations were called using Battenberg54 and structural variants were called by Manta. For WES data copy number aberrations were called using Sclust55. Structural variants were not called for the WES data. RNA-seq data was aligned with STAR aligner and quantified with salmon.
RNAseq data normalization
The MER, ROBUST, GOYA, and Reddy cohorts were reference normalized to a subset of the Discovery data, referred to as the commercial samples, which was fixed as a reference population. To do so, a sample-wise scaling was applied to TPM RNAseq data using the mean of five DLBCL-specific housekeeping genes used in the Nanostring Lymph2Cx assay (ISY1, R3HDM1, TRIM56, UBXN4, and WDR55). After sample-level scaling, each gene was standardized to the reference population by subtracting the reference mean and dividing by the reference standard deviation. Ultimately, the reference fixes all genes to have a mean of 0 and a variance of 1, while all other datasets were transformed to be a gene-wise Z-scoring with respect to the reference population.
Because the REMoDL-B dataset was Illumina BeadArray and not RNAseq data, we applied a self-standardization approach that used the housekeeping scaling step as described above, followed by a gene-wise scaling that explicitly set each gene to have mean 0 and variance 1. This self-standardization approach is suitable for large, representative patient cohorts as applied herein, but could yield unexpected results for a small or non-representative cohort.
The reference normalization approach puts all of the data in a unified numerical space with comparable expression levels and allows for portable models that can be trained in one dataset and applied directly to any other cohort without the need for re-parameterization. It also allows for the normalization of even a single sample, with no requirement for a representative batch, and furthermore, normalized data is never affected by the introduction of new samples. Existing classifiers such as the Reddy COO classifier20 and TME26 classifier14 were adapted to the normalized gene expression space by re-weighting decision thresholds.
In practice, no significant batch effects by dataset were observed in the normalized combined datasets of all cohorts (Supplementary Fig. 23). We also validated that the normalization approach left relevant biological signals intact by comparing gene expression classifiers/signatures applied to the normalized data against orthogonal, non-RNAseq data. These included comparing the Reddy COO classification against the Hans IHC-based method, the double-hit signature classifier12 against FISH calls, and the cell type abundance GSVA scores against cell type marker density from IHC and MIBI. All features derived from the normalized RNAseq data were highly concordant with their corresponding non-RNAseq features.
Statistical Analysis
Differentially expressed significance was calculated by the t-test or Wilcoxon rank-sum test where appropriate. Multiple hypothesis correction was performed with the Benjamini-Hochburg false discovery rate method where noted. All statistical tests were two-tailed. Gene signature scores were calculated using the “ssgsea” method from the GSVA package. Correlation values are calculated as Spearman’s rho. General tests of categorical association are chi-squared (or Fisher where appropriate). The log-rank test is used to compute p-values for survival data. All analysis was performed in R version 4.2.1. Various statistical tests from the stats v3.5.3 R (54) CRAN package were used to check significance of the association of the subpopulations to different variables. Fisher’s exact test for binary data (mutations/CNVs), t-test for continuous variables (GE pathway scores), and global log-rank test for outcome (EFS/OS). Boxplot figures represent the lower, middle, and upper quartiles as the lower box edge, center line, and upper box edge respectively, with whiskers extending to up to 1.5 times the interquartile range. Error bars represent 95% confidence intervals. Statistical analysis was not stratified by (self-reported) sex, but post-hoc analysis did not find significant associations between our findings and sex.
Cell type signatures
Cell type specific signatures were generated from the LM22 matrix which describes 22 functionally defined leukocyte types56. This signature matrix was augmented and tailored to DLBCL by adding an additional cell type representing malignant DLBCL B cells, and was trained on purified cell populations57. Benchmarking deconvolution results using the augmented signature matrix identified high correlation between the abundance of CD20+ cells as measured by IHC. The addition of the DLBCL-specific cell type also significantly reduced the estimated abundance of the unclassifiable “Other” cell type population, which previously accounted for up to 40% of the estimated abundance.
DLBCL classifiers
The Reddy method20 was implemented to call COO from RNAseq data and was shown to have excellent concordance with the gold standard Nanostring method.
The DHIT/DZsig method11,12 was adapted to the normalized RNAseq space by using published gene-level DHITsig coefficients to compute a linear DHITsig score. A threshold for this score was selected in the MER data as the highest threshold that captured all cases known to be double-hit by FISH, which yielded a classifier matching the expected prevalence of DHITsig + cases. This signature was further extended to represent the DZsig classification by adding a third “indeterminate” group with approximately 10% prevalence between the positive and negative groups.
For Ecotyper and Bcell state, we applied the online tool at https://ecotyper.stanford.edu/lymphoma/.
For LME, we applied BostonGene’s package on Github (https://github.com/BostonGene/LME).
For the HMRN classification, we used a total of 115 genetic features which occurred in at least 1% of patients. These features defined binary variables denoting 104 mutations, 4 amplifications (BCL2, MALT1, REL, XPO1), and 7 markers (TP53, TNFRSF14, TNFAIP3, RB1, PRDM1, CDKN2A, CD58) indicating presence of either a homozygous deletion or mutation. We did not have data on HIST1H2BD_S mutation and DDX3X marker.
We applied the LymphGen method using the v1.0 tool available online at https://llmpp.nih.gov/lymphgen/index.php. We followed the documentation including copy number and mutation data where possible, but had a small number of missing input features. We omitted one CNV feature (LOC107986596), and two mutation features (TMEM121 and LOC107986596), out of 104 total features in each. We included synonymous, truncating, and MYD88 L265P annotations for the mutation data. We did not include arm-level copy number data.
To apply a version of the LymphProg classifier to our normalized data, we applied a weighted sum of classifier genes according to the coefficients provided in the literature, and applied the provided score threshold of −0.521, which yielded class prevalences in line with expectations.
The MHG classifier was examined in the REMoDL-B dataset, for which the MHG classifications were publicly available. Although differential signatures of MHG versus other COO subtypes were identified in the literature, we could not find an implementation of the classifier itself to apply to the MER data.
Unsupervised clustering
The clustering input data consisted of normalized RNAseq gene expression features plus feature scores derived from the gene expression data, agnostic to clinical and outcome data. Expression features were restricted to genes in the top quartile of mean expression and variance in TPM space. The derived features consisted of GSVA signature scores19 including the MSigDB Hallmark and C1 pathways, as well as cell type signatures57. While the derived features did share some information content with the raw gene expression features, most features were derived using raw features not selected for clustering. Some presence of redundant signal across the input data types is expected, and indeed relied upon by the latent variable model used.
To better understand the unique signal carried by each input matrix, we calculated the sample-pairwise correlation matrix in each of the four individual feature spaces. We then calculated the Correlation Matrix Distance (CMD) between the correlation matrix in raw gene expression space and the other three correlation matrices, in order to assess whether the correlation structure among patients was repeated across the data types, or if patients showed different patterns of similarity in different feature spaces. This was compared against a null distribution of CMD values when using an equivalent number of raw gene expression features, rather than the engineered features. We found in each case that the derived features showed significantly greater CMD than selected features, indicating that the derived features allow different views of patient similarity not easily captured in raw gene expression space. In short, the engineered signature features provide distinct views of the patients rather than simply representing redundant signal already captured in the gene expression features.
The iClusterPlus method18 was applied to the subset data for multiple choices of K from 2 to 12. This procedure was repeated 200 times, with cluster assignments recorded in each case. The 200 runs were then summarized using a sample-pairwise co-clustering frequency matrix, which was computed as the number of times two samples were assigned to the same cluster, divided by the number of times two samples appeared in the same run. This sample-pairwise matrix was then clustered using hierarchical clustering using the Ward method and 1 minus the co-cluster frequency as the distance metric, in order to obtain one final clustering per choice of K. One value of K was selected by evaluating cluster stability measures including the silhouette metric and co-cluster frequency distribution, with 8 being the being the best-fitting number of clusters (Supplementary Fig. 24).
SubLymE classifier
A generalized linear model (GLM) classifier was trained on the Discovery data using the consensus cluster labels (with A8 samples removed) as the gold standard. Several choices of the elastic net mixing parameter alpha were tested, with the goal of maximizing predictive performance and minimizing model complexity. The regularization parameter lambda was optimized using cross-fold validation and was selected as the minimum value that yielded a misclassification rate within one standard error of the minimum32. Cross-validation results indicated good performance of the classifier training methodology, with 93% accuracy on the training cohort, as well as 81-98% sensitivity/positive predictive value within each cluster individually. Since the training data was normalized to a reference population, the classifier is directly applicable to other datasets normalized to this space, with no need to re-train parameters or thresholds. The SubLymE classifier may be applied to any FFPE RNAseq sample normalized in the same way and will produce a class label for each case (i.e., no case will be unclassified). Although a class label will be produced for any input data, application to poor quality data will produce poor quality results. When applying SubLymE to the previously omitted A8 samples in the Discovery data, most were classified as A4, which was the least reproducible cluster in terms of pathway dysregulation and prevalence.
MHC I/II expression
Differential surface expression of MHC I and II molecules on DLBCL cell lines treated with DMSO or lenalidomide (1 μM) for 3 days was assessed via flow cytometry using anti-human HLA-ABC and anti-human HLA-DR/DP/DQ antibodies (clone G46-2.6 and clone Tu39, respectively, BD Biosciences). No commonly misidentified cell lines were used.
Antigen-specific CD8 T-cell cytotoxicity
DLBCL cell lines selected for the antigen-specific CD8 T cell cytotoxicity were obtained from American Type Culture Collection (ATCC), or Leibniz Institute-DSMZ (Braunschweig, Germany), and maintained in RPMI medium [RPMI-1640 with 10% fetal bovine serum (FBS), supplemented with 2 mmol/L L-glutamine, 1% penicillin/streptomycin and 1 mmol/L sodium pyruvate]. HLA-A2 surface expression was accessed via flow cytometry using antibodies (clone BB7.2, BD Biosciences). HLA-A2 positive, EBV-specific primary human CD8 T cells were obtained from Charles River (donor 213, CMV negative). HLA-A*02:01 EBV BMLF1 peptide was obtained from MBL International. To conduct the cytotoxicity assay, target DLBCL cell lines were labeled with 400 nM of CFSE (Thermo Fisher Scientific) and treated with either DMSO or lenalidomide (1 µM) for 3 days. On the day of co-culture, target cells were labeled with or without 1 ng/ml of EBV peptides for 30 min. EBV-specific effector CD8 T cells were thawed and counted. Co-culture of CD8 T cells with target cells was set up in 96 well round bottom plates at an effector:target ratio of 1:1 overnight. On the following day, supernatants from co-culture samples were collected for Granzyme B secretion via ELISA (Biolegend), while cells were stained with Annexin V and TO-PRO-3 (Thermo Fisher Scientific) and analyzed by flow cytometry. CFSE+ target cells were gated on, followed by gating on Annexin V-, TO-PRO-3- double negative cells as viable target cells. Absolute cell count was obtained using CountBright Absolute Counting Beads (Thermo Fisher Scientific).
Multiplex immunofluorescence in mouse tissue
Mouse spleen samples were fixed in formalin 4% for 72 h and embedded into paraffin blocks. Sequential 4-µm-thick sections were placed on Super Frost slides for standard Hematoxylin–Eosin, IHC and IF staining. Chromogen staining was performed for immunohistochemistry validation of specificity of individual antibodies. Antibodies against B220 (dilution 1:50, PTPRC/1783R, NBP-54578, Novus Biologics), CD8 (dilution 1:200, D4W2Z, 98941S, Cell Signaling Technology), CD4 (dilution 1:50, D7D2Z, 27520, Cell Signaling Technology), KI67 (dilution 1:100, SP6, ab231172, Abcam) were validated as a chromogen and monoplex immunofluorescent staining. IHC and mIF assays were performed on using a VENTANA DISCOVERY ULTRA automated staining instrument, using VENTANA reagents, according to the manufacturer’s instructions. Slides were de-paraffinized using EZ Prep solution (950–102, Roche) and epitope retrieval was accomplished with CC1 solution (950–224, Roche). Slides were incubated with rabbit 1°Abs, diluted in antibody diluent (760-219, Roche), followed by secondary anti Rabbit-HQ (OC 07017812001, Ventana) for chromogen staining or Rabbit-Polymer (non-diluted, ARR1001KT, Ventana) for immunofluorescent staining. For chromogen detection, slides were developed using the Ventana Discovery ChromoMap DAB kit (760-159, Ventana) according to the manufacturer’s instructions. Slides were then counterstained with hematoxylin (790-2208, Ventana), followed by Bluing reagent (760-2037, Ventana). For fluorescent staining and detection, slides were stained with Opal Polaris 7‐Color Automation IHC Kit (NEL871001KT, Akoya Biosciences). We used Opal 480 (1:100), Opal 520 (1:100), Opal 570 (1:100), Opal 620 (1:100), and Opal 690 (dilution 1:50) and Opal 780 (1:25). Fluorescent singleplex staining was performed for each biomarker and compared to the appropriate chromogenic singleplex to assess staining performance. After the staining was completed, the slide was mounted by using Prolong Diamond Antifade Reagent (P36970, Invitrogen). Multispectral images were acquired on the PhenoImager HT system (Vectra Polaris, Akoya Bioscience), analyzed with InForm software (Vectra Polaris, Akoya Bioscience), and the quality report and marker quantification were generated with the phenoptrReports package.
shRNA knockdown
Doxycycline (Dox)-inducible shRNA constructs were generated by Cellecta (Mountain View, CA, USA) using pRSITEP-U6Tet-(sh)-EF1-TetRep-2A-Puro plasmid. Briefly, 293FT cells were co-transfected with lentiviral packaging plasmid mix (Cellecta, Cat# CPCP-K2A) and pRSITEP-shRNA constructs. Viral particle was collected 48 and 72 hrs after transfection and then concentrated with Lenti-X Concentrator (Takara Bio USA). For infections, cells were incubated overnight with concentrated viral supernatants in the presence of 8 µg/ml polybrene. Cells were then washed to remove polybrene the next day. At 48 h post-infection, cells were selected with puromycin (2 µg/ml) for more 1 week before experiments. For knockdown experiments, cells were seeded at 1×105 cells/ml and induced with 20 ng/ml of Dox or DMSO vehicle control. On day 3 of Dox induction, cells were re-seeded in medium with refreshed Dox or DMSO. For proliferation assay, 15,000 cells were seeded in 96 well U-bottom plate followed by measuring cell viability with CellTiter-Glo (Promega) for 5 consecutive days. The remaining cells were seeded at 5×105 cells/ml and incubated for additional 2 days. Cells were then harvested for Western blot analysis. The shRNA target sequences were: shNT: CAACAAGATGAAGAGCACCAA; shTCF4-13: GAGACTGAACGGCAATCTTTC; shTCF4-14: CACGAAATCTTCGGAGGACAA. shMYC-40: CAGTTGAAACACAAACTTGAA; shMYC-42: CCTGAGACAGATCAGCAACAA
Western blotting
Cells were lysed with cell lysis buffer (50 mM TrisHCl pH7.4, 250 mM NaCl, 0.5% Triton X100, 10% glycerol) supplemented with Halt protease/phosphatase inhibitors (Thermo Fisher Scientific, 78443). Cell lysates were subjected to sonication to breakdown nuclei and reduce viscosity caused by released genomic DNA. The protein concentration was measured by a Bradford Protein Assay (Bio-Rad). Samples were diluted to equal concentration followed by with NuPAGE LDS sample buffer and 2-Mercaptoethanol (1.25% final concentration) before boiling at 95 °C for 5 min. Whole-cell lysates were resolved on NuPAG 4-12% Bis-Tris Midi Protein Gels (Invitrogen) and transferred onto nitrocellulose membranes, which were then subjected to blocking in Intercept® (TBS) blocking buffer (LI-COR). Proteins of interest were detected by incubation with the primary antibodies listed below at 4 °C overnight. After washing with 1XTBST, the membrane was incubated with either IRDye 800CW goat anti-rabbit IgG or IRDye 680LT goat anti-mouse IgG secondary antibody (1:10,000) at RT for 1 h. After washing with 1XTBST, bends were visualized by Odyssey Imaging System (LI-COR). Antibody information: TCF4 (Proteintech, 22337-1-AP, 1:1,000), MYC (abcam, ab32072, 1:1,000), GAPDH (Cell Signaling Technology, 2118 L, 1:5,000).
Capillary Western blot analyses
Capillary Western analyses (Supplementary Fig. 16A, B, and F) were performed using the ProteinSimple Jess System (Catalog # 004-650). Protein samples (1 µg/well) were loaded 12-230 kDa separation capillary cartridges (ProteinSimple, SM-W004) and processed according to the manufacture instruction. The antibodies used were identical to those used in the traditional Western blot except for dilution factors. The dilution factors: TCF4 (1: 40), MYC (1:40), and GPADH (1:1,000). Results were analyzed by Compass for SW software (Version 6.2.0).
Multiplexed ion beam imaging (MIBI)
MIBI is performed by staining tissue with a panel of metal-labeled antibodies and then imaging the tissue using time-of-flight secondary ion mass spectrometry (ToF-SIMS). Tumor biopsy slides were stained using standard protocol at Ionpath Inc. (Menlo Park, CA). Expression of markers were quantified at the single cell level using scaled (arcsinh transformed) summed intensities. Thresholds for each marker were initially determined based on the histogram distribution of intensity values across all field of views (FOVs). Samples used in MIBI analysis were taken from the Commercial cohort.
Immunohistochemistry and pathology scoring
Immunohistochemistry (IHC) assay was performed using Bond Polymer Refine Detection Kit on Leica Bond slide autostainer (Leica Microsystems Inc., Buffalo Grove, IL). Briefly, formalin-fixed paraffin-embedded (FFPE) tissues were sectioned at 4 micron and deparaffinized on the Bond autostainer. Antigen retrieval was performed with Epitope Retrieval 2 (ER2, pH 9.0) for 20 min at 100 °C. The slides were blocked for endogenous peroxidase activity with Peroxide Block for 5 min at room temperature. Sections were then incubated with the rabbit monoclonal anti-MYC antibody (Abcam, Catalog No. ab32072) at a 1/200 and 1/1000 dilution, respectively, for 15 min at room temperature. Horseradish peroxidase (HRP) labeled Polymer was used at the instrument’s default condition. The antigen–antibody complex was then visualized with hydrogen peroxide substrate and diaminobenzidine tetrahydrochloride (DAB) chromogen. Slides were counterstained with hematoxylin. MYC immunoreactivity was evaluated on entire tumor sections by a pathologist.
FISH and copy number analysis
For the MER cohort, translocation events in MYC (8q24.1), BCL2 (18q21.2), and BCL6 (3q27) were determined by FISH break apart probes (Abott Laboratories, Des Plaines, IL, USA) on tissue microarray slides. Imaging analysis was completed by a technologist in the Mayo Clinic department of Laboratory Medicine and Pathology. Detailed methods for FISH analysis are previously published58.
Adoptive transfer of eu-myc/hCRBN splenocytes and treatment of recipient mice
Humanized CRBN (hCRBN) C57BL/6 mice were bred with eu-myc C57BL/6 mice. Upon development of lymphoma, spleens were harvested and resulting splenocytes were viably frozen. To transplant diseased splenocytes to hCRBN mice, splenocytes were thawed, washed, and viability determined. Viable splenocytes cells were resuspended in RPMI media (GIBCO) and injected 1×105 lymphoma cells by tail vein into recipient hCRBN mice. Lymphoma cells were allowed to engraft for four days, followed by randomization between vehicle and lenalidomide (30 mg/kg) treatment once daily for via PO for four additional days. Spleens were harvested and subjected to multiplex immunofluorescence with indicated antibodies. Animal research followed the ARRIVE guidelines, using 8-10 week old female mice in groups of 10 We followed AAALAC and NIH guidelines for mouse health and body condition assessment.
RNA extraction, reverse transcription, and real-time PCR
Total RNA was extracted by RNeasy Mini Kit (Qiagen) and reverse transcribed by iScript™ cDNA Synthesis Kit (Bio-rad). Quantitative real-time PCR (qPCR) was conducted on a VillA 7 System using the Power SYBR Green PCR Master Mix (Applied Biosystems). Gene expression values were calculated by normalization to 18S using the comparative CT method. Primers used in the study are listed in Supplementary Table 14,25,27,28,44,51,52.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are provided as a Source Data file, found in the manuscript supplement. The sequencing data for the ROBUST clinical trial have been deposited in the EGA database under accession code EGAD50000000482. The data are available under restricted access, and can be applied for through the BMS FastTrack portal (https://fasttrack.bms.com/, hd-sci-apps@bms.com), which has an expected review time of 2-3 weeks. The ChIPseq data generated in this work are available on GEO under GSE267437. Additional data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
Code availability
Our SubLymE classification code and model weights are available at https://github.com/mattstokesBMS/SubLymE.
References
Teras, L. R. et al. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. Cancer J. Clin. 66, 443–459 (2016).
Sehn, L. & Salles, G. Diffuse large B-cell lymphoma. N. Engl. J. Med. 384, 842–858 (2021).
Scott, D. W. et al. High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements with diffuse large B-cell lymphoma morphology. Blood. J. Am. Soc. Hematol. 131, 2060–2064 (2018).
Alizadeh, A. A. et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000).
Nowakowski, G. S. et al. ROBUST: A phase III study of lenalidomide plus R-CHOP versus placebo plus R-CHOP in previously untreated patients with ABC-type diffuse large B-cell lymphoma. J. Clin. Oncol. 39, 1317 (2021).
Younes, A. et al. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non–germinal center B-cell diffuse large B-cell lymphoma. J. Clin. Oncol. 37, 1285 (2019).
Chapuy, B. et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 24, 679–690 (2018).
Schmitz, R. et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med. 378, 1396–1407 (2018).
Wright, G. W. et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell 37, 551–568 (2020).
Lacy, S. E. et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a haematological malignancy research network report. Blood 135, 1759–1771 (2020).
Alduaij, W. et al. Molecular determinants of clinical outcomes in a real-world diffuse large B-cell lymphoma population. Blood 141, 2493–2507 (2023).
Ennishi, D. et al. Double-hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J. Clin. Oncol. 37, 190 (2019).
Sha, C. et al. Molecular high-grade B-cell lymphoma: defining a poor-risk group that requires different approaches to therapy. J. Clin. Oncol. 37, 202 (2019).
Risueño, A. et al. Leveraging gene expression subgroups to classify DLBCL patients and select for clinical benefit from a novel agent. Blood 135, 1008–1018 (2020).
Kotlov, N. et al. Clinical and biological subtypes of B-cell lymphoma revealed by microenvironmental signatures. Cancer Discov. 11, 1468–1489 (2021).
Steen, C. B. et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell 39, 1422–1437 (2021).
Zhang, M. C. et al. Genetic subtype-guided immunochemotherapy in diffuse large B cell lymphoma: The randomized GUIDANCE-01 trial. Cancer Cell. 41, 1705–1716.e5 (2023).
Mo, Q. et al. Integrative analysis identifies multi-omics signatures that drive molecular classification of uveal melanoma. Cancers 13, 6168 (2021).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 1–15 (2013).
Reddy, A. et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell 171, 481–494 (2017).
Wallden, B. et al. Development of the molecular diagnostic (MDx) DLBCL lymphoma subtyping test (LST) on the nCounter analysis system. J. Clin. Oncol. 33 (2015).
Cerhan, J. R. et al. Cohort profile: the lymphoma specialized program of research excellence (SPORE) molecular epidemiology resource (MER) cohort study. Int. J. Epidemiol. 46, 1753–1754i (2017).
Davies, A. et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): an open-label, randomised, phase 3 trial. Lancet Oncol. 20, 649–662 (2019).
Vitolo, U. et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J. Clin. Oncol. 386, 351 (2017).
Maurer, M. J. et al. Event-free survival at 24 months is a robust end point for disease-related outcome in diffuse large B-cell lymphoma treated with immunochemotherapy. J. Clin. Oncol. 32, 1066 (2014).
Ren, W. W. et al. Genetic and transcriptomic analyses of diffuse large B-cell lymphoma patients with poor outcomes within two years of diagnosis. Leukemia 38, 610–620 (2024).
Wenzl, K. et al. Integrative genomics identifies a high-risk metabolic and TME depleted signature that predicts early clinical failure in DLBCL. Blood 140, 1742–1744 (2022).
Wenzl, K. et al. Multiomic analysis identifies a high-risk metabolic and TME depleted signature that predicts early clinical failure in DLBCL. Blood Cancer J. 14, 100 (2024).
Jain, N. et al. Targetable genetic alterations of TCF4 (E2-2) drive immunoglobulin expression in diffuse large B cell lymphoma. Sci. Transl. Med. 11, eaav5599 (2019).
Elpek, K. G., Lacelle, C., Singh, N. P., Yolcu, E. S. & Shirwan, H. CD4+ CD25+ T regulatory cells dominate multiple immune evasion mechanisms in early but not late phases of tumor development in a B cell lymphoma model. J. Immunol. 178, 6840–6848 (2007).
God, J. M. et al. Elevation of c-MYC disrupts HLA class II–mediated immune recognition of human B cell tumors. J. Immunol. 194, 1434–1445 (2015).
Han, G. et al. 9p21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat. Commun. 12, 1–19 (2021).
Gandhi, A. K. et al. Immunomodulatory agents lenalidomide and pomalidomide co‐stimulate T cells by inducing degradation of T cell repressors I karos and A iolos via modulation of the E 3 ubiquitin ligase complex CRL 4 CRBN. Br. J. Haematol. 164, 811–821 (2014).
Gandhi, A. et al. Dexamethasone synergizes with lenalidomide to inhibit multiple myeloma tumor growth, but reduces lenalidomide-induced immunomodulation of T and NK cell function. Curr. Cancer Drug Targets 10, 155–167 (2010).
Krönke, J. et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305 (2014).
Hagner, P. R. et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood 126, 779–789 (2015).
Hagner, P. R. et al. Interactome of Aiolos/Ikaros reveals combination rationale of cereblon modulators with HDAC inhibitors in DLBCL. Clin. Cancer Res. 28, 3367–3377 (2022).
Carpio, C. B. et al. Avadomide monotherapy in relapsed/refractory DLBCL: safety, efficacy, and a predictive gene classifier. Blood 135, 996–1007 (2020).
Jiang, K. et al. MYC inhibition reprograms tumor immune microenvironment by recruiting T lymphocytes and activating the CD40/CD40L system in osteosarcoma. Cell Death Discov. 8, 1–12 (2022).
Xu-Monette, Z. Y. et al. Clinical features, tumor biology, and prognosis associated with MYC rearrangement and Myc overexpression in diffuse large B-cell lymphoma patients treated with rituximab-CHOP. Mod. Pathol. 28, 1555–1573 (2015).
Collinge, B. et al. The impact of MYC and BCL2 structural variants in tumors of DLBCL morphology and mechanisms of false-negative MYC IHC. Blood 137, 2196–2208 (2021).
Luciano, C., Luca, A., Michael, B. & Francesco, B. DNA copy number changes in diffuse large B cell lymphomas. Front. Oncol. 10, 584095 (2020).
Caro, P. et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 22, 547–560 (2012).
Monti, S. C. et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 22, 359–372 (2012).
Hartert, K. T. et al. Targeting of inflammatory pathways with R2CHOP in high-risk DLBCL. Leukemia 35, 522–533 (2021).
Lu, G. et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 343, 305–309 (2014).
Watson, E. R. et al. Molecular glue CELMoD compounds are regulators of cereblon conformation. Science 378, 549–553 (2022).
Hagner, P. et al. Activity of lenalidomide in mantle cell lymphoma can be explained by NK cell‐mediated cytotoxicity. Br. J. Haematol. 179, 399–409 (2017).
Yang, Y. et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer cell 21, 723–737 (2012).
Zhang, L. H. et al. Lenalidomide efficacy in activated B‐cell‐like subtype diffuse large B‐cell lymphoma is dependent upon IRF 4 and cereblon expression. Br. J. Haematol. 160, 487–502 (2013).
Carrancio, S. et al. CC-99282 is a novel cereblon E3 ligase modulator (CELMoD) agent with enhanced tumoricidal activity in preclinical models of lymphoma. In: American Society of Hematology Annual Meeting. Atlanta, GA (2021).
Tilly, H. et al. Polatuzumab vedotin in previously untreated diffuse large B-cell lymphoma. N. Engl. J. Med. 386, 351–363 (2022).
Bröske, A. M. et al. Pharmacodynamics and molecular correlates of response to glofitamab in relapsed/refractory non-Hodgkin lymphoma. Blood Adv. 6, 1025–1037 (2022).
Nik-Zainal, S. et al. The life history of 21 breast cancers. Cell 149, 994–1007 (2012).
Cun, Y., Yang, T. P., Achter, V., Lang, U. & Peifer, M. Copy-number analysis and inference of subclonal populations in cancer genomes using Sclust. Nat. Protoc. 13, 1488–1501 (2018).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M., & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Cancer Syst. Biol. 1711, 243–259 (2018).
Danziger, S. A. et al. ADAPTS: Automated deconvolution augmentation of profiles for tissue specific cells. PLoS ONE 14, e0224693 (2019).
Krull, J. E. et al. Somatic copy number gains in MYC, BCL2, and BCL6 identifies a subset of aggressive alternative-DH/TH DLBCL patients. Blood Cancer J. 10, 1–8 (2020).
Acknowledgements
The authors thank Maria Wang and Scott Ely for technical assistance to the IHC data generation and interpretation. This work was supported in part by the NIH/NCI grants SPORE-P50 CA97274 (J.R. Cerhan and A.J. Novak), R01 CA212162-01A1 (A.J. Novak and J.R. Cerhan), U01 CA195568 (J.R. Cerhan), and the Predolin Foundation Biobank.
Author information
Authors and Affiliations
Contributions
Conceptualization: S.M.A., T.M.H., G.S.N., J.R.C., A.J.N., A.K.G. Computational analysis and interpretation: M.E.S., S.A.D., F.T., J.P., N.S., V.S., J.P.N., S.L.S., Y.W.A. Preclinical experimentation: Y.N., C.H., L.W., H.C., A.P., P.H. Biological Interpretation: K.W., C.C.H., M.O., M.J.M., R.L.K., B.K.L., A.S. Manuscript drafting: M.E.S., C.C.H., C.H., P.R.H., A.K.G. Review: all authors. Equal contributions: M.E.S., K.W., and C.C.H. contributed equally. A.J.N. and A.K.G. contributed equally.
Corresponding author
Ethics declarations
Competing interests
M.E.S., K.W., C.C.H., A.P., M.O., N.S., Y.N., C.H., L.W., H.C., A.S., P.H., and A.K.G. are employees and/or stakeholders in BMS. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ronald Gartenhaus and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Stokes, M.E., Wenzl, K., Huang, C.C. et al. Transcriptomic classification of diffuse large B-cell lymphoma identifies a high-risk activated B-cell-like subpopulation with targetable MYC dysregulation. Nat Commun 15, 6790 (2024). https://doi.org/10.1038/s41467-024-50830-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50830-y
- Springer Nature Limited