

Abstract
Chromosome rearrangements may distort 3D chromatin architectures and thus change gene regulation, yet how 3D chromatin structures evolve in insects is largely unknown. Here, we obtain chromosome-level genomes for four butterfly species, Graphium cloanthus, Graphium sarpedon, Graphium eurypylus with 2n = 30, 40, and 60, respectively, and Papilio bianor with 2n = 60. Together with large-scale Hi-C data, we find that inter-chromosome rearrangements very rarely disrupted the pre-existing 3D chromatin structure of ancestral chromosomes. However, some intra-chromosome rearrangements changed 3D chromatin structures compared to the ancestral configuration. We find that new TADs and subTADs have emerged across the rearrangement sites where their adjacent compartments exhibit uniform types. Two intra-chromosome rearrangements altered Rel and lft regulation, potentially contributing to wing patterning differentiation and host plant choice. Notably, butterflies exhibited chromatin loops between Hox gene cluster ANT-C and BX-C, unlike Drosophila. Our CRISPR-Cas9 experiments in butterflies confirm that knocking out the CTCF binding site of the loops in BX-C affected the phenotypes regulated by Antp in ANT-C, resulting in legless larva. Our results reveal evolutionary patterns of insect 3D chromatin structures and provide evidence that 3D chromatin structure changes can play important roles in the evolution of traits.
Similar content being viewed by others

Introduction
The fantastic diverse morphological traits of butterflies have been an integral part of ecology and evolutionary biology ever since Darwin’s times1,2,3. Previous studies have demonstrated that the majority of butterflies in the family Papilionidae exhibit a karyotype of 2n = 604. However, our preliminary investigations found significant karyotype variations within the Graphium genus (from 2n = 30 to 40 and 60), suggesting the butterflies of this genus underwent extensive chromosome rearrangement, especially fusion. In addition, our previous C-value estimation revealed that the Graphium genus has a much bigger genome size compared to other butterflies5. All these clues indicate that the Graphium butterflies have experienced extensive genome rearrangements, which may have considerable impact on the 3D chromatin landscapes. Besides, it has been well-known that there are almost no inter-chromosome 3D chromatin interactions in vertebrates, including topologically associating domains (TADs), subTADs and loops6,7,8,9. Our recent studies on muntjac deer, a mammalian group with intensive chromosome fusions, revealed that generally different 3D chromatin structures are conserved among mammals but chromosome rearrangements can result in more 3D chromatin alternations around the breakpoint regions and may have some biological effects in the evolution and adaptation of muntjac deer9. However, in insects, cross-species comparisons of 3D chromatin structures have been restricted in Diptera species with similar karyotypes and few genome rearrangements10,11,12,13,14,15 and their evolutionary patterns in different insect families are largely unknown. Therefore, the butterflies in the Graphium genus are great models for studying patterns and biological effects of 3D chromatin structure evolution in insects.
In this work, we assemble the high-quality chromosome-level genomes of four Papilionidae butterfly species using PacBio HiFi and Hi-C data. Three species are from the genus Graphium with very different karyotypes (2n = 30–60), while Papilio bianor (2n = 60, genome size ~500 Mb) serves as the outgroup control. The genome sizes of the three Graphium species range from 850 Mb to 1200 Mb5,16. Together with large scale Hi-C data, we scrutinize the evolutionary patterns of the 3D chromatin structures along with intensive genome rearrangements at the compartment, TAD, subTAD and chromatin loop levels in insects.
Results
Genome assembly, annotation, and phylogenetic analysis
By combining PacBio sequencing in high-fidelity (HiFi) mode with Hi-C data (Supplementary Tables 1–3), we generated chromosome-level genome assemblies for P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus (Fig. 1a, b, Supplementary Fig. 1). The final genome assemblies are 528.52 Mb with contig N50 of 11.56 Mb for P. bianor, 1227.81 Mb with contig N50 of 3.76 Mb for G. eurypylus, 885.26 Mb with contig N50 of 10.91 Mb for G. sarpedon, and 1107.02 Mb with contig N50 of 4.71 Mb for G. cloanthus (Table 1). The assembled genome sizes are consistent with flow cytometry estimations reported in our previous study (Supplementary Table 4)5. The consensus Quality Value (QV) of the genome assemblies for the three Graphium species and P. bianor ranged from 44.25 to 66.24, indicating very high base accuracy (Table 1). The high quality and continuity of these genome assemblies are reflected not only by their high QV, but also high Benchmarking Universal Single-Copy Orthologs (BUSCO) values (94.4%–98.5%) (Table 1)17. Compared with the previously published P. bianor genome18, the new P. bianor assembly exhibited better synteny with the three Graphium species (Supplementary Fig. 2). Besides, 12 of the 30 chromosomes in our new P. bianor genome assembly have no gap and thus are at the T2T assembly level (Supplementary Data 1, Supplementary Fig. 3), while no such T2T chromosome was obtained in the previous studies18,19.

a Overview of four butterfly genomes. Track A corresponds to chromosome length. Tracks B, C, D, and E represent density distribution of PacBio-HiFi reads, repeat sequences, GC content, protein-coding genes. Track F corresponds to syntenic blocks between P. bianor and other three species. b Interaction map of Hi-C data of G. cloanthus. With numbered chromosomes as coordinates, color of each dot represents log value of interaction intensity of corresponding bin pair of genomes, and interaction intensity increases from white to red. Chr, chromosomes. c The chromosome length (top plot), chromosome TE contents (middle plot), and length of different chromosome-wide TE types (bottom plot) of four butterfly species. d Maximum-likelihood phylogenetic tree constructed based on BUSCO genes of six insect species dated with fossil calibrations. To compute node support, 1000 bootstraps were used, and all nodes showed 100% support. Blue numbers denote divergence time of each node (Mya, million years ago). Scale bar represents 40 Mya. Purple numbers indicate the number of inter-chromosomal rearrangements in this branch. The green bars represent the 95% confidence intervals. e Synteny patterns among genomic regions of three Graphium species and P. bianor. Numbers represent individual chromosomes. Highlighted traits indicate chromosome fission. Source data are provided as a Source data file.
We annotated 37–60% of these genome sequences as repetitive sequences (Fig. 1c, Supplementary Tables 5, 6, Supplementary Note 1) and ~15,000 protein-coding genes per genome with the assistance of transcriptome data from adult individuals of these species (Supplementary Tables 3, 7, 8, Supplementary Note 2). A total of 57%, 44%, 36%, and 47% of P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus was annotatable to be transposable elements (TEs), representing 306 Mb, 551 Mb, 322 Mb, and 526 Mb, respectively (Supplementary Table 6, Supplementary Fig. 4). Among them, the long terminal repeats (LINEs) were annotated as 73 Mb, 292 Mb, 154 Mb, and 268 Mb, and the short terminal repeats (SINEs) were annotated as 8.6 Mb, 13.5 Mb, 25.2 Mb, and 9.79 Mb for P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus, respectively. The estimated activity of these TEs accounts for 153 Mb, 334 Mb, 198 Mb, 317 Mb of P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus, and they were estimated to be active within the last 13 million years (Myr) in the four butterfly species (Supplementary Fig. 5, Supplementary Note 1). Besides, around 87 Mb, 454 Mb, 354 Mb, and 395 Mb of P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus were annotated as neither functional nor repeat sequences, respectively. These regions could be unique sequences that have formed through long-term degradation of TEs, also called “cemetery of TEs” by previous research20,21, indicating that the genome size of the three Graphium species has increased by gradual accumulation of TEs over the past 13 Myr. Previous research indicated that TEs play important roles in regulation of gene expression and genome size22,23,24,25,26. We found the variation of average gene length and intron length was less than the variation of genome size among P. bianor and the three Graphium species (Supplementary Table 7), suggesting that TEs tended to insert in the intergenic regions. Above all, TEs, especially LINEs, is consistent with TEs being the primary contributor to the observed genome size amplification in the three Graphium species (Fig. 1c).
Phylogenetic analysis and divergence time estimation based on genomic data revealed that the three Graphium species form a monophyletic group that diverged from P. bianor approximately 66.6 million years ago (Mya), and the divergence time within the three Graphium species ranges from 8.1 to 27.2 Mya (Fig. 1d). These results provide phylogenetic context for the identification of genome rearrangement events.
Drastic genome rearrangements in Graphium butterflies
We analyzed the chromosome rearrangement events that occurred in Graphium species through the whole-genome alignments with the P. bianor genome. Our chromosome comparisons revealed almost no chromosome rearrangements between G. eurypylus and P. bianor, consistent with the fact that they have the same chromosome number (Fig. 1e). In contrast, genomic collinearity analysis among the three Graphium species showed numerous genome rearrangements among them (Fig. 1e). To clearly identify these genome rearrangement events in the three Graphium species, we determined rearrangement sites (RSs) based on homologous synteny blocks in the three Graphium species with the G. eurypylus genome as the ancestral karyotype. Genomes of G. cloanthus and G. sarpedon were compared to that of G. eurypylus to identify RSs, respectively. To be robust, we only assessed the impact of large-scale rearrangements (length of the rearranged regions >100 kb). Finally, we defined 84 and 84 RSs in G. cloanthus and G. sarpedon, including 15 and 18 inter-chromosome fusion RSs, 69, and 66 intra-chromosome large-scale RSs, and their homologous regions in G. eurypylus (Supplementary Data 2).
Syntenic analysis data indicated that five of the 20 haploid chromosomes of G. sarpedon had not undergone any fusion event, and the remaining 15 were formed by the fusion of ancestral chromosomes (Fig. 1e, Supplementary Data 2, Supplementary Table 9), including seven with telomere-to-telomere fusions from two ancestral chromosomes, two with fusions of two ancestral chromosomes and a fragmented ancestral chromosome, and six with fusion of an ancestral chromosome and a fragmented ancestral chromosome (Fig. 1e, Supplementary Data 2, Supplementary Table 9). For 15 haploid chromosomes of G. cloanthus, only one had not undergone any fusion events, and the remaining 14 were formed by the fusion of ancestral chromosomes (Fig. 1e, Supplementary Data 2, Supplementary Table 9), including one with telomere-to-telomere fusions from three ancestral chromosomes and 13 with telomere-to-telomere fusions from two ancestral chromosomes (Fig. 1e, Supplementary Data 2, Supplementary Table 9). Interestingly, although G. sarpedon and G. cloanthus had undergone extensive inter-chromosome rearrangements and diverged relatively recently (~8.1 Mya), they shared only one chromosome fusion site (Supplementary Table 9). This suggests that most inter-chromosome fusions have evolved independently after speciation within 8.1 Myr between these two species.
Identification of 3D chromatin structures of butterflies revealed by Hi-C
To investigate the 3D chromatin architectures in butterflies, we constructed high-resolution genome-wide chromatin interaction maps for the four butterfly species using large-scale Hi-C data. 177–415 Gb Hi-C data were generated for the four species, respectively (Supplementary Table 3). We mapped Hi-C reads to the correspondent genomes, 4–10 million high-quality read pairs were retained for constructing chromatin interaction maps at resolution of 2150–4400 bp (Supplementary Table 10, Supplementary Fig. 6)27, enabling us to explore 3D chromatin structures at the levels of compartments, TADs, subTADs, and loops28,29,30. By dividing the genome into windows of the same length and counting Hi-C read pairs aligned with each window, we constructed an interaction matrix with different resolutions represented by window size. Using a zoomed-in view of the contact map, we observed a checkerboard pattern of long-range interactions (compartments) at 100 kb resolution, triangles above the diagonal (TADs) at 40 kb resolution, small structures within TAD (subTADs) at 10 kb resolution, and paired points located within some of these TADs (chromatin loops) at 5 kb resolution (Fig. 2a–d) in butterflies, which were consistent with previous studies9,11,29,30,31,32. It is noteworthy that the observed length (TAD at 40 kb) of 3D chromatin structures significantly differs from previous studies on Diptera species (TAD at 5 kb)11,12, which is likely attributed to the larger genome size of the four butterfly species.

a Interaction map of Hi-C data in Chr9 of G. cloanthus showing compartments. The histogram reflects the correspondence between RNA-seq data and the values of the first principal component (PC1). Top: RNA-seq (TPM), the yellow column represents compartment A and the blue column represents compartment B. Middle: PC1 within a 100 kb window, which reflects the degree of interaction frequency, is used to distinguish active (A) and inactive (B) compartments. The red bars represent compartment A, the black bars represent compartment B. Bottom: Hi-C interaction matrix at 100 kb resolution. b Region zoomed-in from Fig. 2a showing TADs (black line) at 40 kb resolution. Top: the insulation score at 40 kb resolution is a quantitative measure used to identify TADs within the genome, with the valley of insulation fraction represents the TAD boundary at 40 kb resolution. Bottom: Hi-C interaction matrix at 40 kb resolution. c Region zoomed-in from Fig. 2b showing subTADs at 10 kb resolution. The black dashed line represents TADs, and the black solid line represents subTADs. Top: Hi-C interaction matrix at 10 kb resolution, bottom: the insulation score at 10 kb resolution is a quantitative measure used to identify subTADs within the TAD, with the valley of insulation fraction represents the subTAD boundary at 10 kb resolution. d Region zoomed-in from Fig. 2c showing chromatin loops at 5 kb resolution. The black frame represents chromatin loops. Source data are provided as a Source Data file.
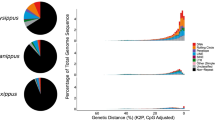
At the 100 kb resolution, we performed compartment analysis by Principal Component Analysis (PCA) (Fig. 2a), a widely used method to identify spatial chromatin compartments9,32,33,34. As our data show, compartment A (active) has significantly higher gene density and gene expression levels compared to compartment B (inactive) (Supplementary Figs. 7 and 8), consistent with previous studies33,34. Using homologous genes as markers, we found that the conservation of the A/B compartments between different butterfly genera (76.6%) was less than that among the three Graphium species (90%) (Fig. 3a, Supplementary Figs. 9 and 10), and the latter was similar to that among species of the mammalian muntjac genus (~90%)9.

a The top plot shows the compartment conservation. Orange: genes in compartment A, blue: genes in compartment B. Bottom-left: the percentage of homologous genes located in Compartment A among all homologous genes. Top-right: the number of conserved (compartment) homologous genes. Bottom-right: the black circles indicate the intersections between different datasets. b Correlation of number of TADs and number of subTADs with genome size of three Graphium and P. bianor genomes, respectively. c Violin plots demonstrate the length distributions of TADs and subTADs in the four species. Top: TAD length. Bottom: subTAD length. Number of TADs and subTADs are shown above each violin-plot. Center and bounds of box correspond to median, 25%, 75% quartiles per group. The whiskers are 1.5*IQR (IQR: the interquartile range between the 25% and 75% percentile). The shape of the violin plots represents data distribution. Statistical significance was determined by a two-tailed Student’s t-test. d The boxplots demonstrate the length distributions of loops across four species. Number of loops are shown above each boxplot. Boxplots depict the median, 25%, 75% quartiles per group. The whiskers are 1.5*IQR (IQR: the interquartile range between the 25% and 75% percentile). Statistical significance was determined by a two-tailed Student’s t-test. e The ratio of conserved TADs, subTADs and chromatin loops in alignment TADs, subTADs and chromatin loops, respectively. The red font indicates the corresponding reference genome used for alignment. Source data are provided as a Source data file.
We then detected TADs and subTADs at 40 kb and 10 kb resolution in accordance to previous researches6,9,32,35,36, respectively, and found 452, 1408, 1031, and 982 TADs, 2448, 6885, 4922, and 4577 subTADs in P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus, respectively (Figs. 2b, c and 3b). These results indicate that the number of TADs and subTADs are positively correlated with genome size and that the average length of TADs, subTADs are much shorter in P. bianor which has much smaller genomic size than G. sarpedon, G. eurypylus, and G. cloanthus (Fig. 3c). In addition, at the 5 kb resolution, we identified a total of 618, 723, 645, and 809 chromatin loops for P. bianor, G. eurypylus, G. sarpedon and G. cloanthus, respectively (Figs. 2d and 3d)27,32. The number of chromatin loops is basically similar in the four butterflies, but the length of chromatin loops is much shorter in P. bianor than in the three Graphium species (Fig. 3d). Subsequently, we performed a comparative analysis on the conservation of TADs, subTADs, and loops among the three Graphium species. Briefly, for each interspecific comparison, genomic coordinates of TADs, subTADs and chromatin loops were lifted over between G. cloanthus and G. sarpedon, G. cloanthus and G. eurypylus, and G. sarpedon and G. eurypylus, respectively. Comparable to their shared genome synteny fraction (58.5%, 24.2%, 23.8%), the successful lift fraction was 47.6%, 34.1%, and 34.2% for TADs, 39.2%, 23.4%, and 25.7% for subTADs, as well as 54.7%, 53.9%, and 36.8% for chromatin loops, respectively (Supplementary Table 11, Supplementary Fig. 11). We found that 65–81% of lifted TADs, 74–78% of lifted subTADs and 52–55% of lifted chromatin loops were conserved in the three Graphium species (Fig. 3e), implying that subTADs may represent a more conserved unit within the 3D chromatin structures. The conservation of lifted TAD fraction was comparable to that observed in muntjac deer (43–73%)9. These findings suggest that in different animal classes the 3D chromatin structures are similarly conserved in syntenic regions among closely related species.
Impact of inter-chromosome fusions on 3D chromatin structures
Previous studies observed that compartment structure was quite conserved along evolution9,32. Upon analyzing the compartment types of genes flanking the inter-chromosomal fusion sites in G. sarpedon and G. cloanthus, and comparing these with the compartment types of corresponding homologous genes in G. eurypylus, we discovered that the conservation of compartment types for homologous genes near inter-chromosomal fusion sites is roughly equivalent (~90%) to the overall conservation level of compartment types for homologous genes across the entire genome (Fig. 3a, Supplementary Fig. 9). Furthermore, we calculated the conservation of compartments in homologous regions near RSs (within 1 Mb) to be about 90% based on genomic alignments, which is consistent with the results obtained using homologous genes (Supplementary Fig. 9). These results suggests that compartment state has been barely affected by inter-chromosome rearrangements. We then compared TADs, subTADs and loops around chromosome fusion site in G. sarpedon and G. cloanthus with the corresponding regions in G. eurypylus. We found the inter-chromosome RSs in G. sarpedon and G. cloanthus were all located at telomeres or TAD boundaries in G. eurypylus, and no chromatin loop spanning across these regions in G. eurypylus was observed. These findings suggest that inter-chromosome fusions almost do not disrupt the pre-existing 3D chromatin structures of ancestral chromosomes. However, we observed the emergence of TADs and even subTADs at some inter-chromosome RSs in the rearranged genomes. In addition, we did not observe telomeric sequences in all the inter-chromosome RSs in G. sarpedon and G. cloanthus (Supplementary Fig. 3, Supplementary Table 9).
We then looked at the compartment states in the flanking regions of these inter-chromosome RSs in G. sarpedon and G. cloanthus. Since it was difficult to determine the exact recombination sites of large rearrangement events, we only evaluated non-gapped inter-chromosome RS regions spanning less than 200 kb in this analysis (Supplementary Data 2, Supplementary Table 9). In G. cloanthus, we observed that the flanking regions of five inter-chromosome RSs were in different compartment types with no new TAD formation, while the flanking regions of seven inter-chromosome RSs were both in compartment A, in which two of them gave rise to new TADs and three RSs formed novel subTADs (Fig. 4a, Supplementary Data 2, Supplementary Table 9). Similarly, in G. sarpedon, we observed that the flanking region of eight inter-chromosome RSs were in different compartments with no new TAD formation, while the flanking region of six inter-chromosome RSs were both in compartment A, in which five of them gave rise to new TADs and one RSs formed a novel subTAD. Interestingly, only the flanking region of one inter-chromosome RSs were both in compartment B, and it has formed a novel TAD across the RSs. These results suggest the inter-chromosome rearrangements barely happened between compartment B, and furthermore, formation of new TADs through inter-chromosome rearrangements requires concordance in the flanking compartment type, particularly when both are in compartment A.

a Combined heatmaps of contact matrix around the fusion sites at 10 kb resolution. Flanking region of RSs of G. cloanthus (left) and their homologous regions in G. eurypylus (right). The circular symbols mark the locations of fusion sites. Top: Flanking region of RSs are in different compartment state. Median: Flanking region of RSs are both in compartment A with no new TAD formation. Bottom: Flanking region of RSs are both in compartment A with new TAD formation. b RSs within TAD or subTAD boundries. c Synteny patterns between genomic regions of four butterfly species showing chromosome rearrangement. Gray line indicates homologous genes, red line indicates Rel, green line and blue line indicate chromosomal rearrangement. d The interaction map of Hi-C data. Black dotted line in the interaction map indicates TADs. Lines of different colors between two interaction maps represent the corresponding homology blocks of Fig. 4c. Top: Region zoomed-in from Fig. 4c showing chromosomal rearrangement between G. eurypylus and G. cloanthus. Bottom: Region zoomed-in from Fig. 4c showing chromosomal rearrangement between G. eurypylus and G. sarpedon. Source data are provided as a Source data file.
Alteration of 3D chromatin structures with intra-chromosome rearrangements
As described above, we identified 66 and 69 long-range (>100 kb) intra-chromosomal rearrangements in G. sarpedon and G. cloanthus, respectively. Besides, we observed a significant correlation between the TE-mediated genome expansion in the three Graphium species (Fig. 1c, Supplementary Table 6) and the increased average lengths and numbers of TADs and subTADs compared to P. bianor (Fig. 3b, c). Therefore, we compared the effects of intra-chromosome rearrangements on 3D chromatin structures from two aspects: long-range (>100 kb) intra-chromosome rearrangements including inversions and translocations, and TE insertions. As for long-range intra-chromosome rearrangements, similar as above, we only evaluated 3D chromatin structures for non-gapped intra-chromosome RS regions spanning less than 200 kb. We found that the conservation level of compartment around large intra-chromosome RSs was comparable to the average compartment conservation level of the whole genome (~90%) (Supplementary Fig. 9), consistent with inter-chromosome rearrangements.
We compared TAD, subTAD, and chromatin loops in the homologous regions of those intra-chromosome RSs in G. eurypylus. We found 40.5% of G. sarpedon and 43.4% RSs of G. cloanthus were mapped into previous TADs and subTADs of G. eurypylus, implying that >40% ancestral TADs and subTADs were disrupted by intra-chromosome rearrangements. The remaining 34.2% and 38.6% of the homologous intra-chromosome RS regions were located at both TADs and subTADs boundaries of G. eurypylus, and 24.1% and 18.1% of the homologous regions of RSs were only located at subTADs boundaries of G. eurypylus (Fig. 4b). Besides, we found six chromatin loops were across the homologous regions of RSs in G. eurypylus (Supplementary Fig. 12). On the other side, we looked at TAD, subTAD and chromatin loops around the intra-chromosome RSs in the rearranged genomes of G. sarpedon and G. cloanthus. We found that 32.3% and 32.1% RSs were located within TADs and subTADs, indicating that they might be newly evolved in G. sarpedon and G. cloanthus, respectively (Fig. 4b). However, we found only one chromatin loop spanning across RSs in G. sarpedon (Supplementary Fig. 12).
We observed the flanking regions of 34 and 36 large intra-chromosome RSs were both in compartment A, and the flanking regions of 17 and 10 intra-chromosome large RSs were both in compartment B, while the flanking regions of 13 and 10 large intra-chromosome RSs had different compartment types in G. sarpedon and G. cloanthus, respectively (Supplementary Data 2), suggesting that compartment A boundaries experienced more chromosomal rearrangement events. Interestingly, we found 21 and 18 large intra-chromosome RSs were located within TADs and subTADs, and nearly all these large intra-chromosome RSs exhibit same compartment types on both sides (Supplementary Data 2). These results suggest that some intra-chromosome rearrangements may lead to significant regulation changes by changing 3D chromatin structures near the RSs. One such remarkable rearrangement case shows that a chromosome inversion event influences an important gene Relish (Rel). Through a comparison of genome collinearity in four butterflies, we discovered that the Rel gene was rearranged in G. sarpedon and G. cloanthus compared to G. eurypylus and P. bianor (Fig. 4c, d). In G. cloanthus, the Rel gene underwent inversion, the loops across the Rel gene disappeared and the subTAD where the Rel gene was located was disrupted. In G. sarpedon, similar to G. cloanthu, the subTAD was disrupted, and the Rel gene was even completely lost (Fig. 4d, Supplementary Figs. 13, 14). The Rel gene encodes a transcription factor that plays key roles in the humoral immune response37,38,39,40.
Given that there are extensive TE insertions in the species as described above, we also looked at rearrangements caused by TE insertions. Previous studies have shown that TEs were enriched in compartment B41. Therefore, we categorized TAD and subTADs based on the compartment types in which they were located and examined impact of compartment types (Fig. 5a, Supplementary Figs. 15 and 16). We found that the proportion of changes in length of the TADs and subTADs within compartment A in G. eurypylus, G. sarpedon, and G. cloanthus were smaller than that of the chromosome size, suggesting that they have been more strongly constrained in compartment A. Consistent with this, we observed a higher level of consistency in the 3D structures in the contiguous regions of compartment A (Fig. 5b). In contrast, the proportion of changes in length of both the TADs and subTADs involved in the “A to B” and “B to B” type transitions in the three Graphium species were higher than that of the chromosome size (Fig. 5b). A notable example is that, by comparing the 3D chromatin structure in P. bianor, we found that TE insertion-mediated chromosomal rearrangements formed a new TAD located in compartment B in three Graphium species. More importantly, several compartments transitioned from compartment B in P. bianor to compartment A in the three Graphium species near this new TAD. Interestingly, an important gene, levan fructotransferase (lft), which regulates TOR signaling for wings in insects42,43, was located in one of these switched compartments (Fig. 5c). The compartment transition might have thereby led to the higher expression of the lft gene in the three Graphium species indicated by our RNA-seq data (Supplementary Table 12). This case suggests that TE insertions can induce compartment switch and change TADs, and thereby influence gene regulation.

a According to the compartment classification of TADs in different species, the length of TADs among the three Graphium species are compared with their corresponding homologous regions in P. bianor, respectively. Number of homologous blocks are shown above each boxplot. Boxplots depict the median, 25%, 75% quartiles per group. The whiskers are 1.5*IQR (IQR: the interquartile range between the 25% and 75% percentile). The red dotted line represents the genome size ratio, respectively. For example, homologous blocks “A to B” in “P. bianor vs G. eurypylus” indicate that TADs are located within compartment B in G. eurypylus, while the corresponding area in P. bianor is located within compartment A. The ‘length ratio’ is calculated by dividing the length of the TAD in G. eurypylus by the length of its corresponding homologous region in P. bianor. b The interaction map of Hi-C data from the genomes of three Graphium species and P. bianor showing conserved 3D chromatin structures located within compartment A. The black frame represents chromatin loops. c The interaction map of Hi-C data from the genomes of P. bianor, G. cloanthus, and G. eurypylus showing TE insertion and compartment change. Source data are provided as a Source data file.
3D chromatin structure evolution in the Hox gene cluster of insects
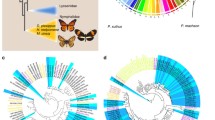
Hox genes play crucial roles in the development of organisms. However, few studies have investigated species differences in the 3D chromatin structure of Hox genes44,45. Here, we compared the 3D chromatin structures around Hox genes among butterflies and fruit flies. We found that the Hox gene clusters in four butterfly species were located within a single TAD, whereas in fruit flies, they are separated into two TADs caused by a 9.6 Mb insertion10,46, forming the Bithorax complex locus (BX-C) and Antennapedia complex locus (ANT-C)47, respectively (Fig. 6a, Supplementary Fig. 17). Moreover, we identified numerous chromatin loops between BX-C and ANT-C loci in butterflies, while no loop between the BX-C and ANT-C loci was observed in flies.

a The interaction map of Hi-C data for Drosophila melanogaster, G. eurypylus, G. sarpedon, and P. bianor. The blue box indicates the interaction between ANT-C and BX-C that is different from Drosophila. The purple circle indicates the chromatin loops between ANT-C and BX-C unique to G. eurypylus, G. sarpedon, and P. bianor. b Schematic diagram of gene regulation and shotgun deletion generating butterflies that are mosaic for different deletion lengths. bp, base pair. c First instar larvae of P. xuthus. CTCF mKO 1: the head shell is asymmetric, and the right T1 leg is missing. CTCF mKO 2: the head shell is asymmetric, and the T1 and T2 leg are missing. CTCF mKO 3: the head shell is asymmetric, and the left T1, T2, and T3 legs are missing. d The three pairs of thoracic legs are abnormal, the tarsus is shortened and thicker compared with wild type. e The termen and apex of the forewing are contractive and smaller than the wild type.
To explore the regulatory role of these chromatin loops between BX-C and ANT-C loci in butterflies, we used Hi-C and genomic data combined with our previous ATAC-seq data48 to identify the boundary insulator motifs of the four chromatin loops which on one side converges to the same location in BX-C (Fig. 6b). Briefly, we performed transcription factor prediction to identify the binding sites of insulator proteins situated on one side of the chromatin loops within the BX-C locus in P. bianor (Supplementary Table 13). Besides, we downloaded the P. bianor ATAC-seq data48 to analyze the cis-regulatory elements (CREs) near the site of the four chromatin loops within the BX-C locus in P. bianor (Supplementary Table 14). Combining the above two results, we found that only one CTCF binding site was predicted with the highest motif score in CRE 1 within one side of the four chromatin loops in the BX-C locus, and the other side of these chromatin loops were located near the Antp gene within the ANT-C locus. Prior studies in Drosophila have demonstrated that CTCF, in conjunction with other proteins, plays a pivotal role in forming the insulator in regulating Hox gene expression49. Moreover, we observed that this CTCF binding site was shared among the four butterfly species and Papilio xuthus50 by sequence alignment (Supplementary Fig. 18). Because we have the gene editing system for P. xuthus in hand, we employed CRISPR-Cas9 genome editing to generate mosaic knockout (mKO) P. xuthus individuals of this CTCF binding site. We hypothesized that the mKO of the CTCF binding site within the BX-C locus in P. xuthus may disrupt interactions and thus regulation between BX-C and ANT-C.
In the gene edited mKOs, we observed high mortality in first-instar larvae, with most of the dead larvae exhibiting leg and head deformities (Fig. 6c, Supplementary Tables 15 and 16), and PCR and sequencing validated that they were mKO at this site (Supplementary Fig. 19). The legless phenotype in some mKO larvae was consistent with a previous study in which Antp was knocked out in silkworm larvae51, suggesting that Antp function was impaired during the butterfly larval development due to the loss of chromatin loops between BX-C and ANT-C. Antp in the ANT-C complex play key roles in leg development52,53,54, and in a small number of mutants which survived to the adult stage, we also observed deformities in the legs of the adults, including shortening of all legs (Fig. 6d, Supplementary Table 16, Supplementary Fig. 20). In some adult mutants, we also observed reduction in forewing size (Fig. 6e), which aligns with previous phenotypes observed when Antp was knocked out in silkworms51 (Supplementary Table 16, Supplementary Fig. 20). To further investigate the underlying causes of these phenotypic changes, we conducted Hi-C sequencing for the P. xuthus 1st-instar larvae of both CTCF mKO and wild type. Interestingly, the results show that the mutants have a sharp decline in Hi-C reads depth near the knockout sites than the wild types (Supplementary Fig. 21a), suggesting that there is a remarkable reduction in interaction frequency near the knockout sites of the mutant larvae. By comparing the Hi-C interaction heatmaps of the mutant and wild-type larvae, we observed that the chromatin loop connecting the ANT-C near the Antp gene with the BX-C loci, clearly visible in wild-type larvae, but is weakened in the mutant larvae as indicated by reduced heatmap intensity (Supplementary Fig. 21b). Furthermore, we conducted quantitative real-time PCR (qRT-PCR) experiments, and consistent with the Hi-C analysis results, the qRT-PCR result shows that the expression level of the Antp gene decreased by ~30% (Supplementary Fig. 21c, Supplementary Table 17). These results suggest that the chromatin loops between BX-C and ANT-C complexes have important regulatory functions in the development of butterfly phenotypes, and changes in Hox 3D chromatin architectures can influence morphological evolution across insect taxa. Besides, in accordance with methodologies delineated in prior studies50,55, we executed next-generation sequencing for three CTCF mKO mutants, respectively, and undertook evaluations for off-target loci in the three mutants, revealing no conspicuous off-target phenomena (Supplementary Note 3).
Discussion
In this study, combining PacBio-HiFi data and Hi-C technology, we successfully assembled the high-quality chromosome-level genomes of three Graphium species with drastic karyotype difference (G. cloanthus, G. sarpedon, and G. eurypylus), and updated our previously published genome of P. bianor. These four butterfly genomes not only lay foundation for investigating 3D chromatin structures, but also provide important resources for understanding evolution of 3D chromatin structures in insects. We revealed that the drastic chromosome number reduction in G. sarpedon and G. cloanthus mainly resulted from independent chromosome fusion events after they diverged around 8.1 million years ago, which is significantly different from the previously observed gradual fusions in muntjac deer9. In muntjac deer, chromosomal fusion has progressively reduced the ancestral karyotype from 2n = 70 to 2n = 46 in Muntjak reevesi and to 2n = 8/9 in Muntjak crinifrons, in which species with different karyotypes share many fusion points9. In contrast, in butterflies, G. sarpedon and G. cloanthus only share one fusion point despite they have similar chromosome numbers, suggesting that the rest of the chromosomal rearrangements in these butterflies were from independent fusion events after species divergence (Fig. 1e, Supplementary Data 2, Supplementary Table 9). And we also discovered that the genome size amplification in the three Graphium butterflies was primarily driven by TE insertions. Previous studies have shown a depletion of LINEs at TAD boundaries and an enrichment of SINEs at TAD boundaries56. Longer TAD and subTAD lengths and more LINE insertion in the three Graphium butterflies indicate that LINE expansions might have led to the elongation of TAD and subTAD lengths, whereas SINE expansions may cause appearance of more TADs and subTADs because SINEs usually locate at their boundaries. Similar to the expanded length of TADs and subTADs, the enlarged chromatin loops length was also possibly caused by LINE insertions.
With large-scale Hi-C data, we found the 3D chromatin structures including compartments, TADs, subTADs and chromatin loops were all present in four butterfly species, as previously shown in dipteran species10,11. The proportions of interspecifically conserved compartments, TADs, subTADs and loops in the three Graphium species are 90%, 65–81%, 74–78%, and 52–55% (Fig. 3e, Supplementary Table 11), respectively. Although TADs and subTADs exhibit conservation in syntenic regions within the genus Graphium diverged of nearly 27 Mya, this process is accompanied by many chromosome rearrangements among the three Graphium species. Our results show that new TADs and subTADs could be formed when the flanking regions of RSs are in the same type, especially between compartment As. The finding of new TADs formation across inter-chromosome RSs in butterflies significantly differed from that of muntjac deer in our previous study9, where no TAD were found across fusion sites. In muntjac deer, where chromosome rearrangement occurred primarily through telomere and centromere fusion, ancient fusion sites exhibited increased chromosomal interactions across these regions and possessed fewer telomeric and centromeric sequences compared to newer fusion sites9, suggesting that the loss of repetitive sequences over time may have facilitated the evolution of new TADs at these locations. Therefore, the difference of 3D chromatin evolutionary patterns after extensive chromosome fusions between muntjac deer and butterflies may be due to the longer evolutionary time after chromosomal rearrangements in butterflies (~8 Myr) compared to muntjac deer (~3 Myr), which lent butterflies more time to lose the repetitive sequences at the fusion site and thus made it easier to form new TADs and subTADs. Furthermore, unlike in muntjac deer, we did not find telomeric or centromeric sequences at the fusion sites in G. sarpedon and G. cloanthus, and therefore, we were not able to infer why chromosome fusion has recurrently happened in the butterfly genus. The difference in chromosome structure, with the feature of holocentric chromosomes in butterflies57 and monocentric chromosomes in muntjac deer9, may affect the fusion mechanism and formation of new TADs and subTADs, which is awaited to be investigated in the future.
Our results also showed that chromosome fusions barely disrupted previous 3D chromatin structures around the fusion sites, and RSs of most inter-chromosome and more than 60% intra-chromosome rearrangements are located at the TADs and subTADs boundaries, as reported in dipteran species and mammals7,11,58. It seems that subTADs, usually containing several genes and regulatory elements, exist as a relatively stable entity within TADs and less affected by chromosome rearrangements, as shown by the higher conservation level of subTADs than TADs (Fig. 3e). Aligning with this, previous studies on Drosophila also found that disrupting TADs does not influence coordinated gene expression in most cases14.
Interestingly, we observed intra-chromosome rearrangements disrupted more than 50% of TADs, 30% of subTADs, and several loops, and many new TADs, subTADs, and loops near the RSs have also been evolved. We found three remarkable such cases that 3D chromatin landscapes were obviously altered by intra-chromosome rearrangements. The first case is the 3D chromatin structure around the Rel gene, which regulates the antibacterial response. Interestingly, the host plants of G. sarpedon and G. cloanthus are mainly Lauraceae, which contain antimicrobial compounds. In contrast, the host plants of G. eurypylus and P. bianor are Annonaceae and Rutaceae, respectively, which do not contain bactericidal compounds59. These correlations suggest that G. eurypylus and P. bianor could use the Rel pathway to resist pathogens, but loss or downregulation of Rel in G. sarpedon and G. cloanthus may reflect the fact that they can use antimicrobial compounds in their hosts. Therefore, alterations of the 3D chromatin structure around the Rel gene may be correlated to the host specificity of these butterflies.
The second case is the lft gene, which regulates TOR signaling for wings in insects42. Previous study had observed that RNA interference (RNAi) targeting the lft gene led to the reduction of cross-vein length in Drosophila melanogaster45. A TE insertion near the lft gene resulted in compartment switch and TAD split. It is likely that the compartment shift from B to A increased the expression level of the lft gene in the three Graphium species (Supplementary Table 12), and thereby may account for the venation differences observed between P. bianor and the three Graphium species.
The third example is the Hox gene clusters. We found that there are a large number of loops between BX-C and ANT-C in butterflies, but not in Drosophila in which there is a 9.6 Mb insertion between BX-C and ANT-C loci. We identified a CTCF site in the butterfly BX-C but not in Drosophila. After we disrupted the CTCF sequence and thereby the loops in butterfly, we observed leg and wing abnormal larvae and adults, showing phenotypes resembling Antp, a key gene in the ANT-C locus, knockout silkworms51. Furthermore, Hi-C sequencing of the mKO mutant and wild type larvae confirmed the significant disappearance of the loop between BX-C and Antp gene. Consistently, qRT-PCR experiment data demonstrated a decrease in the expression of Antp gene in the mKO mutant larvae compared the wild-type larvae. These results intrigued a possibility that the absence of loop interactions between ANT-C and BX-C in Drosophila may be correlated with the evolution of legless phenotype in dipteran larvae.
The three cases of genome rearrangement-caused 3D chromatin structure changes provide the evidence that alternations of 3D chromatin structures by chromosome rearrangements can play important roles in the evolution of animal traits. These findings underscore the importance of 3D chromatin structures in the evolution of organisms and therefore have explored a new direction for future evolutionary genetics studies.
Methods
Butterfly sample collection and sequence strategy
This research was approved by the Committee for Animal Experiments of the Institute of Zoology, Chinese Academy of Science, China (SMKX-SQ-20200408-041). Butterfly sample collection was supported by Biodiversity Conservation Program of the Ministry of Ecology and Environment, China (China BON-Butterflies) and by the Program of Yunnan Forestry and Grassland Administration (2022GF258D-11).
Live adults of Papilio bianor, Graphium eurypylus, Graphium sarpedon, and Graphium cloanthus were collected from natural populations (Supplementary Table 1) and transported to the laboratory in plastic bags. The samples were then rapidly frozen in liquid nitrogen and stored at −80 °C until use.
For P. bianor, one female adult was sequenced using the Sequel II system (PacBio) in high-fidelity (HiFi) mode, one female adult was used for Hi-C sequencing, and one female adult was used for RNA-seq sequencing. For G. eurypylus, two male adults were sequenced using the PacBio in HiFi mode, two male adults were used for Hi-C sequencing, and one male adult was used for RNA-seq sequencing. For G. sarpedon and G. cloanthus, two female adults were sequenced using the PacBio in HiFi mode, two female adults were used for Hi-C sequencing, and one female adult was used for RNA-seq sequencing, respectively.
Library construction and PacBio sequencing
For P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus, genomic DNA was extracted from whole body using the standard cetyltrimethylammonium bromide method60, and sequenced using the PacBio in HiFi mode. SMRTbell library construction and sequencing were performed at Novogene (Tianjin, China) for G. eurypylus and Berry Genomics (Beijing, China) for P. bianor, G. sarpedon, and G. cloanthus, respectively, following the official protocols of PacBio for preparing ~20-kb SMRTbell libraries. After obtaining the sequence data, we processed the raw information with SMRT Link (version 8.0 https://github.com/PacificBiosciences/pbcommand), using the CCS method with default parameters. To filter out mitochondrial genome contamination, we removed sequences from HiFi reads that were aligned to the mitochondria using minimap2 (version 2.17-r974-dirty)61. For each read, if the alignment length accounted for more than 90% of the read, this read was filtered out before assembly.
Hi-C experiment and library construction
For Hi-C sequencing, we followed the standard protocol described previously with minor modifications. In brief, adult tissues from P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus were used to isolate cells. The cells were cross-linked and then homogenized by tissue lysis, and digested with the restriction enzyme MboI overnight. The proximal chromatin DNA was re-ligated using a ligation enzyme. The nuclear complexes were reversely cross-linked by incubation with proteinase K at 65 °C. DNA was purified using phenol-chloroform extraction, and biotin was removed from non-ligated fragment ends using T4 DNA polymerase. The ends of sheared fragments (300–500 base pairs) were repaired using a mixture of T4 DNA polymerase, T4 polynucleotide kinase, and Klenow DNA polymerase. Biotin-labeled Hi-C samples were specifically enriched using streptavidin C1 magnetic beads. After adding A-tails to the fragment ends and ligating Illumina paired-end (PE) sequencing adapters, Hi-C sequencing libraries were amplified by PCR (12–14 cycles) and sequenced on the Illumina NovaSeq sequencing platform at Novogene (Tianjin, China).
Genome assembly and chromosome construction
PacBio HiFi reads were used to perform de novo genome assembly for P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus using both Hifiasm (version 0.16.0-r369 with default parameters)62 and Canu (version 2.0 with parameters corresponding to different genome sizes estimated by flow cytometry)63. We chose the better results that were closer to the estimated genome size and had a higher N50 value by comparing the outputs of the two software programs. For the four butterfly species, one set of the two haploid contigs with longer length from the Hifiasm output was used for further assembly. For G. eurypylus, G. sarpedon, and G. cloanthus, because the HiFi reads from two adult individuals were mixed respectively, the haplotypes were purged using purge_dups (version 1.2.3)64. Finally, Hi-C sequencing data were used to anchor the assembled contigs into pseudochromosome molecules. For G. eurypylus, G. sarpedon, and G. cloanthus, the Hi-C data were first mapped to the purged genome assembly using Juicer (version 1.6)65. For P. bianor, the Hi-C data were first mapped to the raw genome assembly from the Hifiasm output using Juicer (version 1.6). Next, chromosome-level assembly was generated using 3D-DNA (version 180922)66 with default parameters. To further improve chromosome-scale assembly, manual review and refinement of the candidate assembly were performed using Juicebox (version 1.6) Assembly Tools.
Assembly quality evaluation
We evaluated the quality of the genome assemblies using five methods. First, we compared the genome sizes of the four assemblies to flow cytometry estimations. We measured G. cloanthus genome size (C-values) using flow cytometry. The genome size of G. cloanthus was found to be comparable to the flow cytometry estimation, as were the genome sizes of P. bianor, G. eurypylus, and G. sarpedon5. Second, we assessed genome completeness and consensus quality value (QV) using Merqury67. The QV score refers to a measure of the confidence in the accuracy of a consensus sequence in sequencing data. Third, we performed a BUSCO17 assessment of the genome sequences using the lepidoptera_odb10 database. Fourth, we mapped PacBio-HiFi reads to the assemblies using minimap268 and calculated the mapping ratios using samtools (version 1.9)69. Finally, we examined the telomeric regions in the four genomes using telomeric-identifier and searched for centromeric regions using BLAST (version 2.10.0)70.
Repeat annotation, LTR retrotransposon analysis, and gene annotation
We firstly detected and masked the repeats for the four butterfly species using RepeatModeler (version 1.0.4) and RepeatMasker following the previous publications9,20. Then we used both de novo and homology-based method to annotate genes following the previous publications9,20. Finally, we performed the gene function annotation by aligning their protein sequence to the SwissProt, KEGG and TrEMBL databases using BLAST software (version 2.10.0)70. The detailed steps for these can be found in Supplementary Note 1, 2.
Phylogenetic tree construction and divergence time estimation
The phylogenetic relationship of the four butterflies and outgroup species was reconstructed using two methods: coalescent and concatenation. For the concatenation method, we identified 4702 single-copy BUSCO genes from six species (including four butterflies, Bombyx mori, and Helicoverpa armigera) using BUSCO517. We extracted protein multiple sequence alignments (MSAs) of each single-copy BUSCO gene using Prank71 and converted the alignments into corresponding CDS MSAs. These were then concatenated to generate a super-gene alignment. We extracted fourfold degenerate sites (4dTV) from the remaining orthologs and used them to reconstruct a maximum-likelihood species tree using IQ-TREE2 (version 2.1.2)72.
For the coalescent method, we inferred a maximum-likelihood gene tree for each putative single-copy Busco gene group using IQ-TREE2 (version 2.1.2)72 and used ASTRAL (version 5.7.3)73 to infer species trees from the gene trees. We used MCMCTREE in the PAML package (version 4.8)74 to estimate divergence times based on 4dTV sites, using time calibration points from the TimeTree database and fossil records. The common ancestral node of H. armigera and B. mori was constrained to 63.4–122.1 Mya, while the common ancestral node of G. sarpedon and P. bianor was constrained to 49.2–62.8 Mya.
Comparative genomes and gene synteny analysis
For P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus, we used MCScanX75 with default parameters to identify syntenic blocks. We constructed chromosome-scale syntenic block plots and dot-plots using the python version of MCScan (https://github.com/tanghaibao/mcscan) and Circos76. Cactus (version v1.2.3)77 was used to perform genome alignments in four butterfly species.
Identification of recombination sites (RSs)
We determined RSs based on the alignment results of multiple species and single-copy homologous gene alignment using a previously described method with minor modifications9,11. Due to the collinearity and similar karyotypes between P. bianor and G. eurypylus, we only calculated the RSs intervals (>100 kb) for G. eurypylus-G. sarpedon and G. eurypylus-G. cloanthus. Briefly, for each alignment group, sequence alignment results were extracted from multi-species alignments obtained from Cactus (version v1.2.3)77 using LAST software (version 1061)78. Next, MAF-formatted files were processed to extract alignment information into bed format. Then we calculated RSs using Perl scripts following pervious research79. These Perl scripts determine the type and position of chromosome rearrangements based on the alignment information of upstream and downstream sequences. Then, we filtered out short RSs (less than 5 kb) that might have resulted from misalignment. For the remaining structure variations (SVs), we calculated the distance between adjacent RSs and filtered RSs with distances less than 100 kb. Collinear homologous gene blocks for two corresponding species were determined using blast (version 2.10.0)70, OrthoFinder (version 2.5.4)80, and MCScanX75. Finally, we intersected the RSs obtained above with gene collinear blocks. RSs located within gene collinear blocks were filtered as evidence of conflicting alignment relationships and gene collinearity.
Construction of normalized contact matrix
For P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus, high-quality Hi-C data were pre-processed using HiC-Pro (version 2.11.4)81 as described in a previous research9. In Brief, Hi-C reads were mapped to the reference genome using Bowtie282, and unmapped, singleton, and multihit reads were filtered out. The remaining reads were assigned to MboI restriction fragments, and valid read pairs were obtained by filtering out dangling ends, self-circles, ligation products, dumped pairs, and PCR artifacts. The genome was divided into specific size bins and valid pairs were counted per bin. We generated raw contact matrices at different resolutions (100 kb, 40 kb, 20 kb, 10 kb, 5 kb), which were then normalized using the iterative correction and eigenvector decomposition (ICE) method83. The valid pair files were transformed into .hic files using the pre command in Juicer (version 1.6)65 and then converted into .cool files at different resolutions using hicexplorer81 software. We defined and calculated the Hi-C map resolution as described in a previous method27. In brief, the map resolution is defined as the smallest bin size where 80% of bins have at least 1000 contact reads. The Hi-C data visualization in the article was generated using the Juicebox software.
Comparison of compartment A/B
To compare the A/B compartments of the four butterfly species, we assigned A/B compartments for the four butterfly species following published studies9,32. Briefly, Using the eigenvector command in Juicer (version 1.6)65, we identified the compartments based on KR-normalized matrices in “.hic” format at 100 kb resolution. This process involves the use of Principal Component Analysis (PCA), where the sign (positive or negative) of the first principal component (PC1) distinguishes chromatin regions into two distinct spatial compartments. The compartment characterized by higher gene density and higher gene expression was identified as the A compartment, while the other was designated as the B compartment.
We used homologous genes to determine changes in compartments between different species at two levels: changes in chromosome size and RSs. For chromosome size change, we used P. bianor as a reference and compared P. bianor-G. eurypylus, P. bianor-G. sarpedon, and P. bianor-G. cloanthus. For each homologous genes, if they were located in the same compartment type, we considered these compartments to be conserved; otherwise, they were considered non-conserved. For RSs, to eliminate interference introduced by genome amplification, we compared the compartments of G. eurypylus-G. sarpedon and G. eurypylus-G. cloanthus. For each RSs, we compared the compartments at the flank region of the RSs.
We also calculated the conservation of compartments in homologous regions near RSs (within 1 Mb) based on the results of genomic alignments by Cactus (version v1.2.3)77 following previous research with minor modifications9. Briefly, we utilized Cactus to identify homologous intervals among G. eurypylus, G. sarpedon, and G. cloanthus. Subsequently, given the relatively distant divergence time, we converted each 100 kb (compartment resolution) homologous windows near RSs (within 1 Mb) from G. cloanthus and G. sarpedon to G. eurypylus. Next, we intersected the converted windows from G. cloanthus and G. sarpedon with G. eurypylus. If more than 50% of a converted window from G. cloanthus and G. sarpedon interacted with any window of G. eurypylus, these two windows in the corresponding two species were defined as homologous windows. Finally, homologous windows located in the same compartment type across species were defined as conserved, while the others were marked as non-conserved.
TAD and subTAD calling
To compare the TADs and sub-TADs of different species, we first used the hicExplorer (version 3.0)84, insulation score85 and Domain callers6 to detect TAD and sub-TAD boundaries for the four butterfly genomes based on the normalized contact matrix at 40 kb and 10 kb resolution, respectively. Visual assessment suggested that the insulation scores85 (cworld-dekker, https://github.com/dekkerlab/cworld-dekker, with default parameters) method at 40 kb resolution and the hicExplorer software (based on the insulation score method, with default parameters) at 10 kb resolution provided the best results. Briefly, the insulation score method identifies TADs by calculating interaction insulation across genomic regions. This score measures the extent to which a genomic segment inhibits interactions across its boundaries. Regions with lower insulation scores indicate higher internal interactions, typically representing the interiors of TADs. In contrast, sharp increases in insulation scores often delineate TAD boundaries, signifying areas with diminished cross-boundary interactions. Then we used the above two software programs and their respective parameters to calculate TADs and sub-TADs, respectively.
Comparison of TAD and subTAD
We primarily investigated the effects of chromosomal rearrangements and changes in chromosome size on TADs. For TADs, we defined conserved and species-specific TADs as described in previous work with minor modifications32. Briefly, we performed comparative analyses of three groups, including G. cloanthus-G. sarpedon, G. cloanthus-G. eurypylus, and G. sarpedon-G. eurypylus, where the first genome mentioned in each group served as the reference genome and the second genome as the query genome. We used the UCSC LiftOver tool (with parameter -minMatch = 0.5) to convert genomic coordinates from one reference genome to another86. We lifted over query genome TAD boundaries (80 kb upstream and downstream of TAD boundaries region) to reference genome. Due to differences in sequences between species, TADs with one or both boundaries that were not aligned were filtered out. If two corresponding boundaries/boundaries between species in comparison was less than 80 kb (2 bins), these two boundaries were defined as conserved boundaries in the two species. Boundaries with a distance more than or equal 80 kb (2 bins) were defined as non-conserved boundaries. TADs flanked by conserved boundaries on both sides were defined as conserved TADs, while those flanked by species-specific boundaries on at least one side were defined as non-conserved TADs.
For sub-TADs, we defined conserved and species-specific sub-TADs using the methods above with minor modifications (the lift region of TAD boundaries was 20 kb upstream and downstream region). Briefly, if two corresponding boundaries/boundaries between species in comparison was less than 20 kb (2 bins), these two boundaries were defined as conserved boundaries in the two species. Boundaries with a distance more than or equal to 20 kb were defined as non-conserved boundaries.
For changes in chromosome size, the three Graphium species were used as reference genome, respectively, and we compared P. bianor vs G. eurypylus, P. bianor vs G. sarpedon, and P. bianor vs G. cloanthus. For RSs, to eliminate interference introduced by genome amplification, we only compared G. eurypylus vs G. sarpedon and G. eurypylus vs G. cloanthus.
Comparison of loops
For P. bianor, G. eurypylus, G. sarpedon, and G. cloanthus, chromatin loops were calculated using Hi-C computational UnbiasedPeakSearch (HiCCUPS) with 5 kb resolutions as described in the previous study65. Loops located at centromeric and telomeric regions were removed. For chromatin loops, we transformed the coordinates of both sides of the loops of three groups, including G. cloanthus-G. sarpedon, G. cloanthus-G. eurypylus, and G. sarpedon-G. eurypylus, where the first genome mentioned in each group served as the reference genome and the second genome as the query genome using liftOver86 software, respectively. Chromatin loops with one or both sides that were not aligned due to sequence differences between species were filtered out. If the distance of each side of chromatin loops in two species was less than 10 kb, this kind of chromatin loop was defined as a conserved loop. If the distance was greater than or equal to 10 kb, this kind of loop was defined as a non-conserved loop between the corresponding two species.
ATAC data and motif analysis
ATAC data of P. bianor were downloaded from our previous research48. Index of the P. bianor was built using bwa (version 0.7.17-r1198-dirty)87 and clean reads were aligned to the reference genome using Bowtie282. These reads were then filtered for high quality (MAPQ ≥ 13), we also removed reads that were not properly paired and with PCR duplicates by picard (version 2.25.7 https://broadinstitute.github.io/picard/). All peak calling was performed with MACS2 (version 2.1.0)88 using “–call-summits–nomodel –shift −100 –extsize 200”. The motif database was extracted from JASPAR89. Then, motif enrichment analysis was performed for ATAC-seq peaks using the fimo (version 5.3.0) from the MEME suite90. Only motifs with an enrichment p-value smaller than 1e−5 and q-value less than 0.01 were considered.
CRISPR/Cas9 mosaic shotgun CRE deletions
We selected the CTCF binding site in CRE 1 to be incised from the genome using CRISPR/Cas9. We designed three sgRNAs for the CTCF binding sites (Supplementary Table 14), following the motif N20NGG91, and purchased sgRNAs from Tsingke (Beijing Tsingke Biotech Co., Ltd). This “shotgun” approach of injecting multiple sgRNAs produce a range of different deletion lengths in and around individual CREs (Supplementary Table 15) in order to screen for different possible sequence-specific functions of the element. Mated females of P. xuthus were stimulated by placing a host plant (Zanthoxylum piperitum) in their cage for oviposition. Eggs were collected after 0.5 h, washed with sterile water for three times, glued to a slide and injected using capillary needles. All larva hatched from injected eggs were fed on host leaves (Zanthoxylum piperitum), reared at 27 °C, 16 h light/25 °C, 8 h darkness and kept 80% relative humidity. All the hatched larvae were carefully raised but most of them could not finish their larval developments because their abnormal heads, thorax and segments. The final number of injected eggs and surviving caterpillars are in the Supplementary Table 16. Our CRISPR mosaic “shotgun deletion” strategy uses three pooled sgRNAs to create indels of different sizes at targeted loci. We used Sanger sequencing to confirm the correct targeting of sgRNAs in vivo. DNA was extracted from whole bodies (or bodied with wing removed) of butterflies that showed mutant phenotypes using T1ANamp Genomic DNA Kit (Tiangen, Beijing, China). We then amplified target sequence using PCR with specific primer pairs (Supplementary Table 16), and then conducted TA cloning followed the methodology described in previous studies88. The PCR products with the expected size target fragment were sent to Sangon Biotech corporation for Sanger sequencing, and the results were viewed in IGV software to check the knock-out target sites (Supplementary Figs. 19 and 20).
Quantitative real-time PCR (qRT-PCR)
For qRT-PCR, we used qRT‐PCR to confirm the accuracy of the cDNA sequence. Total RNA was extracted from the 1st‐instar larvae using TRIzol Reagent (Invitrogen). We synthesized first‐strand cDNA using the PrimeScriptTM RT reagent Kit (Takara), then amplified full‐length cDNA sequences of Antp genes with gene‐corresponding primers. The qPCR was performed for Antp genes with gene‐corresponding primers (Px_Antp_2_F: CATGCCCTACCCCAGGTTTC, Px_Antp_2_R: CCCATACCCGTCAGACTTGG) using SYBR Green Real‐time PCR Master Mix (Takara) based on default conditions. β‐actin (F: GCACCACACCTTCTACAATGAGC; R: ATCCCTCATAGATGGGCACCGT) was used as the reference gene. For each qPCR and each treatment, we conducted three replicates and each replication includes 20 1st-instar larvae. The relative expression levels were calculated using the 2−ΔΔCt method. SPSS and GraphPad Prism software were used for statistical analyses of the expression patterns of Antp gene based on one‐way ANOVA followed by Tukey’s test.
RNA-Seq and data analysis
For each of four species, RNA was extracted from the whole body of one adult individual excluding intestinal tract and the libraries were constructed using the Illumina TruSeq RNA Library Preparation Kit and sequenced on the Illumina HiSeq 4000 platform with pair-end 150 bp reads. After filtering low-quality bases and sequence adapters, the clean RNA sequencing data were mapped to each genome using the hisat2. High-quality mapping reads were extracted using samtools (version 1.9)69 and PCR duplicates were filtered using PicardTools (version 2.20.2) (http://broadinstitute.github.io/picard/). The remaining reads were used to calculate the expression level of genes using stringtie (version 2.1.4)92 with TPM used for quantification of expression levels.
Data availability
All sequencing data that support the findings of this study and genome assemblies are available at National Center for Biotechnology Information (NCBI) under accession number PRJNA1009405. The Hi-C data of Drosophila used in this study are available in the NCBI Gene Expression Omnibus (GEO) under accession number GSE101317. The ATAC-seq data of P. bianor used in this study are available in the NCBI under accession number PRJNA728516. The Hi-C matrix data, genome annotation file and genome assemblies for G. cloanthus, G. sarpedon, G. eurypylus, and P. bianor are also deposited in Figshare database (https://doi.org/10.6084/m9.figshare.25702434.v2). Source data are provided with this paper.
Code availability
All code and software sources used in this study are publicly available on the Internet as described in the “Methods” with corresponding citations of references and Source data file.
References
Wallace, A. R. On the phenomena of variation and geographical distribution as illustrated by the Papilionidae of the Malayan region. Trans. Linn. Soc. Lond. 25, 1–71 (1865).
Wallace, A. R. Contributions to the Theory of Natural Selection: A Series of Essays (Macmillan, 1870).
Poulton, E. B. The Colours of Animals: Their Meaning and Use, Especially Considered in the Case of Insects. (Kegan Paul, 1890).
Maeki, K. & Makino, S. Chromosome numbers of some Japanese Rhopalocera. Lepid. N. 7, 36–38 (1953).
Liu, G. C. et al. Genome size variation in butterflies (Insecta, Lepidotera, Papilionoidea): a thorough phylogenetic comparison. Syst. Entomol. 45, 571–582 (2020).
Dixon, J. R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012).
Krefting, J., Andrade-Navarro, M. A. & Ibn-Salem, J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biol. 16, 87 (2018).
Fudenberg, G. & Pollard, K. S. Chromatin features constrain structural variation across evolutionary timescales. Proc. Natl Acad. Sci. USA 116, 2175–2180 (2019).
Yin, Y. et al. Molecular mechanisms and topological consequences of drastic chromosomal rearrangements of muntjac deer. Nat. Commun. 12, 6858 (2021).
Wang, Q., Sun, Q., Czajkowsky, D. M. & Shao, Z. Sub-kb Hi-C in D. melanogaster reveals conserved characteristics of TADs between insect and mammalian cells. Nat. Commun. 9, 188 (2018).
Lukyanchikova, V. et al. Anopheles mosquitoes reveal new principles of 3D genome organization in insects. Nat. Commun. 13, 1960 (2022).
Torosin, N. S., Anand, A., Golla, T. R., Cao, W. & Ellison, C. E. 3D genome evolution and reorganization in the Drosophila melanogaster species group. PLoS Genet. 16, e1009229 (2020).
Liao, Y., Zhang, X., Chakraborty, M. & Emerson, J. J. Topologically associating domains and their role in the evolution of genome structure and function in Drosophila. Genome Res. 31, 397–410 (2021).
Ghavi-Helm, Y. et al. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet. 51, 1272–1282 (2019).
Li, L. et al. Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol. Cell 58, 216–231 (2015).
Maeki, K. Notes on the W-Chromosome of the butterfly, Graphium sarpedon (Papilionidae, Lepidoptera). P. Jpn Acad. B. 57, 371–373 (1981).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Lu, S. et al. Chromosomal-level reference genome of Chinese peacock butterfly (Papilio bianor) based on third-generation DNA sequencing and Hi-C analysis. GigaScience 8, giz128 (2019).
He, J. W. et al. High-quality reference genomes of swallowtail butterflies provide insights into their coloration evolution. Zool. Res. 43, 367–379 (2022).
Wang, K. et al. African lungfish genome sheds light on the vertebrate water-to-land transition. Cell 184, 1362–1376.e1318 (2021).
Sirijovsky, N., Woolnough, C., Rock, J. & Joss, J. M. NfCR1, the first non-LTR retrotransposon characterized in the Australian lungfish genome, Neoceratodus forsteri shows similarities to CR1-like elements. J. Exp. Zool. 304, 40–49 (2005).
Fueyo, R., Judd, J., Feschotte, C. & Wysocka, J. Roles of transposable elements in the regulation of mammalian transcription. Nat. Rev. Mol. Cell Biol. 23, 481–497 (2022).
Gu, Z. et al. Enrichment analysis of Alu elements with different spatial chromatin proximity in the human genome. Protein cell 7, 250–266 (2016).
Chuong, E. B., Elde, N. C. & Feschotte, C. Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 18, 71–86 (2017).
Choudhary, M. N. et al. Co-opted transposons help perpetuate conserved higher-order chromosomal structures. Genome Biol. 21, 16 (2020).
Choudhary, M. N. K., Quaid, K., Xing, X., Schmidt, H. & Wang, T. Widespread contribution of transposable elements to the rewiring of mammalian 3D genomes. Nat. Commun. 14, 634 (2023).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Emerson, D. J. et al. Cohesin-mediated loop anchors confine the locations of human replication origins. Nature 606, 812–819 (2022).
Bhat, K. H. et al. An Igh distal enhancer modulates antigen receptor diversity by determining locus conformation. Nat. Commun. 14, 1225 (2023).
Zhang, H. et al. Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature 576, 158–162 (2019).
Phillips-Cremins, J. E. et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295 (2013).
Luo, X. et al. 3D Genome of macaque fetal brain reveals evolutionary innovations during primate corticogenesis. Cell 184, 723–740.e721 (2021).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Wu, P. et al. 3D genome of multiple myeloma reveals spatial genome disorganization associated with copy number variations. Nat. Commun. 8, 1937 (2017).
Krietenstein, N. et al. Ultrastructural details of mammalian chromosome architecture. Mol. Cell 78, 554–565.e7 (2020).
Lebeau, B. et al. 3D chromatin remodeling potentiates transcriptional programs driving cell invasion. Proc. Natl Acad. Sci. USA 119, e2203452119 (2022).
Hedengren, M. et al. Relish, a central factor in the control of humoral but not cellular immunity in Drosophila. Mol. Cell 4, 827–837 (1999).
Liu, Y. et al. Inflammation-induced, STING-dependent autophagy restricts Zika virus infection in the Drosophila brain. Cell Host Microbe 24, 57–68 (2018).
Stöven, S., Ando, I., Kadalayil, L., Engström, Y. & Hultmark, D. Activation of the Drosophila NF-kappaB factor Relish by rapid endoproteolytic cleavage. EMBO Rep. 1, 347–352 (2000).
Goto, A. et al. The kinase IKKβ regulates a STING- and NF-κB-dependent antiviral response pathway in Drosophila. Immunity 49, 225–234.e4 (2018).
Lu, J. Y. et al. Homotypic clustering of L1 and B1/Alu repeats compartmentalizes the 3D genome. Cell Res. 31, 613–630 (2021).
Mao, Y., Kucuk, B. & Irvine, K. D. Drosophila lowfat, a novel modulator of Fat signaling. Development 136, 3223–3233 (2009).
Hogan, J., Valentine, M., Cox, C., Doyle, K. & Collier, S. Two frizzled planar cell polarity signals in the Drosophila wing are differentially organized by the Fat/Dachsous pathway. PLoS Genet 7, e1001305 (2011).
Marlétaz, F. et al. The little skate genome and the evolutionary emergence of wing-like fins. Nature 616, 495–503 (2023).
De, S. et al. A high-quality Ixodes scapularis genome advances tick science. Nat. Genet. 55, 301–311 (2023).
Negre, B., Ranz, J. M., Casals, F., Cáceres, M. & Ruiz, A. A new split of the Hox gene complex in Drosophila: relocation and evolution of the gene labial. Mol. Biol. Evol. 20, 2042–2054 (2003).
Scott, M. P. & Weiner, A. J. Structural relationships among genes that control development: sequence homology between the Antennapedia, Ultrabithorax, and fushi tarazu loci of Drosophila. Proc. Natl Acad. Sci. USA 81, 4115–4119 (1984).
Wan, W. T. et al. Genome-wide survey of open chromatin regions in two swallowtail butterflies Papilio machaon and P. bianor. Arch. Insect Biochem. Physiol. 111, e21952 (2022).
Kyrchanova, Olga et al. Architectural protein Pita cooperates with dCTCF in organization of functional boundaries in Bithorax complex. Development 144, dev.149815 (2017).
Li, X. et al. Outbred genome sequencing and CRISPR/Cas9 gene editing in butterflies. Nat. Commun. 6, 8212 (2015).
Fang, C. et al. The Hox gene Antennapedia is essential for wing development in insects. Development 149, dev199841 (2022).
Brena, C., Chipman, A. D., Minelli, A. & Akam, M. Expression of trunk Hox genes in the centipede Strigamia maritima: sense and anti-sense transcripts. Evol. Dev. 8, 252–265 (2006).
Glicksman, M. A. & Brower, D. L. Misregulation of homeotic gene expression in Drosophila larvae resulting from mutations at the extra sex combs locus. Dev. Biol. 126, 219–227 (1988).
Zhang, H. et al. Expression patterns of the homeotic genes Scr, Antp, Ubx, and abd-A during embryogenesis of the cricket Gryllus bimaculatus. Gene Exp. Pattern. 5, 491–502 (2005).
Chen, H. et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 11, 1–11 (2020).
Kentepozidou, E. et al. Clustered CTCF binding is an evolutionary mechanism to maintain topologically associating domains. Genome Biol. 21, 5 (2020).
Pazhenkova, E. A. & Lukhtanov, V. A. Whole-genome analysis reveals the dynamic evolution of holocentric chromosomes in Satyrine butterflies. Genes 14, 437 (2023).
Renschler, G. et al. Hi-C guided assemblies reveal conserved regulatory topologies on X and autosomes despite extensive genome shuffling. Genes Dev. 33, 1591–1612 (2019).
Parham, S. et al. Antioxidant, antimicrobial and antiviral properties of herbal materials. Antioxidants 9, 1309 (2020).
Healey, A. et al. A simple method for extracting next generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods 10, 1–8 (2014).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Nurk, S. et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30, 1291–1305 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. 10, 421 (2009).
Löytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 1079, 155–170 (2014).
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Zhang, C., Rabiee, M., Sayyari, E. & Mirarab, S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19, 15–30 (2018).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Armstrong, J. et al. Progressive Cactus is a multiple-genome aligner for the thousand-genome era. Nature 587, 246–251 (2020).
Kielbasa, S. M., Wan, R., Sato, K., Horton, P. & Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493 (2011).
Chen, L. et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 364, eaav6202 (2019).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Imakaev, M. et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003 (2012).
Wolff, J. et al. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 48, W177–W184 (2020).
Crane, E. et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244 (2015).
Hinrichs, A. S. et al. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 34, D590–D598 (2006).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Castro-Mondragon, J. A. et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 50, D165–D173 (2022).
Grant, C. E., Bailey, T. L. & Noble, W. S. FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011).
Liu, G. et al. Genome-wide identification and gene-editing of pigment transporter genes in the swallowtail butterfly Papilio xuthus. BMC Genomics 22, 120 (2021).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 278 (2019).
Acknowledgements
We thank Wenting Wan, Jinwu He, Jianfeng Huang, Chuyang Mao from the Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China for their grateful help in assistance with the experiments. We thank Jianan Xu from Yunnan Agricultural University, Kunming, China for his grateful help in sample collection. We thank local Forestry Department and National Reserve of China for permission to undertake field surveys and for other assistance. This study was supported by grants from the National Natural Science Foundation of China (32070482), Yunnan Provincial Science and Technology Department (Talent Project of Yunnan: 202105AC160039), Biodiversity Conservation Program of the Ministry of Ecology and Environment, China (China BON-Butterflies) and the Program of Yunnan Forestry and Grassland Administration (2022GF258D-11) to X.Y.L.; New Cornerstone Foundation to W.W.
Author information
Authors and Affiliations
Contributions
X.Y.L. and W.W. conceived and designed the project. B.T.Z., W.W., and X.Y.L. drafted the manuscripts. B.T.Z., P.H., Y.Y., W.W., and X.Y.L. revised the manuscript. Z.C. and Z.W.D. collected and prepared the samples, help in assistance with the experiment. P.H. and G.C.L. designed and performed all experiments. B.T.Z., Z.Z.T., Z.H.L., and G.H. performed the data analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Steven van Belleghem and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, B., Hu, P., Liu, G. et al. Evolutionary patterns and functional effects of 3D chromatin structures in butterflies with extensive genome rearrangements. Nat Commun 15, 6303 (2024). https://doi.org/10.1038/s41467-024-50529-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50529-0
- Springer Nature Limited