

Abstract
Real-time genomics through nanopore sequencing holds the promise of fast antibiotic resistance prediction directly in the clinical setting. However, concerns about the accuracy of genomics-based resistance predictions persist, particularly when compared to traditional, clinically established diagnostic methods. Here, we leverage the case of a multi-drug resistant Klebsiella pneumoniae infection to demonstrate how real-time genomics can enhance the accuracy of antibiotic resistance profiling in complex infection scenarios. Our results show that unlike established diagnostics, nanopore sequencing data analysis can accurately detect low-abundance plasmid-mediated resistance, which often remains undetected by conventional methods. This capability has direct implications for clinical practice, where such “hidden” resistance profiles can critically influence treatment decisions. Consequently, the rapid, in situ application of real-time genomics holds significant promise for improving clinical decision-making and patient outcomes.
Similar content being viewed by others


Introduction
The World Health Organization has declared antibiotic resistance one of the ten most severe global health threats1, with resistant infections leading to higher mortality and morbidity due to delayed or inappropriate therapy2. The rapid and accurate identification of resistant bacterial pathogens could facilitate the earlier administration of appropriate therapy, decreasing the mortality rate and infection- and treatment-related morbidity3.
Real-time genomics, powered by nanopore sequencing technology, offers the potential to expedite pathogen identification and antibiotic resistance profiling directly within clinical settings4,5. The portability of this technology, coupled with its capability for real-time analysis, enables cost-efficient adaptive applications, where as much genomic data as needed can be directly obtained on-site to reach minimum certainty thresholds for making timely and clinically relevant predictions6. However, for real-time genomics to be integrated into routine clinical practice, its accuracy in predicting antibiotic resistance must be directly compared with that of established diagnostic approaches7. While several proof-of-concept studies have showcased the feasibility of using nanopore sequencing for rapid infectious disease diagnostics in clinical settings5,6,7,8,9, it remains to be proven that real-time genomics can outperform established diagnostics in detecting clinically relevant resistance.
Nanopore sequencing’s capability to produce long reads can be leveraged to create highly accurate, near-complete genome assemblies for strain-level identification and de novo detection of bacterial pathogens and their antibiotic resistance profiles10. This is particularly pertinent for complex infections, where clinically established methods for taxonomic bacterial identification (e.g., MALDI-TOF mass spectrometry) and resistance profiling (e.g., VITEK2) might lack resolution, but where rapid, targeted therapy can be particularly beneficial for patient outcome.
Here, we show the power of in situ real-time genomics in a clinical setting through the example of a Klebsiella pneumoniae infection for which real-time genomics-based resistance predictions—in contrast to clinically established diagnostics—could identify a novel antibiotic resistance gene variant located on low-abundance plasmids. This finding has significant implications for clinical decision-making and potentially for patient outcomes, illustrating the transformative potential of integrating real-time genomic analysis into clinical practice.
Results
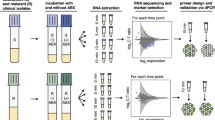
We conducted a comparative analysis of the performance between clinically established diagnostics and real-time genomics-based predictions using bacterial isolates from the same infection case. Our established diagnostic methods included MALDI-TOF MS for taxonomic bacterial identification and VITEK2 for antibiotic resistance profiling (Methods; Fig. 1). The case study involved an immunocompromised patient at the University Hospital rechts der Isar in Germany, who presented with a fever and was initially treated with the carbapenem antibiotic Meropenem.

After incubation and primary pathogen identification, pure bacterial isolates are recovered through sub-culturing, followed by pathogen and resistance profiling of the isolates. While the clinically established workflow can take up to 52 h after subculturing, the real-time genomic workflow delivers the first data after ~1.5 h and can be employed in an adaptive manner to create the necessary amount of data. Created with Biorender.com.
This comparative approach allowed us to directly assess the accuracy and speed of real-time genomic predictions against traditional methods in a real-world clinical scenario, highlighting the potential advantages of genomic technologies in rapid and accurate pathogen identification and resistance prediction.
Clinically established diagnostics
Initially, an endotracheal aspirate sample was collected (“pre-treatment” sample), from which clinically established diagnostics identified a carbapenem-resistant K. pneumoniae isolate, with K. pneumoniae carbapenemase (KPC) as the putative resistance-conferring mechanism (Methods; Fig. 2). KPCs can hydrolyze a variety of beta-lactam antibiotics, including carbapenems11. Consequently, Ceftazidime-Avibactam (CAZ-AVI) is recommended as a treatment option12 due to its efficacy against such resistant strains; in this specific case, following the diagnostics that also predicted CAZ-AVI susceptibility, the treatment was promptly switched from Meropenem to CAZ-AVI (Methods; Fig. 2). After initial clinical improvement, the patient’s condition deteriorated under CAZ-AVI therapy, and a subsequent blood culture taken from the patient (“post-treatment” sample) grew a K. pneumoniae isolate which now showed restored in-vitro carbapenem susceptibility but CAZ-AVI resistance (Fig. 2). Notably, while new KPCs variants that confer resistance to CAZ-AVI have been documented13,14,15, the clinically established diagnostics failed to detect any carbapenemase in the post-treatment isolate. Consequently, Meropenem was reintroduced as part of the treatment regimen. Despite these measures, the patient passed away shortly afterwards (Fig. 2).

The patient was firstly treated with Meropenem. K. pneumoniae bacterial isolates of the first positive patient sample (endotracheal aspirate) were subjected to VITEK2 for general resistance testing and additional tests for CAZ-AVI resistance and KPC detection (R resistant, S susceptible; KPC + /-: absence or presence of KPC; Methods); the diagnostics led to a change in the antibiotic treatment to CAZ-AVI after three days. After clinical deterioration, the second isolate (from blood culture) showed reversed antibiotic resistance test results. While Meropenem was subsequently administered, the patient passed away shortly after. After completion of the routine diagnostics, we used real-time genomics to sequence DNA from the pre-and post-CAZ-AVI treatment bacterial isolates using the portable nanopore sequencing device Mk1b (Methods). Both isolates were sequenced for 15 h, and the first isolate was sequenced for another 8 h to simulate the potential of adaptive sequencing in the clinical setting (Methods). Created with Biorender.com.
Real-time genomics diagnostics
To explore the potential of real-time genomics in antibiotic resistance prediction, we simulated its application for this clinical case as follows. We applied rapid nanopore shotgun sequencing (Oxford Nanopore Technologies) on both the pre-and post-treatment K. pneumoniae bacterial isolates (Methods) using the portable Mk1b sequencing device and rapid barcoding library preparation (Fig. 2; Supplementary Table 1; Methods). We processed the raw nanopore data through high-accuracy basecalling, de novo genome assembly, species identification, and antibiotic resistance prediction using EPI2ME’s Antimicrobial Resistance protein homolog model16 (Methods; Supplementary Data 1). Our analysis correctly identified K. pneumoniae in the pre-and post-treatment isolates as the causative pathogen. In the pre-treatment isolate, we detected accurate (>90%) blaKPC-2 gene copies (n = 40; Table 1), confirming the Meropenem resistance observed by clinically established diagnostics (Fig. 2)17. For the post-treatment isolate, we identified numerous copies of the blaKPC-14 (n = 44; Table 1), which had previously been recognized as one of the few KPC subtypes that confer CAZ-AVI resistance while potentially restoring in-vitro carbapenem susceptibility14. Hence, the genomics-based resistance prediction not only aligned with the resistance patterns identified by traditional diagnostics but also pinpointed blaKPC-14 as the putative resistance-conferring mechanism (Table 1).
Crucially, our real-time genomics approach also detected a single copy of the blaKPC-14 resistance gene in the pre-treatment isolate. Although this one copy would not have been sufficient to predict CAZ-AVI resistance initially, this case served as a valuable example to simulate the adaptive nature of real-time genomics applications in the clinic, where a sample can be sequenced as long as necessary to obtain the necessary minimum data for reliable genomics-based predictions. For this purpose, we conducted additional sequencing of a technical replicate of the pre-treatment isolate for another eight hours (Methods; Fig. 2; Supplementary Table 1), which resulted in the identification of four more highly accurate copies of blaKPC-14 (Table 1). Remarkably, a second blaKPC-14 gene copy would have already been detected after two hours of additional sequencing and would have rapidly indicated the potential of CAZ-AVI resistance (Fig. 3; Methods).

The run started with one blaKPC-14 copy number which was detected in the first sequencing run, and all additional blaKPC-14 copies were detected within the first seven hours of this second sequencing run.
Epidemiological and functional analyses
We successfully created de novo assemblies of one complete chromosome and three complete circular plasmids from both the pre- and post-treatment isolate (Fig. 4; Methods). Core-genome multilocus sequence typing (cgMLST; Methods)18,19 revealed that both isolates were of the emerging high-risk sequence type ST14720. Single-linkage clustering analysis identified no close relatives within the 50-allele threshold typically used for cgMLST clustering (Methods), indicating that our bacterial isolates are genetically distinct from globally known ST147 genomes.

Top: The assemblies are annotated by respective contig length; the IncN plasmid is highlighted by the blue square. Additional non-circular contigs are visualized in grey color. Created with Bandage v0.9034. Bottom: Functional annotation and visualisation of the IncN plasmids highlighting open reading frames (ORF) and plasmid functionality genes (integration/excision, transfer, replication/recombination/transfer) from the mobileOG-db (Methods). The alignment to the respective other IncN plasmid (pre- vs. post-treatment sample) is shown in grey color. Created with ProkSee.com35.
Functional annotation of the assembled plasmid genomes revealed that the blaKPC-2 (pre-treatment isolate) and blaKPC-14 (post-treatment isolate) gene were located on IncN plasmids, which were 99.7%-identical according to sequence alignments (E-score 0, Bit-Score >1.461e + 5; Methods; Fig. 4). Additionally, both IncN plasmids shared key plasmid features (relaxase type: MOBF, mpf type: MPF_T, orit type: MOBF), and were predicted to be conjugative21,22.
We further inferred a copy-number of three and four for the IncN plasmids relative to the bacterial chromosome in the pre-and post-treatment isolates, respectively (Methods). To assess the changes in the abundance of blaKPC-14 gene between pre- and post-treatment isolates, we further normalized the blaKPC-14 copy-numbers against the most abundant resistance gene (blaTEM-4) detected on the IncN plasmid (Methods). We observed that the normalized abundance of blaKPC-14 increased from 0.56% to 26.6% following CAZ-AVI exposure.
Upon submission of the post-treatment bacterial isolate to the German National Reference Center for Gram-negative bacteria, the KPC resistance gene that we initially defined as blaKPC-14 was identified as a previously undocumented KPC subtype and subsequently named blaKPC-159 (NCBI sequence ID: OQ450354.1). To confirm this, we utilised the BLASTn tool and established that blaKPC-159 shows 99.9% similarity in nucleotide sequence (875/876 bases) with the query sequence of blaKPC-14 (Methods), thus, leading to the classification of blaKPC-159 as blaKPC-14.
Discussion
The application of real-time genomics to this patient’s case underscores the considerable potential for using this technology to rapidly and accurately profile complex bacterial infections in the clinical setting. Our findings suggest that the shift in in-vitro antibiotic resistance was likely due to a complex infection involving the same K. pneumoniae lineage with a low-abundance blaKPC-14-carrying IncN plasmid which became dominant due to its evolutionary selective advantage under CAZ-AVI exposure. We have shown that nanopore sequencing could have unveiled the CAZ-AVI resistance that phenotypic methods failed to detect, thereby influencing the therapeutic approach, such as the early administration of alternative antibiotics or combination therapy23. Further, identifying the blaKPC gene as the underlying CAZ-AVI resistance mechanism would have directly informed clinical infection prevention protocols, reducing the risk of between- and within-patient KPC transmission. The fast, adaptive, and in situ nature of antibiotic resistance profiling by nanopore sequencing would have surpassed current clinical practice in accurately informing clinical management.
Importantly, our genomic data also enabled us to trace changes in antibiotic resistance to variations in the copy-number of plasmids within the patient’s initial infection. The presence of multiple plasmids in an infection raises public health concerns as it can accelerate the emergence of resistance under selection pressures24. This has previously been described as a key factor in the rapid emergence of resistance to the last-resort antibiotic CAZ-AVI under drug pressure24. The anticipated increase in antibiotic resistance and the limitations of current diagnostic methods to fully assess complex infections pose significant challenges to effective antibiotic treatment strategies.
While our analysis provides evidence for the added value of real-time genomics for complex bacterial infections, two significant limitations to our work remain. Firstly, focusing on bacterial isolates may have limited our ability to fully understand the microbial diversity of the patient’s infection. A metagenomic sequencing approach using direct patient samples, rather than cultured bacteria, could have identified non-culturable organisms that might have influenced the antibiotic resistance profile7. Secondly, our analysis was confined to just two bacterial isolates from the same patient over an extended period. A larger sample size and more frequent sampling might have provided more detailed insights into how plasmid selection evolved in response to the antibiotic selection pressure. Unfortunately, due to the retrospective nature of this study, we were unable to adjust the sample size or obtain direct patient samples.
Current clinically established diagnostics are often too slow to promptly inform clinical management and require substantial initial investments in technology5,6,7. Our study showcases how cost-efficient, rapid real-time genomics can outperform established diagnostics in accuracy for predicting antibiotic resistance. Further research is needed to transition from phenotypic resistance testing to genomics-based predictions fully. However, we already now anticipate great potential for combining the advantages of real-time genomic technology with clinically established approaches for antibiotic resistance profiling in the hospital setting. We further envision that the ongoing improvements in sequencing accuracy and the relatively low investment required for nanopore sequencing technology25 offer promising prospects for managing complex infection cases worldwide, particularly in low- and middle-income settings where advanced diagnostic equipment may not be readily available.
Methods
Clinically established workflow for species identification and antibiotic susceptibility testing
All ethical approval for the following clinical research was given by the ethics committee of the Technical University of Munich, Germany (2023-575-W-NP, 2022-611-S-KH). Informed patient consent was waived as samples were taken under routine diagnostics. This research conforms to the principles of the Helsinki Declaration.
Initially, the clinical samples were plated out on BD® Columbia Blood and MacConkey agar plates (Beck Dickinson GmbH, Heidelberg, Germany) and incubated for approximately 16–24 h. Following subculture, species identification was done from a single colony forming unit (CFU) of pure bacterial isolates using Matrix-assisted laser desorption ionization time-of-flight mass spectrometry instructions (MALDI-TOF MS, Bruker Daltronics GmbH, Leipzig Germany), as per the manufacturer’s instructions.
Antibiotic susceptibility testing was performed using VITEK2 (BioMérieux, Marcy l’Etoile, France). For this, up to three bacterial CFUs were transferred to a saline tube to generate a homogenous suspension with a density equivalent to 0.5 McFarland. Subsequently, Minimum Inhibitory Concentrations (MICs in mg/L) of the two isolates were determined using the VITEK2 gram-negative (AST-GN69) card, and the results interpreted according to the European Committee on Antimicrobial Susceptibility Testing (EUCAST) guidelines26 (Supplementary Table 2).
Given that the AST-GN69 card does not detect the presence of carbapenemases or measure MICs for CAZ-AVI, we additionally performed the following tests. For isolates with antibiotic susceptibility profiles indicative of carbapenem resistance, we identified carbapenemases with a multiplex immunochromatography assay consisting of lateral flow assays (O.K.N.V.I Resist-527). This test detects the presence of Oxa-48, NDM, VIM, IMP-carbapenemases and the most prevalent KPC subtypes (e.g., KPC-2, KPC-3)28,29. CAZ-AVI MIC was measured with the Liofilchem® MIC test strip (MTSTM) (LIOFILCHEM s.r.l., Roseto degli Abruzzi, Italy), which contains Ceftazidime concentrations ranging from 0,016–256 μg/ml with a fixed Avibactam concentration of 4 μg/ml. Similar to the VITEK2 MIC measurements, up to three bacterial CFUs were transferred to a sterile saline tube to form a homogenised suspension with a density equivalent to 0.5 McFarland. This suspension was then plated out on a BD® Muller Hinton Agar using sterile cotton-tipped swabs and incubated with a CAZ-AVI Liofilchem® MTSTM for 16 h. MICs were interpreted following EUCAST guidelines (Supplementary Table 2). After completing these diagnostic steps, five to ten CFUs of each isolate were stored at −80 °C for future use.
Real-time genomic data generation
The stored isolates were thawed and grown overnight at 37 °C on BD® Columbia blood agar plates. In the first round of sequencing, we isolated DNA from ten CFUs of the pre-and post-treatment isolates using an automated magnetic-bead-based DNA purification approach through the Maxwell® RSC Blood DNA extraction protocol for Gram-negative bacteria with the Promega Maxwell®RSC 48 Instrument (Promega Corporation, Madison, USA). DNA concentrations were measured using the QubitTM (Thermo Fischer Scientific, Waltham, USA) dsDNA HS kit according to the manufacturer’s instructions. Nanopore sequencing libraries of both samples were generated using the SQK-RBK004 Rapid Barcoding Kit and sequenced on an Oxford Nanopore Technologies MinION MK1b device with R9.4.1 flow cells for 15 h28. For the second round of sequencing of the pre-treatment isolate, we subcultured the stored isolate again, generating a technical replicate of the pre-treatment isolate. We then extracted DNA from circa 50 CFUs and sequenced the extracted DNA for 8 h (Supplementary Fig. 1).
Real-time genomic data analysis
An overview of the real-time genomic data analysis is presented in Supplementary Fig. 1. All computational analyses were conducted on a portable laptop with an 8 GB NVIDIA GeForce RTX 4070 GPU, 16 GB 5200 MHz RAM, and an Intel i7-13800H CPU with 14 cores and 20 threads.
The raw nanopore data was basecalled using Guppy v6.3.2, using the “High-accuracy” model (r9.4.1_450bps_hac). We used Porechop v0.2.3 (https://github.com/rrwick/Porechop) to trim the adapter sequences and filtered out low-quality reads (Q < 9) and short sequences (< 200 bp) using Nanofilt v2.8.0 (https://github.com/wdecoster/nanofilt). Sequencing summaries were generated using NanoStat v1.6.0 (https://github.com/wdecoster/nanostat)30.
Subsequent analyses involved the EPI2ME Fastq Antimicrobial Resistance (v2023.04.26–1808834) workflow, which includes quality control of the filtered and trimmed reads, taxon identification via the WIMP (What’s in My Pot; v2023.06.13-1865548) workflow, based on the NCBI RefSeq database and Centrifuge31, and resistance gene identification using the Comprehensive Antibiotic Resistance Database (CARD)16. We retained only the resistance gene detections identified by the workflow’s protein homolog model, which is the most conservative model of the Fastq Antimicrobial Resistance workflow16, and excluded resistance genes with detection accuracy below 90% according to the protein homolog model to minimize the false positive rate.
Subsequently, we created de novo assemblies using Flye v2.9.1 (https://github.com/fenderglass/Flye)10,28. These were polished using Minimap2 v2.1832 and Racon v1.5 (https://github.com/isovic/racon). We assessed assembly coverage using SAMtools depth v1.19.2 (https://github.com/samtools/samtools)33. We then analyzed our de novo assemblies using the Pathogenwatch v2.3.118 platform which integrates Kleborate19 for Klebsiella species complex assignments and identification of acquired virulence factors and recognized resistance markers. We additionally used Pathogenwatch for core genome multi-locus sequence typing (cgMLST)18,19 of our assemblies based on the Life Identification Number (LIN) code scheme for the assignment of sublineages and clonal groups.
Plasmid detection and annotation
We visualized our assembly graphs using Bandage v0.9034 (Fig. 4), and identified the chromosomal and plasmid genomes. The plasmid functional annotation was done using MOB-suite v3.1.8 and visualized using the mobileOG-db17 implemented in ProkSee35 (Fig. 4). We used the MOB-typer modules from the Mob-suite program22 to identify key mobilization genes (relaxase), origin of transfer (oriT), mate-pair formation (MPF).
To estimate plasmid copy-number, we calculated the ratio of plasmid replicon sequencing depth to the sequencing depth of the respective chromosomal contig36. To accurately estimate the normalized abundance of specific resistance genes per plasmid, we extracted contig-specific read IDs using SAMtools v1.19.233, retrieved the respective sequencing reads from the processed fastq files using SeqKit v2.8.037, and calculated the copy-number ratio of the resistance gene of interest in comparison with the most abundant resistance gene identified on the same plasmid.
Reference center annotation
The post-treatment bacterial isolate was submitted to the German National Reference Center for Gram-negative bacteria (https://memiserf.medmikro.ruhr-uni-bochum.de/nrz), where our blaKPC-14 gene variant was identified as a previously undetected CAZ-AVI-resistant KPC subtype with reduced carbapenem-hydrolysing activity using short-read whole-genome sequencing (Illumina MiSeq). This KPC subtype is now registered as blaKPC-159 (NCBI sequence ID: OQ450354.1).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The nanopore sequencing data generated in this study have been deposited in the NCBI database (https://www.ncbi.nlm.nih.gov, SRA submission: PRJNA1041345).
Code availability
All computational scripts are available https://github.com/Genomics4OneHealth/AMR_nanopore/releases/tag/v0.
References
EClinicalMedicine. Antimicrobial resistance: a top ten global public health threat. EClinicalMedicine 41, 101221 (2021).
Murray, C. J. L. et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
Alberto, L., Marshall, A. P., Walker, R. & Aitken, L. M. Screening for sepsis in general hospitalized patients: a systematic review. J. Hospital Infection 96, 305–315 (2017).
Taxt, A. M., Avershina, E., Frye, S. A., Naseer, U. & Ahmad, R. Rapid identification of pathogens, antibiotic resistance genes and plasmids in blood cultures by nanopore sequencing. Sci. Rep. 10, 7622 (2020).
Harris, P. N. A. et al. Rapid nanopore sequencing and predictive susceptibility testing of positive blood cultures from intensive care patients with sepsis. Microbiol. Spectr. 12 https://doi.org/10.1128/spectrum.03065-23 (2024).
Bloomfield, M. et al. Oxford nanopore next generation sequencing in a front-line clinical microbiology laboratory without on-site bioinformaticians. Pathology https://doi.org/10.1016/j.pathol.2023.07.014 (2023).
Charalampous, T. et al. Routine Metagenomics Service for ICU Patients with Respiratory Infection. Am. J. Respir. Crit. Care Med. 209, 164–174 (2024).
Whittle, E. et al. Optimizing Nanopore Sequencing for Rapid Detection of Microbial Species and Antimicrobial Resistance in Patients at Risk of Surgical Site Infections. mSphere 7 https://doi.org/10.1128/msphere.00964-21 (2022).
Charalampous, T. et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat. Biotechnol. 37, 783–792 (2019).
Foster-Nyarko, E. et al. Nanopore-only assemblies for genomic surveillance of the global priority drug-resistant pathogen, Klebsiella pneumoniae. Microb. Genom 9, (2023).
Hammoudi Halat, D. & Ayoub Moubareck, C. The Current Burden of Carbapenemases: Review of Significant Properties and Dissemination among Gram-Negative Bacteria. Antibiotics 9, 186 (2020).
Tumbarello, M. et al. Ceftazidime-Avibactam Use for Klebsiella pneumoniae Carbapenemase–Producing K. pneumoniae Infections: A Retrospective Observational Multicenter Study. Clin. Infect. Dis. 73, 1664–1676 (2021).
Wu, Y. et al. Identification of a KPC Variant Conferring Resistance to Ceftazidime-Avibactam from ST11 Carbapenem-Resistant Klebsiella pneumoniae Strains. Microbiol Spectr 10 https://doi.org/10.1128/spectrum.02655-21 (2022).
Niu, S. et al. A Ceftazidime-Avibactam-Resistant and Carbapenem-Susceptible Klebsiella pneumoniae Strain Harboring bla KPC-14 Isolated in New York City. mSphere 5, e00775–20 (2020).
Shi, Q. et al. Multiple Novel Ceftazidime-Avibactam-Resistant Variants of bla KPC-2 -Positive Klebsiella pneumoniae in Two Patients. Microbiol Spectr. 10, e0171421 (2022).
Alcock, B. P. et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res 51, D690–D699 (2023).
Mehta, S. C. et al. KPC-2 β-lactamase enables carbapenem antibiotic resistance through fast deacylation of the covalent intermediate. J. Biol. Chem. 296, 100155 (2021).
Argimón, S. et al. Rapid Genomic Characterization and Global Surveillance of Klebsiella Using Pathogenwatch. Clin. Infect. Dis. 73, S325–S335 (2021).
Lam, M. M. C. et al. A genomic surveillance framework and genotyping tool for Klebsiella pneumoniae and its related species complex. Nat. Commun. 12, 4188 (2021).
Peirano, G., Chen, L., Kreiswirth, B. N. & Pitout, J. D. D. Emerging Antimicrobial-Resistant High-Risk Klebsiella pneumoniae Clones ST307 and ST147. Antimicrob. Agents Chemother. 64, e01148–20 (2020).
Brown, C. L. et al. mobileOG-db: a Manually Curated Database of Protein Families Mediating the Life Cycle of Bacterial Mobile Genetic Elements. Appl Environ. Microbiol 88, e0099122 (2022).
Robertson, J. & Nash, J. H. E. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Micro. Genom. 4, e000206 (2018).
Lombardo, D., Ambretti, S., Lazzarotto, T. & Gaibani, P. In vitro activity of imipenem-relebactam against KPC-producing Klebsiella pneumoniae resistant to ceftazidime-avibactam and/or meropenem-vaborbactam. Clin. Microbiol. Infect. 28, 749–751 (2022).
Bianco, G., Boattini, M., Iannaccone, M., Cavallo, R. & Costa, C. Bloodstream infection by two subpopulations of Klebsiella pneumoniae ST1685 carrying KPC-33 or KPC-14 following ceftazidime/avibactam treatment: considerations regarding acquired heteroresistance and choice of carbapenemase detection assay. J. Antimicrobial Chemother. 75, 3075–3076 (2020).
Urban, L. et al. Real‐time genomics for One Health. Mol. Syst. Biol. 19, e11686 (2023).
The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters, version 13.1, https://www.eucast.org/clinical_breakpoints (2023).
Hong, J., Kang, D. & Kim, D. Performance Evaluation of the Newly Developed In Vitro Rapid Diagnostic Test for Detecting OXA-48-Like, KPC-, NDM-, VIM- and IMP-Type Carbapenemases: The RESIST-5 O.K.N.V.I. Multiplex Lateral Flow Assay. Antibiotics 10, 460 (2021).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Completing bacterial genome assemblies with multiplex MinION sequencing. Micro. Genom. 3, e000132 (2017).
Bernabeu, S., Bonnin, R. A. & Dortet, L. Comparison of three lateral flow immunochromatographic assays for the rapid detection of KPC, NDM, IMP, VIM and OXA-48 carbapenemases in Enterobacterales. J. Antimicrobial Chemother. 77, 3198–3205 (2022).
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Kim, D., Song, L., Breitwieser, F. P. & Salzberg, S. L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729 (2016).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352 (2015).
Grant, J. R. et al. Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 51, W484–W492 (2023).
Wick, R. R., Judd, L. M., Wyres, K. L. & Holt, K. E. Recovery of small plasmid sequences via Oxford Nanopore sequencing. Microb Genom 7 https://doi.org/10.1099/mgen.0.000631 (2021).
Shen, W., Le, S., Li, Y. & Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One 11, e0163962 (2016).
Acknowledgements
We thank the laboratory technicians and physicians at the Institute of Medical Microbiology at the Technical University Munich for their support with phenotypic species identification, susceptibility testing, and sample storage and retrieval. We thank Sam Chorlton for helpful discussions on the BugSeq platform. We thank Niels Pfennigwerth from the German National Reference Center for Gram-Negative Bacteria for his work on identifying the novel KPC-159 subtype. We thank Dr. Youssef Hamway for his help with laboratory work and conceptualization of the study. This project was funded by a Helmholtz Principal Investigator Grant awarded to L.U. DNA extraction was funded by C.P.dC.’s DZIF grant TTU 03.818_01, and initial clinical work by C.P.dC.’s Bavarian Ministry of Commerce grant 47-6665 g/1311/2-MW-2012-0008. Figures 1, 2 and Supplementary Fig. 1 were created with Biorender.com, Fig. 4 was created with ProkSee.com and Bandage.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
E.S. and L.U. conceptualised the project. E.S., L.U. and E.F.N. designed the experiments. E.S., T.R., and A.P. conducted the real-time sequencing experiments under L.U.’s supervision. E.S., T.R., and S.V. conducted the bioinformatic analysis including data curation, formal analysis, and visualisation, under L.U.’s and E.F.N.’s supervision. N.G. consulted on genomic data analysis and interpretation and edited manuscript drafts. N.W., and C.P.dC. provided samples and laboratory equipment for phenotypic testing. C.C. supervised and synthesized the clinical case, as well as the routine and reference laboratory diagnostics. E.S. and L.U. wrote the manuscript with input from all co-authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sauerborn, E., Corredor, N.C., Reska, T. et al. Detection of hidden antibiotic resistance through real-time genomics. Nat Commun 15, 5494 (2024). https://doi.org/10.1038/s41467-024-49851-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-49851-4
- Springer Nature Limited