

Abstract
The NuA3 complex is a major regulator of gene transcription and the cell cycle in yeast. Five core subunits are required for complex assembly and function, but it remains unclear how these subunits interact to form the complex. Here, we report that the Taf14 subunit of the NuA3 complex binds to two other subunits of the complex, Yng1 and Sas3, and describe the molecular mechanism by which the extra-terminal domain of Taf14 recognizes the conserved motif present in Yng1 and Sas3. Structural, biochemical, and mutational analyses show that two motifs are sandwiched between the two extra-terminal domains of Taf14. The head-to-toe dimeric complex enhances the DNA binding activity of Taf14, and the formation of the hetero-dimer involving the motifs of Yng1 and Sas3 is driven by sequence complementarity. In vivo assays in yeast demonstrate that the interactions of Taf14 with both Sas3 and Yng1 are required for proper function of the NuA3 complex in gene transcription and DNA repair. Our findings suggest a potential basis for the assembly of three core subunits of the NuA3 complex, Taf14, Yng1 and Sas3.
Similar content being viewed by others


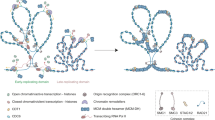
Introduction
The histone acetyltransferase complex NuA3 (nucleosomal acetylation of H3) mediates responses to environmental stresses and is implicated in transcription and cell cycle regulation1,2. The canonical NuA3 complex acetylates histone H3, generating primarily the epigenetic mark H3K14ac3, and consists of five core subunits: the catalytic subunit Sas3 (something about silencing 3) and the nonenzymatic subunits Nto1 (NuA three ORF1), Eaf6 (Esa1p associated factor 6), Yng1 (yeast inhibitor of growth protein 1) and Taf14 (transcription initiation factor subunit 14) (Fig. 1a). The Yng1 and Taf14 subunits contain histone binding modules, the so-called readers of PTMs in histone proteins4,5. The plant homeodomain (PHD) finger of Yng1 binds to (or reads) trimethylated lysine 4 of H3 (H3K4me3)6,7,8,9,10, whereas the YEATS (Yaf9, ENL, AF9, Taf14, Sas5) domain of Taf14 recognizes acylated lysine 9 of H3 (H3K9acyl)11,12,13,14 (Fig. 1b). Previous studies showed that the association of the NuA3 complex with chromatin depends in part on Yng1 and Taf141,6,15, however, little is known about the NuA3 complex assembly and how the complex subunits are linked and cooperate directing its enzymatic activity at specific genomic sites.

a Schematic of the yeast histone acetyltransferase NuA3 complex, which catalyzes acetylation of lysine 14 in histone H3 (H3K14ac). b Domain architecture of Taf14 and Yng1. H3K4me3, H3K9acyl and DNA, the known ligands for the PHD finger of Yng1, the YEATS domain of Taf14, and the linker of Taf14, respectively, are indicated. c Overlayed 1H,15N HSQC spectra of Taf14FL recorded in the presence of increasing amounts of Yng1EBM peptide. The spectra are color coded according to the protein:peptide molar ratio. d MST binding curve for the interaction of Taf14FL with Yng1EBM. The Kd value represents average of three independent measurements ± SEM, and error bars represent SEM for each point. n = 3. e Tryptophan fluorescence binding curves for the interaction of Taf14FL with Yng1EBM. The Kd value represents average of three independent measurements, with error calculated as SD between the runs. f Superimposed 1H,15N HSQC spectra of Taf14ET recorded in the presence of increasing amount of Yng1EBM. The spectra are color coded according to the protein:peptide molar ratio. g Tryptophan fluorescence binding curves for the interaction of Taf14ET with Yng1EBM. The Kd value represents average of three independent measurements, with error calculated as SD between the runs. h, i Superimposed 1H,15N HSQC spectra of Taf14ET (h) or Taf14YEATS (i) recorded in the presence of increasing amounts of Yng1EBM-PHD (h) or Yng1EBM peptide (i). The spectra are color coded according to the protein:ligand molar ratio. The apparent Kd values in (d, e and g) were calculated using a 1:1 stoichiometry. Source data are provided as a Source Data file.
Genetic studies have shown that the interaction of Yng1 with H3K4me3 helps to recruit the NuA3 complex to promoters of actively transcribed target genes, where this complex acetylates H3K14, facilitating transcription initiation6. Yng1 is a small 219-residue protein whose C-terminal PHD finger recognizes H3K4me3 through a canonical ‘aromatic cage’ dependent binding mechanism6. The Yng1 region N-terminal to the PHD finger remains uncharacterized, though AlphaFold modeling (Uniprot) predicts that it may adopt an α-helical fold, likely due to similarity with the N-terminal regions of homologous human inhibitor of growth proteins ING4 and ING516,17.
Taf14 is a core component of several chromatin associating complexes that regulate gene expression, DNA damage response and cytoskeletal organization in yeast cells. These include the general transcription factors complexes TFIID and TFIIF, the ATP-dependent chromatin-remodeling complexes INO80, SWI/SNF and RSC, and the acetyltransferase complex NuA31,18,19,20,21,22,23. Along with the H3K9acyl-binding YEATS domain, Taf14 contains the C-terminal extra-terminal (ET) domain that associates with co-factors in RSC and TFIID complexes, and a short region with DNA-binding activity that links the YEATS and ET domains, hence named a linker24,25,26 (Fig. 1b).
Sas3 belongs to the MYST (MOZ, Ybf2/Sas3, Sas2, TIP60) family of histone acetyltransferases (HATs), distinguished by their catalytic MYST domain and is a yeast counterpart of the human HATs MOZ and MORF27. The central MYST domain of Sas3 is followed by a long stretch of aspartate and glutamate residues which are essential in the association with the transcriptional elongation factor Spt161. Although the selectivity of the MYST domain of Sas3 toward acetylation of H3 and particularly H3K14 has been established in vitro and in vivo1,3, Sas3 remains the least well understood MYST family member.
In this study, we show that the Taf14 subunit of the NuA3 complex interacts with EBMs from other two complex subunits, Yng1 and Sas3, and describe the molecular mechanism by which dimeric EBM is recognized by the ET domain of Taf14. Our findings demonstrate that both interactions of Taf14 with Sas3 and Yng1 are required for proper function of the NuA3 complex in gene transcription and DNA repair and suggest a model for the assembly of the three core subunits of the NuA3 complex.
Results and Discussion
Yng1EBM is recognized by Taf14ET
Taf14 has been shown to bind other subunits in the TFIID and RSC complexes through its ET domain24,25, therefore we explored the idea that Taf14 has a similar function in the NuA3 complex. We analyzed amino acid sequences of the NuA3 core subunits and identified the LLLKI sequence of Yng1 (aa 116-120) as a potential ligand of Taf14. To determine whether Taf14 binds to Yng1, we carried out NMR, microscale thermophoresis (MST), and fluorescence experiments. For NMR, we produced full length uniformly 15N-labeled Taf14 (Taf14FL) and recorded its 1H,15N heteronuclear single quantum coherence (HSQC) spectra. Highly dispersed amide resonances of Taf14FL indicated that the apo-state is folded. Titration of unlabeled Yng1 peptide (aa 113-124 of Yng1) into the Taf14FL sample led to substantial chemical shift perturbations (CSPs) (Fig. 1c and Supplementary Fig. 1). A number of resonances corresponding to the unbound state of Taf14FL decreased in intensity and disappeared, and concomitantly, another set of resonances that corresponds to the bound state of Taf14FL gradually appeared. This pattern of CSPs is characteristic of the slow exchange regime on the NMR time scale and indicates tight binding, which was confirmed by measuring binding affinity of Taf14FL to the Yng1 peptide. The apparent dissociation constant (Kd) determined by MST and tryptophan fluorescence was found to be 1.2 μM and 1.1 μM, respectively (Fig. 1d, e).
We next examined individual domains of Taf14, i.e. YEATS (Taf14YEATS) and ET (Taf14ET), by NMR to establish their roles in the interaction with Yng1. Gradual addition of the Yng1 peptide into 15N-labeled Taf14ET caused CSPs that were on par with CSPs observed in Taf14FL, indicating that Taf14ET binds to this sequence of Yng1, from here on referred to as the ET-binding motif (Yng1EBM) (Fig. 1f and Supplementary Fig. 1). However, a 0.6 μM binding affinity of Taf14ET to Yng1EBM implied a tighter binding compared to the binding of Taf14FL to Yng1EBM (Fig. 1g). These data support the previous observations that while an isolated Taf14ET is more freely available for the interactions with ligands, it is less freely available in the context of the full-length protein due to inhibitory contacts with the linker region of Taf14 (Fig. 1e)25,26.
Full length Yng1 expressed in E. coli formed inclusion bodies, however we were able to produce a soluble construct containing EBM coupled to the PHD finger of Yng1 (aa 100-219 of Yng1, Yng1EBM-PHD) and tested it by NMR. Because similar patterns of CSPs were observed in 1H,15N HSQC spectra of Taf14ET upon titration with either Yng1EBM-PHD or Yng1EBM, we concluded that the PHD finger of Yng1 does not affect binding of Taf14ET to Yng1EBM (Fig. 1h and Supplementary Fig. 2). No CSPs were induced in 15N-labeled Taf14YEATS by Yng1EBM, implying that Taf14YEATS is also not involved in this interaction (Fig. 1i and Supplementary Fig. 2).
Structural basis for the Taf14-Yng1 complex formation
To gain insight into the binding mechanism, we co-crystallized Taf14ET and Yng1EBM and determined the structure of the Taf14ET-Yng1EBM complex refining it to a 1.9 Å resolution (Fig. 2 and Supplementary Table 1). In the crystal structure, Taf14ET forms a head-to-toe dimer, which creates a deep, elongated binding groove for dimeric Yng1EBM, and mass photometry data indicate that the Taf14FL-Yng1EBM complex can also form a dimer in solution (Supplementary Fig. 3). Each Taf14ET monomer, colored light blue and wheat, folds into a three-helix bundle and a two-stranded anti-parallel β-sheet linking helices α2 and α3 (Fig. 2a and Supplementary Fig. 4). The α1 helices of both Taf14ET monomers (α1 and α1’) are packed against each other in an antiparallel manner. The α1-α1’ dimerization interface of Taf14ET is stabilized through van der Waals and hydrophobic contacts. The side chains of the symmetry-related F184 residues in α1 and α1’ are buried in the dimer interface, and although the aromatic rings are not ideally parallel to produce π stacking, they are only ~ 4.5 Å apart.

a Ribbon diagram of the Taf14ET-Yng1EBM complex structure. Dimeric Taf14ET is colored light blue (monomer 1) and wheat (monomer 2), and two monomers of dimeric Yng1EBM are colored yellow and red. b The Taf14ET-Yng1EBM complex with monomer 2 depicted as a surface model colored according to its electrostatic potential (positive potential, blue; negative potential, red). c Ribbon diagram of the Taf14ET-Yng1EBM complex with two molecules of Yng1EBM shown as sticks. Residues involved in the interaction between Taf14ET and Yng1EBM are labeled. Dashed lines represent hydrogen bonds. d Residues in two Yng1EBM are shown as sticks and labeled. Dashed lines represent hydrogen bonds and salt bridges.
Dimeric Yng1EBM forms a long two-stranded anti-parallel β-sheet (one β strand is colored yellow, and another is red), which pairs with the β2 and β2’ strands from Taf14ET monomers and is restrained through extensive intermolecular interactions (Fig. 2b, c and Supplementary Fig. 4). Symmetric β-sheet contacts are observed between the backbone amides of L122, I120 and L118 of each Yng1EBM strand and G218, F220 and I222 of the respective β2 and β2’ strands of Taf14ET (Fig. 2c). Additional hydrogen bonds and electrostatic interactions, engaging residues of the Taf14ET monomer 1 (light blue) and Yng1EBM (yellow), further stabilize the complex. The side chain amino groups of K115, K119 and K124 of Yng1EBM form salt bridges with the carboxyl groups of D223, E219 and E191 of Taf14ET, and the side chain amino group of K123 of Yng1EBM shares a hydrogen bond with the backbone carbonyl group of E216 of Taf14ET. Another hydrogen bond is formed between the backbone amide of L116 of Yng1EBM and the side chain hydroxyl group of Y225 of Taf14ET. A distinct hydrogen bonding network links the Taf14ET monomer 2 (wheat) and Yng1EBM (red): the hydroxyl group of Y225 of Taf14ET is restrained by the backbone amide of L116 of Yng1EBM, and the side chain amide of N121 of Yng1EBM is engaged with the carboxyl group of E219 of Taf14ET. Nine residues in each Yng1EBM strand, from E113 to N121, form a long β-sheet, allowing for a large number of hydrophobic, electrostatic and polar contacts within the Taf14-Yng1 complex (Fig. 2d).
Critical role of hydrophobic contacts
The Yng1EBM-binding groove of the Taf14ET dimer is large and hydrophobic, and two molecules of Taf14ET almost encircle the dimeric Yng1EBM (Fig. 3a). The side chains of L117, L118, I120 and L122 from both strands of Yng1EBM are oriented toward and essentially buried in the hydrophobic groove. To assess the importance of the hydrophobic interface residues in the complex formation, we replaced L116, L118 and I120 of Yng1EBM individually with aspartate, as well as F220, I222 and L224 of Taf14ET individually with arginine and tested the mutated proteins and peptides by NMR (Fig. 3b–g and Supplementary Fig. 5). An almost negligible CSPs in 1H,15N HSQC spectra of wild type Taf14ET upon addition of I120D Yng1EBM indicated that I120 is required for this interaction, and CSPs induced by L118D Yng1EBM or L116D Yng1EBM revealed weak interactions characterized by Kds of 430 μM and 180 μM, respectively (Fig. 3g). Such a considerable decrease in binding activity of Taf14ET suggested that the hydrophobic contacts are the driving force for this complex formation. Mutation of either F220, I222 or L224 in Taf14ET caused protein unfolding, pointing to a critical role of these hydrophobic residues in structural stability of Taf14.

a Electrostatic surface potential of the Taf14ET-Yng1EBM complex, with blue and red colors representing positive and negative charges, respectively. Dimeric Yng1EBM is shown as yellow and red β strands. (b-f) NMR assays to monitor interactions of Taf14ET with the indicated mutants of Yng1EBM. Overlayed 1H,15N HSQC spectra of Taf14ET recorded in the presence of increasing amounts of the mutated Yng1EBM peptides are shown in (b, c, and e). The spectra are color coded according to the protein:peptide molar ratio. Binding curves used to determine Kds by NMR are shown in (d, f). The Kd value represents average of CSPs observed in three amides ± SD. g Binding affinities of Taf14 proteins to the indicated ligands measured by aMST, bfluorescence, and cNMR. nb, no binding (h) Domain architecture of Yng1, a subunit of the NuA3 complex, and of Yng2, a subunit of the NuA4 complex. EBM sequence in Yng1 and the corresponding sequence in Yng2 are shown. The apparent Kd values in (d, f, and g) were calculated using a 1:1 stoichiometry. Source data are provided as a Source Data file.
We note, that human ING4 or homologous ING5 is dimeric16,17, and either is present in the human HAT complexes as a native subunit28. Despite the high degree similarity to Yng1, homologous Yng2, a well-known component of the yeast NuA4 complex, has not been detected in the yeast NuA3 complex. A lack of EBM in the Yng2 sequence may account for the inability of Yng2 to interact with Taf14, therefore precluding Yng2 to be part of the NuA3 complex (Fig. 3h).
Yng1EBM enhances DNA binding activity of Taf14
As Taf14 was found to bind DNA via its linker region25, we tested whether the interaction with Yng1EBM impacts the DNA binding function of Taf14FL using electrophoretic mobility shift assays (EMSA). We mixed increasing amounts of Taf14FL with 601 Widom DNA without and with Yng1EBM and resolved the reaction mixtures on a 8% native polyacrylamide gel. As shown in Fig. 4a and Supplementary Fig. 6, Taf14FL shifts the free 601 DNA band, indicating formation of the Taf14FL-DNA complex. The presence of Yng1EBM enhanced binding of Taf14FL to DNA, supporting previous observations that the DNA binding activity of Taf14 is regulated by Taf14ET25. Because of its association with Taf14ET, the linker is not freely accessible in the context of full-length protein but is released for DNA binding once Taf14ET is ligand-bound25. Our data indicate that Yng1EBM outcompetes the linker for binding to Taf14ET. Unexpectedly, Yng1EBM increased the DNA binding activity of the isolated Taf14ET as well (Fig. 4b and Supplementary Fig. 6). This increase could be due to the extended positively charged patch formed by the dimeric Taf14ET-Yng1EBM complex on the side opposite to the Yng1EBM-binding groove (Fig. 4c, outlined by green dots). We then examined whether the interactions of Taf14ET with Yng1EBM and of Taf14YEATS with crotonylated H3K9 (H3K9cr) affect each other. The binding affinity of Taf14FL to Yng1EBM in the presence of H3K9cr (Kd of 0.9 μM) indicated that the two functions of Taf14FL are independent (Fig. 4d). Together, these results suggest that the formation of the dimeric complex of Taf14FL can bridge two EBM-containing proteins in an antiparallel manner, enhances DNA binding activity of Taf14FL, and does not impede binding of Taf14YEATS to H3K9cr (Fig. 4e).

a, b EMSAs of 601 DNA in the presence of increasing amounts of Taf14FL (a) or Taf14ET (b) with or without Yng1EBM. DNA:protein ratio is shown below gel images. Experiments in (a, b) were performed once and twice, respectively, see also Supplementary Fig. 6. c Rotated views of the Taf14ET-Yng1EBM complex shown in Fig. 3a and colored according to its electrostatic potential (positive potential, blue; negative potential, red). An extended positively charged patch on the surface is outlined by green oval. d Binding curves used to determine apparent Kd for the interaction of Taf14FL with Yng1EBM in the presence of H3K9cr peptide at a 1:10 Taf14FL:H3K9cr molar ratio by tryptophan fluorescence (using a 1:1 stoichiometry). The apparent Kd value represents average of three independent measurements, with error calculated as SD between the runs. e A model for the formation of the complex between two molecules of Taf14 and two proteins containing EBM. H3K9acyl and DNA, the known ligands of the YEATS domain of Taf14, and the linker of Taf14, respectively, are indicated. f Overlap in genes with promoters bound by Taf14 and Yng1. g GO and KEGG category enrichments of genes with promoters co-occupied by Taf14 and Yng1. Circle size represents the approximate number of genes in the enrichment pathway. The exact number of genes is shown for the top pathway in each GO/KEGG category. h Genomic distribution of Taf14, Yng1, and Sas3 binding sites. Source data are provided as a Source Data file.
Taf14, Yng1 and Sas3 localize to gene promoters
To explore the functional cooperation of Taf14 and Yng1, we analyzed ChIP-exo data from Saccharomyces cerevisiae TAP-tagged strains in BY4741 background previously reported for these proteins29. We found that Yng1 occupies promoters of 71 genes, and remarkably 70 of these gene promoters were co-bound by Taf14 (Fig. 4f). These Yng1-Taf14 co-bound genes were markedly enriched for ribosomal functionality and regulators of translation (Fig. 4g and Supplementary Data 1), suggesting that Taf14 and Yng1 co-regulate aspects of these biological processes. Further analysis of genomic occupancies revealed that Taf14, Yng1, and the catalytic subunit of the NuA3 complex, Sas3, each bound nearly exclusively to promoters, with their binding sites mapping to 1096, 71, and 9 unique genes, respectively (Fig. 4h). Furthermore, 6 gene promoters bound by Sas3 were also co-bound by Yng1 and Taf14, suggesting that Sas3, Yng1 and Taf14 cooperate in transcriptional regulation in vivo.
Sas3EBM is recognized by Taf14ET
The catalytic subunit of the NuA3 complex, Sas3, contains the LKVRI sequence (aa 114-118) that might also serve as EBM for Taf1424 (Fig. 5a). Indeed, Taf14FL associated with the Sas3 peptide (aa 111-122, Sas3EBM) with the binding affinity of 21 μM as measured by tryptophan fluorescence (Fig. 5b). The interaction between Taf14ET and Sas3EBM was further confirmed by NMR. Sas3EBM caused large CSPs in 1H,15N HSQC spectra of 15N-labeled Taf14ET, and the Kd value for the Taf14ET-Sas3EBM interaction was found to be 18 μM (Fig. 5c, d and Supplementary Fig. 7). We note that although the binding affinity of Taf14FL or Taf14ET to Sas3EBM was moderately weaker than that of toward Yng1EBM, it was still in the range of typical affinities exhibited by chromatin associating proteins. Much like hydrophobic contacts in the Taf14-Yng1 complex, the hydrophobic contacts were required for the formation of the Taf14-Sas3 complex. The single point substitutions V116D and I118D in Sas3EBM abrogated binding of Taf14ET, and L114D mutation in Sas3EBM substantially reduced this interaction (Kd = 540 μM) (Fig. 5e–h and Supplementary Fig. 7).

a Domain architecture of Yng1, Taf14 and Sas3. Sequence alignment of Yng1EBM and Sas3EBM is shown on the right. b Binding curves used to determine Kd for the interaction of Taf14FL with Sas3EBM by tryptophan fluorescence. The Kd value represents average of three independent measurements, with error calculated as SD between the runs. c Superimposed 1H,15N HSQC spectra of Taf14ET recorded in the presence of increasing amounts of Sas3EBM peptide. The spectra are color coded according to the protein:peptide molar ratio. d Binding curves used to determine Kd for the interaction of Taf14ET with Sas3EBM by tryptophan fluorescence. The Kd value represents average of three independent measurements, with error calculated as SD between the runs. e–h NMR assays to monitor interactions of Taf14ET with the indicated mutants of Sas3EBM. Overlayed 1H,15N HSQC spectra of Taf14ET recorded in the presence of increasing amounts of the mutated Sas3EBM peptides are shown in (e, g, h). The spectra are color coded according to the protein:peptide molar ratio. Binding curves used to determine Kd by NMR are shown in (f). The Kd value represents average of CSPs observed in three amides ± SD. The apparent Kd values in (b, d and f) were calculated using a 1:1 stoichiometry. Source data are provided as a Source Data file.
The finding that two Taf14ET bring together two EBMs suggests a possibility for the formation of the hetero-dimeric complex containing one EBM from Yng1 and another EBM from Sas3 (Fig. 6a). Notably, amino acid sequences of Yng1EBM and Sas3EBM have a high degree of charge-charge and hydrophobicity complementarity when aligned in an antiparallel manner (Fig. 6b). The AlphaFold2-predicted models of the Taf14ET-Yng1EBM and Taf14ET-Sas3EBM complexes suggest that, in the hetero-tetrameric complex, Sas3EBM can occupy the same position as one of Yng1EBM in the crystal structure of the dimeric Taf14ET-Yng1EBM complex (Fig. 6c). Although additional experimental studies are needed to confirm this arrangement, circular dichroism (CD) spectra show that the mixture of Yng1EBM and Sas3EBM in a 1:1 ratio stabilizes the structure of the complex with Taf14ET to the same extent as individual Yng1EBM and Sas3EBM stabilize it (Supplementary Fig. 8). Furthermore, the DNA binding activity of Taf14ET was augmented by either the mixture of EBMs or Yng1EBM in EMSA assays (Supplementary Fig. 9 and Fig. 4b), and comparable molecular mass of Taf14FL was measured in solution in the presence of Yng1EBM or the mixture of EBMs by mass photometry (Supplementary Fig. 3).

a A model for the formation of the complex between two molecules of Taf14 and two EBMs, one EBM from Yng1 and another EBM from Sas3. The ligand of Taf14YEATS, H3K9acyl, is indicated. b Sequence alignment of Yng1EBM and Sas3EBM presented in an antiparallel manner. Dotted lines represent complementary charge-charge contacts. c Overlay of the AlphaFold2-predicted models of the Taf14ET-Yng1EBM (salmon) and Taf14ET-Sas3EBM (gold) complexes with the structure of Taf14ET-Yng1EBM (dimeric Yng1EBM is colored red and yellow as in Fig. 2). d 0.5 OD600 of the indicated yeast strains were 5-fold serially diluted on synthetic complete (SC) medium lacking Uracil (SC -Ura) or SC -Ura plates supplemented with 100 µg/ml 6-azauracil (6-AU) and grown for 4 days. e 0.5 OD600 of the indicated yeast strains were 5-fold serially diluted onto YPD-containing plates supplemented with either 100 mM hydroxyurea (HU), 0.03% methanesulfonate (MMS), or 5 µg/ml Zeocin and grown at 30 °C for 3 days.
Binding of Taf14 to Yng1EBM and Sas3EBM is required for the functional NuA3 complex
To evaluate the biological consequences of the Taf14-Sas3 and Taf14-Yng1 interactions within the NuA3 complex, we generated Saccharomyces cerevisiae TAP-tagged strains in BY4741 background harboring the mutations I118D in Sas3 and I120D in Yng1 that disrupt binding of Taf14. The WT and modified cells were then plated on media containing various compounds or growth conditions to discern drug and growth sensitivity phenotypes associated with the loss of binding of Taf14 to Yng1 or Sas3 in the NuA3 complex (Fig. 6d, e and Supplementary Fig. 10). Given the role of NuA3 in transcription elongation30, we first investigated the impact of elimination of the direct Taf14-Sas3 and Taf14-Yng1 contacts on sensitivity to the transcription elongation inhibitor 6-azauracil (6-AU). As shown in Fig. 6d, strains containing point mutations in either Sas3 or Yng1 that prevent Taf14 association exhibited strong sensitivity to 6-AU, with a near loss of growth observed for the Sas3 I118D mutation. These data reveal that both interactions of Taf14 with Sas3 and Yng1 are required for the proper activity of the NuA3 complex in gene transcription, and elimination of either abrogates this function. The same trend albeit less pronounced was observed under genotoxic agents that induce DNA damage and/or replication stress, such as hydroxyurea (HU) and methyl methanesulfonate (MMS). In these conditions, the strain containing the Sas3 I118D mutation was more sensitive than the strain containing the Yng1 I120D mutation. Interestingly, neither mutant was sensitive to Zeocin but Sas3 I118D was temperature sensitive and showed a severe growth defect at both 37 °C and 16 °C (Supplementary Fig. 10). These phenotypes underscore the importance of the Sas3-Taf14 and Yng1-Taf14 interactions in the biological activities of the NuA3 complex and suggest that the NuA3 complex is essential in maintaining the integrity of DNA replication and DNA repair processes within the cell.
In conclusion, this work underscores the ability of the Taf14 subunit of the NuA3 complex to interact with EBMs from other two complex subunits, Yng1 and Sas3, and demonstrates that both interactions are required for the transcriptional and DNA repair activities of the NuA3 complex. Our structural and functional analyses suggest a model in which the Taf14 subunit may bridge Yng1 and Sas3 through the formation of a heterodimeric assembly. Future cryo-EM structural studies will be important to fully evaluate this model and determine the mechanisms by which Sas3, Yng1 and Taf14 are engaged with other two core subunits of the NuA3 complex and with chromatin.
Methods
Protein expression and purification
The S. cerevisiae Taf14FL (aa 1-244) (UniProt code P35189), Taf14YEATS (aa 1-137), Taf14ET (aa 168-243) and Yng1EBM-PHD (aa 100-219) (UniProt code Q08465) were cloned into pGEX-6P1, pET22b or pCIOX vectors. The F220R, I222R and L224R mutants of Taf14ET were generated using QuikChange II site directed mutagenesis kit (Agilent). Sequences were confirmed by DNA sequencing. All proteins were expressed in Escherichia coli Rosetta-2 (DE3) pLysS cells grown in either Terrific Broth, Luria Broth or in minimal media supplemented with 15NH4Cl (Sigma). Protein production was induced with 0.3 mM IPTG for 18 h at 16 °C. Bacteria were harvested by centrifugation and lysed by sonication. GST-fusion proteins were purified on glutathione agarose 4B beads (Thermo Fisher Sci). The GST-tag was cleaved with either PreScission or tobacco etch virus (TEV) protease. After cleavage, Taf14FL and Taf14YEATS retained the pGEX-6P1vector-derived residues Gly and Pro. His-fusion proteins were incubated with Ni-NTA resin (ThermoFisher), washed and eluted with a gradient of imidazole. When necessary, removal of the His-tag was completed during an overnight dialysis, while incubating the protein with ULP1 protease. Proteins were further purified by size exclusion chromatography (SEC) and concentrated in Millipore concentrators (Millipore).
MST
Microscale thermophoresis (MST) experiments were performed using a Monolith NT.115 instrument (NanoTemper). All experiments were performed using SEC purified His-Taf14ET and His-Taf14FL in a buffer containing 20 mM Tris-HCl (pH 7.5), 150 mM NaCl and 2.5 mM DTT or 0-0.2 mM TCEP as described. To monitor binding between Taf14ET or Taf14FL and Yng1EBM peptide Ac-EPKLLLKINLKK-NH2 (Synpeptide), His-tagged proteins were labeled using a His-Tag Labeling Kit RED-tris-NTA (2nd Generation, Nanotemper), and the concentration of each labeled protein was kept at 10 nM. Dissociation constants were determined using a direct binding assay, in which increasing amounts of unlabeled Yng1EBM were added stepwise. Protein concentration was calculated by measuring the UV absorbance at 280 nm, and peptide concentration was calculated based on the weight of the lyophilized powder. The measurements were performed at 40% LED and medium MST power with 3 s steady state, up to 20 s laser on time and 1 s off time. The apparent Kd values were calculated using MO Affinity Analysis software (NanoTemper) using a 1:1 stoichiometry. Figures were generated in GraphPad PRISM.
Fluorescence spectroscopy
Spectra were recorded at 25 °C on a Fluoromax-3 spectrofluorometer (HORIBA). The samples containing 1 μM Taf14ET or Taf14FL in 50 mM Tris-HCl (pH 6.8), 150 mM NaCl and 1 mM DTT buffer or 50 mM Tris-HCl (pH 7.5), 150 mM NaCl and 0.2 mM TCEP buffer and progressively increasing concentrations of Yng1EBM Ac-EPKLLLKINLKK-NH2 or Sas3EBM Ac-SEELKVRIKYDS-NH2 peptides (Synpeptide) were excited at 295 nm. Emission spectra were recorded between 330 and 360 nm with a 0.5 nm step size and a 1 s integration time. The apparent Kd values were determined using a nonlinear least-squares analysis and a 1:1 stoichiometry as described31 and the equation:
where [L] is concentration of the peptide, [P] is concentration of the protein, ΔI is the observed change of signal intensity and ΔImax is the difference in signal intensity of the free and bound states of the protein. Kd values were averaged over three separate experiments, and error was calculated as the standard deviation between the runs.
NMR experiments
Nuclear magnetic resonance experiments were performed at 298 K on Varian INOVA 900 MHz (for Taf14FL) and 600 MHz (for Taf14ET and Taf14YEATS) spectrometers equipped with cryogenic probes as described32. The NMR samples contained 0.05–0.2 mM uniformly 15N-labeled Taf14 proteins in 50 mM Tris-HCl (pH 6.8–7.5), 150 mM NaCl, 0–5 mM DTT or 1x PBS (pH 6.6–7.5), supplemented with up to an additional 100 mM NaCl, 0–5 mM DTT or 0.2 mM TCEP and 8-10% D2O. Binding was characterized by monitoring chemical shift changes in 1H,15N HSQC spectra of the proteins induced by the addition of wild type or mutated Yng1EBM (aa 113-124), Sas3EBM (aa 111-122) and Yng1EBM-PHD (aa 100-219). NMR data were processed and analyzed using NMRPipe as previously described32, with the apparent Kds being determined by a nonlinear least-squares analysis and a 1:1 stoichiometry in GraphPad Prism using the equation:
Where [L] is the concentration of the peptide, [P] is concentration of the protein, \(\Delta \delta\) is the observed chemical shift change, and \(\Delta \delta\)max is the normalized chemical shift change at saturation. Normalized chemical shift changes were calculated using the equation \(\delta=\sqrt{{\left(\Delta \delta H\right)}^{2}+\,{(\Delta \delta N/5)}^{2}}\) (3), where \(\Delta \delta\) is the change in chemical shift in parts per million (ppm).
X-ray crystallography
Taf14ET was concentrated to ~2 mM in 50 mM Tris (pH 7.5), 150 mM NaCl. The protein was incubated with Yng1EBM peptide (Synpeptide) at a molar ratio of 1:2.5, for 16 h at 4 °C. The protein-peptide mixture was concentrated to 10 mg/ml. Crystals were grown using sitting-drop vapor diffusion method at 23 °C by mixing 0.3 µL of protein with 0.3 µL of well solution composed of 40% PEG 400, 0.1 M Tris pH 8.5, 0.2 M LiSO4. X-ray diffraction data were collected on a beamline 4.2.2 at Advance Light Source. The diffraction data were processed using the iMosflm program 633, then scaled by Aimless Pointless in the CCP4 software suite 734. The structure was determined by molecular replacement using the program PHASER35, the Taf14ET structure (PDB ID: 7UHE) was used as a search model. The models from the molecular replacement were built using Crystallographic Object-Oriented Toolkit program 936 and subsequently subjected to refinement using PHENIX software37. Crystallography diffraction data collection and refinement statistics are summarized in Supplementary Table 1.
DNA purification and preparation
Double stranded DNA containing the 147 bp 601 Widom DNA sequence in the pJ201 plasmid (32x) was transformed into DH5α cells. The plasmids containing the 601 sequence were amplified in LB media and extracted using the QIAprep® Spin Miniprep kit (QIAGEN). Amplification of double stranded 601 Widom DNA was performed by polymerase chain reaction (PCR) using Q5® High-Fidelity 2X master mix (New England Biolab). PCR product was verified by agarose gel and cleaned up by MinElute® Gel Extraction Kit (QIAGEN).
EMSA
EMSAs were performed by mixing increasing amounts of Taf14 proteins with 0.25 pmol/lane of 601 DNA in 25 mM Tris-HCl pH 7.5, 25-100 mM NaCl, 0.2 mM ethylenediaminetetraacetic acid (EDTA), 10% glycerol and 1 mM DTT in 10 µL reaction volume. The Taf14 proteins were either preincubated with Yng1EBM at a 1:2.5 molar ratio or with Yng1EBM and Sas3EBM at a 1:1:1 molar ratio for a minimum of 1 h on ice or used without peptides prior to addition of DNA. Reaction mixtures were incubated at 4 °C for 10 min (2-2.5 µl of loading dye was added to each sample) and loaded onto an 8% native polyacrylamide gel. Electrophoresis was performed in 0.5x Tris-borate-EDTA (TBE) at 80–120 V on ice. The gels were stained with SYBR Gold (Thermo Fisher Sci) and visualized by Blue LED (UltraThin LED Illuminator-GelCompany).
Mass photometry
Mass photometry experiments were conducted using a Refeyn TwoMP mass photometer (Refeyn Ltd, Oxford, UK) to monitor the formation of the dimeric states for apo and EBM-bound Taf14FL. Coverslips (24 mm×50 mm, Thorlabs Inc.) and silicon gaskets (Grace Bio-Labs) were cleaned with several rinses of ultrapure water and HPLC-grade isopropanol and then dried with a clean air stream. Measurements were performed at room temperature in buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM TCEP). 50 nM β-amylase in the same buffer was used as calibration standard (56 kDa, 112 kDa, and 224 kDa). Samples for measurements were prepared by rapid in-drop dilution. Before each measurement, 19 µl of buffer were added to a well, and the focus was set and locked. 1 µl of Taf14FL sample in the absence and presence of Yng1EBM (1:1 molar ratio) or the mixture of Yng1EBM and Sas3EBM (1:1:5 molar ratio) was added to the drop for a final concentration of 100 nM. Samples were briefly mixed, and movies were captured immediately for 60 s (2800 frames) using AcquireMP software (version 2024 R1; Refeyn Ltd). Data were processed using DiscoverMP (version 2024 R1; Refeyn Ltd).
Circular dichroism
Circular dichroism spectra were acquired on a Jasco J-815 spectropolarimeter at 25 °C over the range of 190–250 nm in 1 nm steps with 2 s integration time, 1 nm bandwidth, 50 nm/min scan speed, and 1 mm cell length. Spectra were collected using a 1 mm cuvette. The Taf14 samples contained 12 µM Taf14ET without and with 12 µM Sas3EBM, 12 µM Yng1EBM, or the mixture of 6 µM Sas3EBM and 6 µM Yng1EBM in 25 mM sodium phosphate and 50 mM sodium fluoride buffer pH 7.5. The peptide samples contained 12 µM Sas3EBM, 12 µM Yng1EBM, or the mixture of 6 µM Sas3EBM and 6 µM Yng1EBM in the same buffer.
ChIP-exo analysis
ChIP-exo peak files for Taf14, Yng1 and Sas3 generated using Saccharomyces cerevisiae TAP-tagged strains in BY4741 background were downloaded from the gene expression omnibus, accession number GSE147927 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101099]29. ChIPseeker was used to obtain genomic distributions and gene annotations of binding sites in peak list for each factor. gProfiler38 was then used to identify GO and KEGG enrichments in the list of genes with promoters co-bound by Taf14 and Yng1.
Site-directed mutagenesis to generate BY4741 yng1 I120D-TAP and BY4741sas3 I118D-TAP
Genomic DNA of the respective TAP-tagged wild type Saccharomyces cerevisiae strains under study (Supplementary Fig. 10) was used to PCR amplify full-length YNG1-TAP and SAS3-TAP from the respective strains, and these fragments were cloned into pTOPO vector for site-directed mutagenesis. For site-directed mutagenesis, the oligonucleotides pairs were designed to make the indicated amino acid codon change in the respective genes of interest. Following the QuickChangeTM standard protocol as recommended by manufacturer (Agilent), the codon changes, I120D in Yng1 and I118D in Sas3, were confirmed by sequencing. The mutated yeast fragments, yng1 I120D-TAP and sas3 118D-TAP, were excised from the pTOPO vector backbone by restriction enzyme digestion, and then purified. Yeast cells BY4741 yng1(I120D) and BY4741 sas3(I118D) were transformed with the purified gene fragments as previously described39. Following selection and genotype confirmation, the respective genomic integrants were validated by sequencing.
Cell spotting assays
Saccharomyces cerevisiae strains (Supplementary Fig. 10) were cultivated in 10 ml YPD media overnight. For cell spotting assays, cells at 0.5 OD600 were subjected to fivefold serial dilutions and spotted onto the designated plates for the indicated temperatures and days. For 6-AU studies, strains were spotted on synthetic complete (SC) media lacking uracil (Ura), or SC -Ura plates supplemented with 6-azauracil (6-AU) at 100 µg/ml. All other treatments were performed using YPD-containing plates supplemented with either 0.03% MMS, 100 mM HU, or 5 µg/ml Zeocin plates at the indicated days or temperature.
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism 8 software. MST data are presented as average of three independent measurements ± SEM, and tryptophan fluorescence data are presented as average of three independent measurements ± SD. The apparent Kd values were calculated using a 1:1 stoichiometry.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Coordinates and structure factors have been deposited in the Protein Data Bank under the accession code 8U77. All other relevant data supporting the key findings of this study are available within the article and its Supplementary Information. Source data are provided with this paper.
References
John, S. et al. The something about silencing protein, Sas3, is the catalytic subunit of NuA3, a yTAF(II)30-containing HAT complex that interacts with the Spt16 subunit of the yeast CP (Cdc68/Pob3)-FACT complex. Genes Dev. 14, 1196–1208 (2000).
Howe, L. et al. Yng1p modulates the activity of Sas3p as a component of the yeast NuA3 histone acetyltransferase complex. Mol. Cell. Biol. 22, 5047–5053 (2002).
Howe, L. et al. Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev. 15, 3144–3154 (2001).
Taverna, S. D., Li, H., Ruthenburg, A. J., Allis, C. D. & Patel, D. J. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14, 1025–1040 (2007).
Musselman, C. A., Lalonde, M. E., Cote, J. & Kutateladze, T. G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227 (2012).
Taverna, S. D. et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. cell 24, 785–796 (2006).
Peña, P. V. et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442, 100–103 (2006).
Martin, D. G. et al. The Yng1p plant homeodomain finger is a methyl-histone binding module that recognizes lysine 4-methylated histone H3. Mol. Cell. Biol. 26, 7871–7879 (2006).
Shi, X. et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 442, 96–99 (2006).
Shi, X. et al. Proteome-wide analysis in saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J. Biol. Chem. 282, 2450–2455 (2007).
Shanle, E. K. et al. Association of Taf14 with acetylated histone H3 directs gene transcription and the DNA damage response. Genes Dev. 29, 1795–1800 (2015).
Andrews, F. H., Shanle, E. K., Strahl, B. D. & Kutateladze, T. G. The essential role of acetyllysine binding by the YEATS domain in transcriptional regulation. Transcription 7, 14–20 (2016).
Andrews, F. H. et al. The Taf14 YEATS domain is a reader of histone crotonylation. Nat. Chem. Biol. 12, 396–398 (2016).
Klein, B. J. et al. Yaf9 subunit of the NuA4 and SWR1 complexes targets histone H3K27ac through its YEATS domain. Nucleic Acids Res. 46, 421–430 (2018).
Martin, D. G., Grimes, D. E., Baetz, K. & Howe, L. Methylation of histone H3 mediates the association of the NuA3 histone acetyltransferase with chromatin. Mol. Cell. Biol. 26, 3018–3028 (2006).
Culurgioni, S. et al. Crystal structure of inhibitor of growth 4 (ING4) dimerization domain reveals functional organization of ING family of chromatin-binding proteins. J. Biol. Chem. 287, 10876–10884 (2012).
Ormaza, G. et al. The tumor suppressor ING5 is a dimeric, bivalent recognition molecule of the histone H3K4me3 mark. J. Mol. Biol. 431, 2298–2319 (2019).
Poon, D. et al. Identification and characterization of a TFIID-like multiprotein complex from saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 92, 8224–8228 (1995).
Henry, N. L. et al. TFIIF-TAF-RNA polymerase II connection. Genes Dev. 8, 2868–2878 (1994).
Shen, X. Preparation and analysis of the INO80 complex. Methods Enzymol. 377, 401–412 (2004).
Cairns, B. R., Henry, N. L. & Kornberg, R. D. TFG/TAF30/ANC1, a component of the yeast SWI/SNF complex that is similar to the leukemogenic proteins ENL and AF-9. Mol. Cell. Biol. 16, 3308–3316 (1996).
Kabani, M., Michot, K., Boschiero, C. & Werner, M. Anc1 interacts with the catalytic subunits of the general transcription factors TFIID and TFIIF, the chromatin remodeling complexes RSC and INO80, and the histone acetyltransferase complex NuA3. Biochem. Biophys. Res. Commun. 332, 398–403 (2005).
Dahan, O. & Kupiec, M. The Saccharomyces cerevisiae gene CDC40/PRP17 controls cell cycle progression through splicing of the ANC1 gene. Nucleic acids Res. 32, 2529–2540 (2004).
Chen, G. et al. Taf14 recognizes a common motif in transcriptional machineries and facilitates their clustering by phase separation. Nat. Commun. 11, 4206 (2020).
Klein, B. J. et al. Taf2 mediates DNA binding of Taf14. Nat. Commun. 13, 3177 (2022).
Nguyen, M. C., Wang, D., Klein, B. J., Chen, Y. & Kutateladze, T. G. Differences and similarities in recognition of co-factors by Taf14. Biochim. Biophys. Acta Gene Regul. Mech. 1866, 194961 (2023).
Lafon, A., Chang, C. S., Scott, E. M., Jacobson, S. J. & Pillus, L. MYST opportunities for growth control: yeast genes illuminate human cancer gene functions. Oncogene 26, 5373–5384 (2007).
Doyon, Y. et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell 21, 51–64 (2006).
Badjatia, N. et al. Acute stress drives global repression through two independent RNA polymerase II stalling events in saccharomyces. Cell Rep. 34, 108640 (2021).
Gilbert, T. M. et al. A PWWP domain-containing protein targets the NuA3 acetyltransferase complex via histone H3 lysine 36 trimethylation to coordinate transcriptional elongation at coding regions. Mol. Cell. Proteom.: MCP 13, 2883–2895 (2014).
Tencer, A. H. et al. Molecular basis for nuclear accumulation and targeting of the inhibitor of apoptosis BIRC2. Nat. Struct. Mol. Biol. 30, 1265–1274 (2023).
Gatchalian, J. et al. Dido3 PHD modulates cell differentiation and division. Cell Rep. 4, 148–158 (2013).
Battye, T. G., Kontogiannis, L., Johnson, O., Powell, H. R. & Leslie, A. G. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D., Biol. Crystallogr. 67, 271–281 (2011).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Reimand, J., Kull, M., Peterson, H., Hansen, J. & Vilo, J. g:Profiler-a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35, W193–W200 (2007).
Gietz, R. D. & Schiestl, R. H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 (2007).
Acknowledgements
This work was supported in part by NIH grants HL151334, CA252707, GM125195, GM135671 and AG067664 to T.G.K., GM118760 and CA221306 to S.D.T., GM126900 to B.D.S, and CA279317 to M.A.B. S.D.T. research is supported in part by a seed grant from the Vice Chancellor for Research and Innovation at UAMS. Mass photometry experiments were performed in the Shared Instruments Pool (RRID: SCR_018986) in the Department of Biochemistry at the University of Colorado Boulder. CU Anschutz NMR spectrometers are supported by NIH grants P30CA046934 and S10 OD025020.
Author information
Authors and Affiliations
Contributions
M.C.N., H.R., A.R., P.W., D.C.B., B.J.K. and T.M.G. performed experiments and together with A.H.E., G.Z., M.A.B., B.D.S., S.D.T. and T.G.K. analyzed the data and prepared figures. T.G.K. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
B.D.S. is a co-founder of EpiCypher, Inc. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Francisco Blanco and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, M.C., Rostamian, H., Raman, A. et al. Molecular insight into interactions between the Taf14, Yng1 and Sas3 subunits of the NuA3 complex. Nat Commun 15, 5335 (2024). https://doi.org/10.1038/s41467-024-49730-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-49730-y
- Springer Nature Limited